Effects of GrandFusion Diet on Cognitive Impairment in Transgenic Mouse Model of Alzheimer’s Disease

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Animals
2.2. Diet Treatment
2.3. Immunohistochemistry Staining
2.4. Aβ Peptide and NGF Analysis
2.5. CTFβ and sAβPPα Analyses
2.6. ELISA Analysis
2.7. Morris Water Maze Test
2.8. Statistical Analysis
3. Results
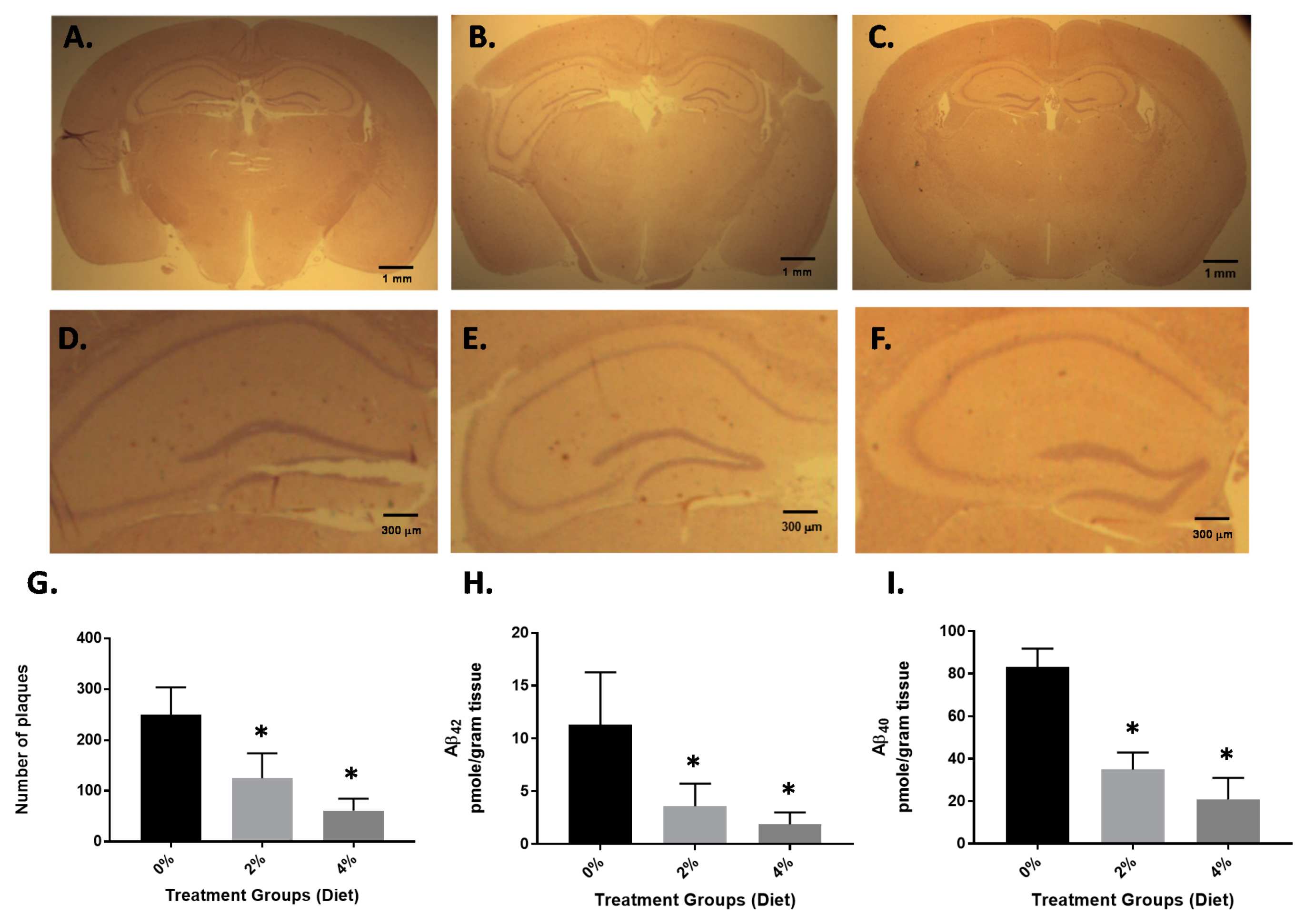
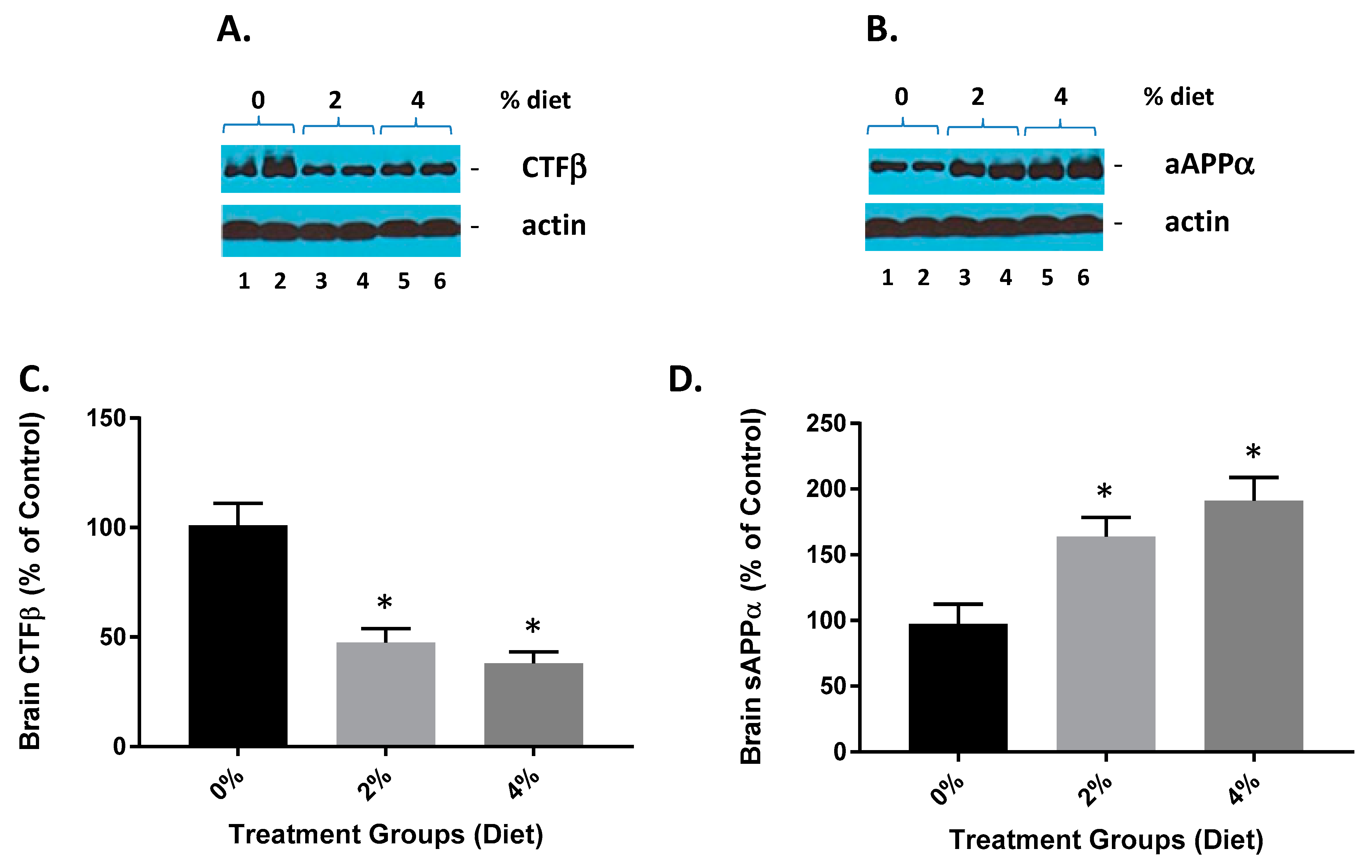
3.1. Impact of GF Diet on Amyloid Pathology in the APP/PS-1 Mice
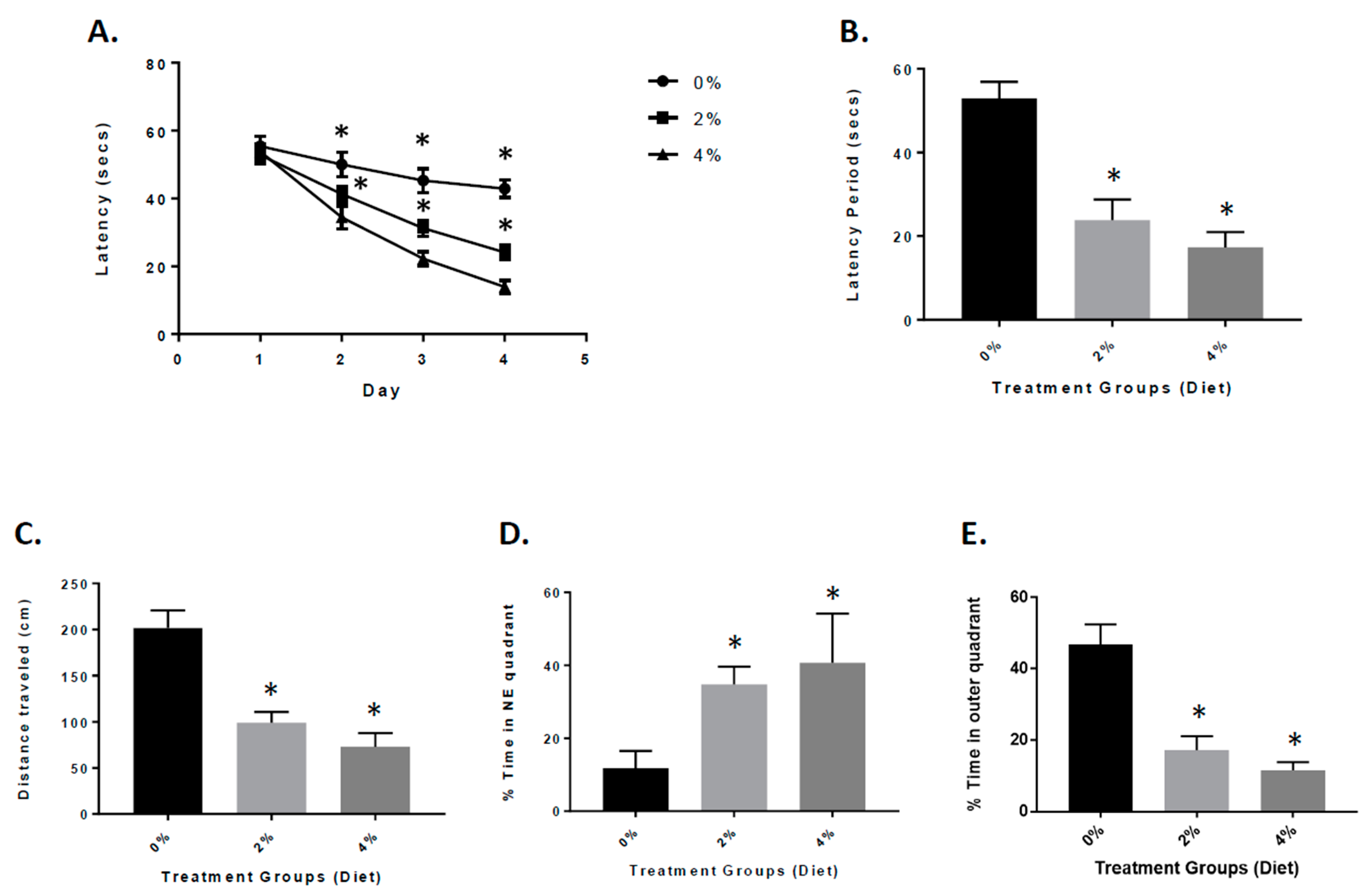
3.2. Attenuation of Neurological Deficits in APP/PS-1 Mice during GF Diet Treatment
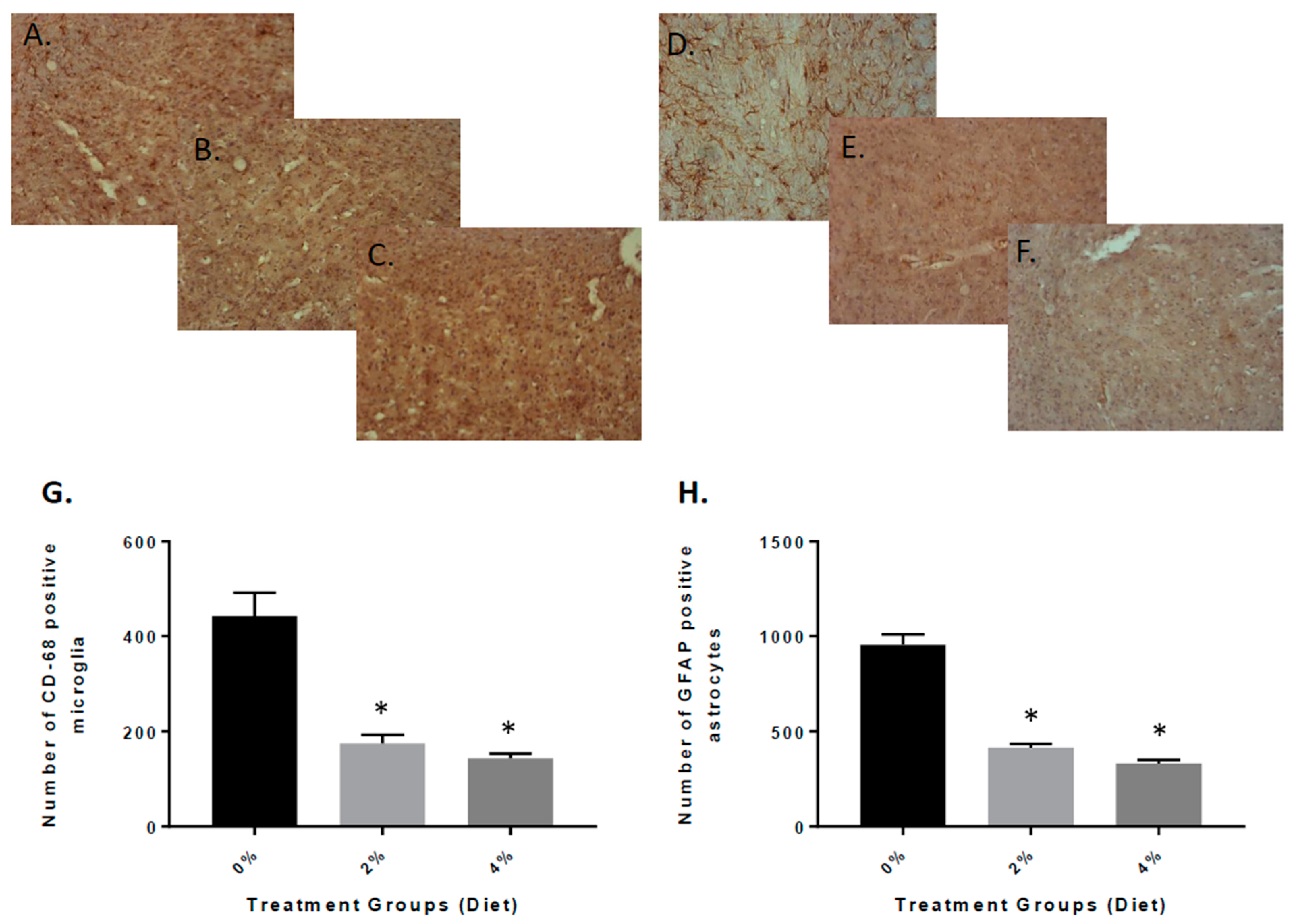
3.3. Reduced Inflammation in APP/PS-1 Mice with GF Diets
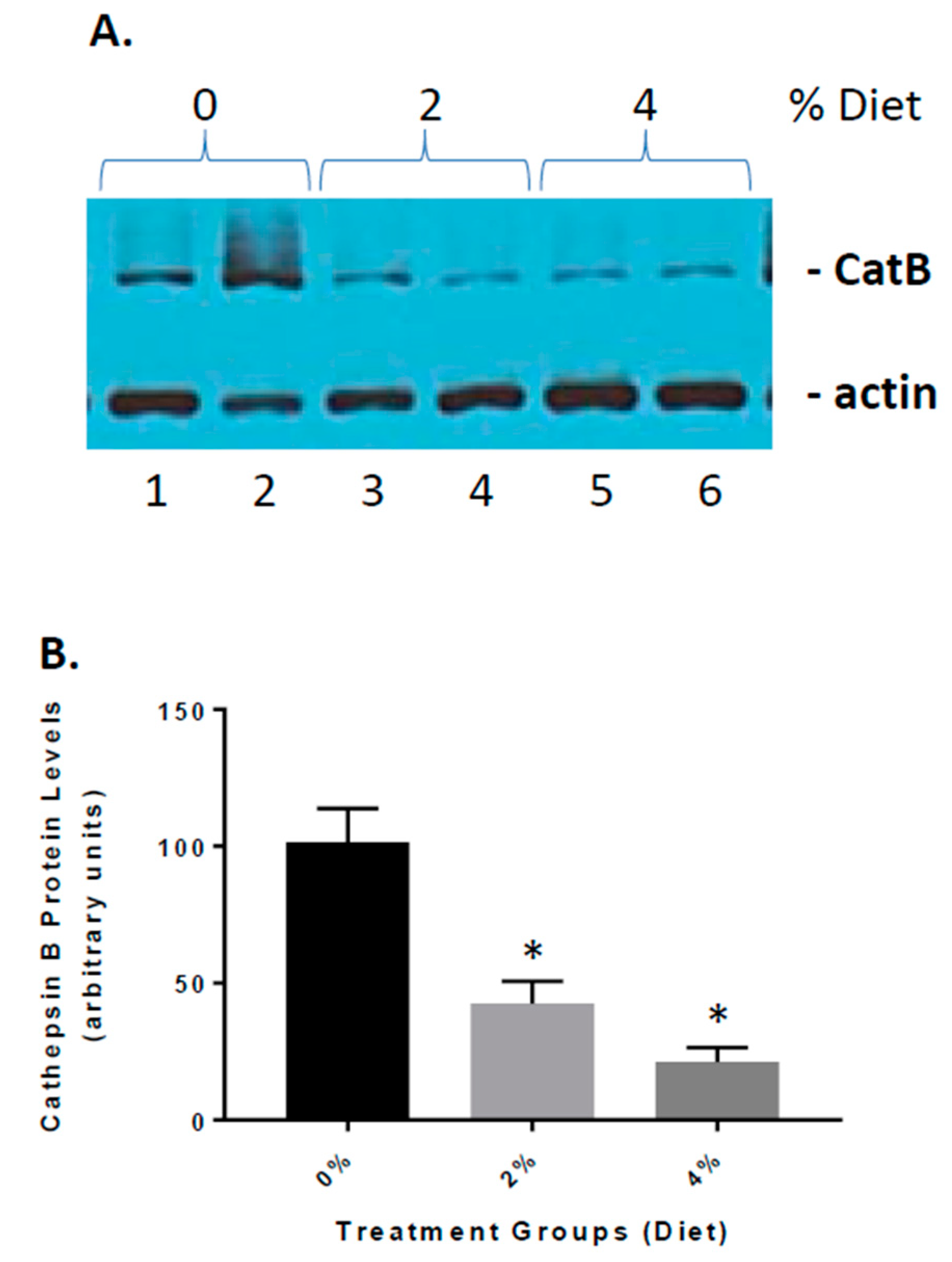
3.4. Reduced Cathepsin B Expression in GF Diet-Treated APP/PS-1 Mice
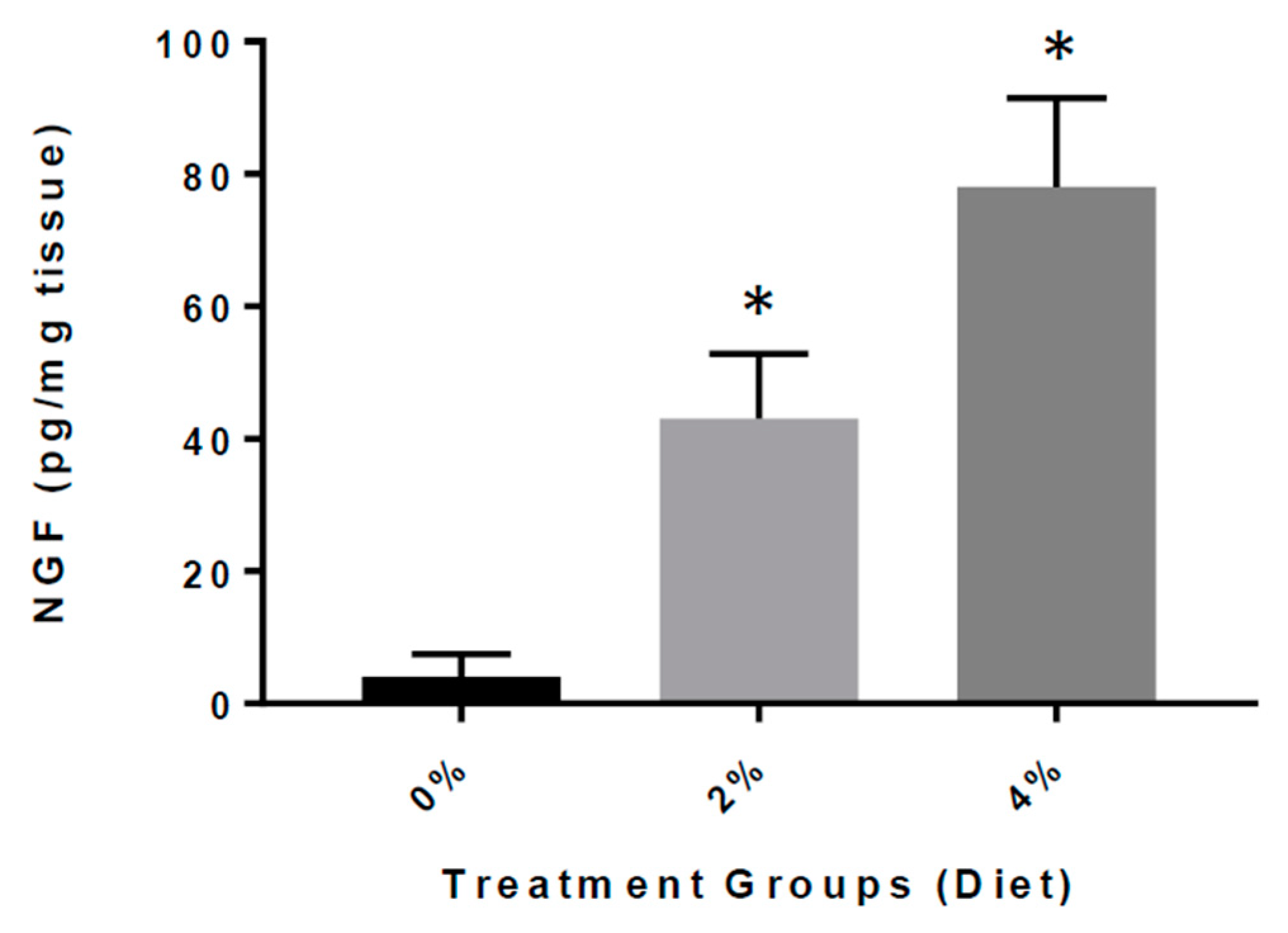
3.5. Increased Expression of NGF in GF Diet-Treated Mice
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Mayeux, R. Clinical practice. Early Alzheimer’s disease. N. Engl. J. Med. 2010, 362, 2194–2201. [Google Scholar] [CrossRef] [PubMed]
- Castellani, R.J.; Plascencia-Villa, G.; Perry, G. The amyloid cascade and Alzheimer’s disease therapeutics: Theory versus observation. Lab. Investig. 2019, 99, 958–970. [Google Scholar] [CrossRef] [PubMed]
- Guarino, A.; Favieri, F.; Boncompagni, I.; Agostini, F.; Cantone, M.; Casagrande, M. Executive Functions in Alzheimer Disease: A Systematic Review. Front. Aging Neurosci. 2019, 10, 437. [Google Scholar] [CrossRef] [PubMed]
- Sharma, P.; Tripathi, A.; Tripathi, P.N.; Prajapati, S.K.; Seth, A.; Tripathi, M.K.; Srivastava, P.; Tiwari, V.; Krishnamurthy, S.; Shrivastava, S.K. Design and development of multitarget-directed N-Benzylpiperidine analogs as potential candidates for the treatment of Alzheimer’s disease. Eur. J. Med. Chem. 2019, 167, 510–524. [Google Scholar] [CrossRef]
- Selkoe, D.J. Developing preventive therapies for chronic diseases: Lessons learned from Alzheimer’s disease. Nutr. Rev. 2007, 65, S329–S343. [Google Scholar] [CrossRef]
- Holmes, C.; Cunningham, C.; Zotova, E.; Woolford, J.; Dean, C.; Kerr, S.; Culliford, D.; Perry, V.H. Systemic inflammation and disease progression in Alzheimer disease. Neurology 2009, 73, 768–775. [Google Scholar] [CrossRef] [Green Version]
- Solfrizzi, V.; Agosti, P.; Lozupone, M.; Custodero, C.; Schilardi, A.; Valiani, V.; Santamato, A.; Sardone, R.; Dibello, V.; Di Lena, L.; et al. Nutritional interventions and cognitive-related outcomes in patients with late-life cognitive disorders: A systematic review. Neurosci. Biobehav. Rev. 2018, 95, 480–498. [Google Scholar] [CrossRef]
- Serrano-Pozo, A.; Growdon, J.H. Is Alzheimer’s Disease Risk Modifiable? J. Alzheimers Dis. 2019, 67, 795–819. [Google Scholar] [CrossRef]
- Alkhouli, M.F.; Hung, J.; Squire, M.; Anderson, M.; Castro, M.; Babu, J.R.; Al-Nakkash, L.; Broderick, T.L.; Plochocki, J.H. Exercise and resveratrol increase fracture resistance in the 3xTg-AD mouse model of Alzheimer’s disease. BMC Complement Altern. Med. 2019, 19, 39. [Google Scholar] [CrossRef] [Green Version]
- Hill, E.; Goodwill, A.M.; Gorelik, A.; Szoeke, C. Diet and biomarkers of Alzheimer’s disease: A systematic review and meta-analysis. Neurobiol. Aging 2018, 76, 45–52. [Google Scholar] [CrossRef]
- Broom, G.M.; Shaw, I.C.; Rucklidge, J.J. The ketogenic diet as a potential treatment and prevention strategy for Alzheimer’s disease. Nutrition 2019, 60, 118–121. [Google Scholar] [CrossRef] [PubMed]
- Muñoz Fernández, S.S.; Lima Ribeiro, S.M. Nutrition and Alzheimer Disease. Clin. Geriatr. Med. 2018, 34, 677–697. [Google Scholar] [CrossRef] [PubMed]
- Scarmeas, N.; Anastasiou, C.A.; Yannakoulia, M. Nutrition and prevention of cognitive impairment. Lancet Neurol. 2018, 17, 1006–1015. [Google Scholar] [CrossRef]
- Yu, J.; Zhu, H.; Gattoni-Celli, S.; Taheri, S.; Kindy, M.S. Dietary supplementation of GrandFusion(®) mitigates cerebral ischemia-induced neuronal damage and attenuates inflammation. Nutr. Neurosci. 2016, 19, 290–300. [Google Scholar] [CrossRef] [PubMed]
- Yu, J.; Zhu, H.; Perry, S.; Taheri, S.; Kindy, M.S. Daily supplementation with GrandFusion® improves memory and learning in aged rats. Aging 2017, 9, 1041–1054. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yu, J.; Zhu, H.; Taheri, S.; Mondy, W.; Perry, S.; Kindy, M.S. Impact of nutrition on inflammation, tauopathy, and behavioral outcomes from chronic traumatic encephalopathy. J. Neuroinflamm. 2018, 15, 277. [Google Scholar] [CrossRef] [PubMed]
- Yu, J.; Zhu, H.; Taheri, S.; Perry, S.; Kindy, M.S. The Effect of Diet on Improved Endurance in Male C57BL/6 Mice. Nutrients 2018, 10, 1101. [Google Scholar] [CrossRef] [Green Version]
- Yu, J.; Zhu, H.; Taheri, S.; Monday, W.L.; Perry, S.; Kindy, M.S. Reduced Neuroinflammation and Improved Functional Recovery after Traumatic Brain Injury by Prophylactic Diet Supplementation in Mice. Nutrients 2019, 11, 299. [Google Scholar] [CrossRef] [Green Version]
- Hook, G.; Yu, J.; Toneff, T.; Kindy, M.; Hook, V. Brain pyroglutamate amyloid-β is produced by cathepsin B and is reduced by the cysteine protease inhibitor E64d, representing a potential Alzheimer’s disease therapeutic. J. Alzheimers Dis. 2014, 41, 129–149. [Google Scholar] [CrossRef] [Green Version]
- Jankowsky, J.L.; Fadale, D.J.; Anderson, J.; Xu, G.M.; Gonzales, V.; Jenkins, N.A.; Copeland, N.G.; Lee, M.K.; Younkin, L.H.; Wagner, S.L.; et al. Mutant presenilins specifically elevate the levels of the 42 residue beta-amyloid peptide in vivo: Evidence for augmentation of a 42-specific gamma secretase. Hum. Mol. Genet. 2004, 13, 159–170. [Google Scholar] [CrossRef] [Green Version]
- Savonenko, A.; Xu, G.M.; Melnikova, T.; Morton, J.L.; Gonzales, V.; Wong, M.P.; Price, D.L.; Tang, F.; Markowska, A.L.; Borchelt, D.R. Episodic-like memory deficits in the APPswe/PS1dE9 mouse model of Alzheimer’s disease: Relationships to beta-amyloid deposition and neurotransmitter abnormalities. Neurobiol. Dis. 2005, 18, 602–617. [Google Scholar] [CrossRef] [PubMed]
- Hook, V.Y.; Kindy, M.; Hook, G. Inhibitors of cathepsin B improve memory and reduce Abeta in transgenic Alzheimer’s disease mice expressing the wild-type, but not the Swedish mutant, betasecretase AβPP site. J. Biol. Chem. 2008, 283, 7745–7753. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Elvington, A.; Atkinson, C.; Zhu, H.; Yu, J.; Takahashi, K.; Stahl, G.L.; Kindy, M.S.; Tomlinson, S. The alternative complement pathway propagates inflammation and injury in murine ischemic stroke. J. Immunol. 2012, 189, 4640–4647. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- El-Amouri, S.S.; Zhu, H.; Yu, J.; Gage, F.H.; Verma, I.M.; Kindy, M.S. Neprilysin: An enzyme candidate to treat Alzheimer’s disease. Am. J. Pathol. 2008, 172, 1342–1354. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yu, J.; Gattoni-Celli, M.; Zhu, H.; Bhat, N.R.; Sambamurti, K.; Gattoni-Celli, S.; Kindy, M.S. Vitamin D3-Enriched Diet Correlates with a Decrease of Amyloid Plaques in the Brain of APP Transgenic Mice. J. Alz. Dis. 2011, 25, 295–307. [Google Scholar]
- Zhang, L.; Fu, X.H.; Yu, Y.; Shui, R.H.; Li, C.; Zeng, H.Y.; Qiao, Y.L.; Ni, L.Y.; Wang, Q. Treatment with CA-074Me, a Cathepsin B inhibitor, reduces lung interstitial inflammation and fibrosis in a rat model of polymyositis. Lab. Investig. 2015, 95, 65–77. [Google Scholar] [CrossRef] [Green Version]
- Kochanek, K.D.; Murphy, S.L.; Xu, J.; Tejada-Vera, B. Deaths: Final Data for 2014. Natl. Vital Stat. Rep. 2016, 65, 1–122. [Google Scholar]
- Karran, E.; De Strooper, B. The amyloid cascade hypothesis: Are we poised for success or failure? J. Neurochem. 2016, 2, 237–252. [Google Scholar] [CrossRef]
- Holtzman, D.M.; Mandelkow, E.; Selkoe, D.J. Alzheimer disease in 2020. Cold Spring Harb. Perspect. Med. 2012, 2, a011585. [Google Scholar] [CrossRef]
- Hung, S.Y.; Fu, W.M. Drug candidates in clinical trials for Alzheimer’s disease. J. Biomed. Sci. 2017, 24, 47. [Google Scholar] [CrossRef]
- Atri, A. The Alzheimer’s Disease Clinical Spectrum: Diagnosis and Management. Med. Clin. North Am. 2019, 103, 263–293. [Google Scholar] [CrossRef] [PubMed]
- Liang, J.; Li, J.; Jia, R.; Wang, Y.; Wu, R.; Zhang, H.; Hang, L.; Xu, Y. Identification of the optimal cognitive drugs among Alzheimer’s disease: A Bayesian meta-analytic review. Clin. Interv. Aging 2018, 13, 2061–2073. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Querfurth, H.W.; LaFerla, F.M. Alzheimer’s disease. N. Engl. J. Med. 2010, 362, 329–344. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Walsh, D.M.; Selkoe, D.J. A beta oligomers—A decade of discovery. J. Neurochem. 2007, 101, 1172–1184. [Google Scholar] [CrossRef] [PubMed]
- Selkoe, D.J. The therapeutics of Alzheimer’s disease: Where we stand and where we are heading. Ann. Neurol. 2013, 74, 328–336. [Google Scholar] [CrossRef] [PubMed]
- Selkoe, D.J.; Hardy, J. The amyloid hypothesis of Alzheimer’s disease at 25 years. EMBO Mol. Med. 2016, 8, 595–608. [Google Scholar] [CrossRef]
- Li, S.; Shankar, G.M.; Selkoe, D.J. How do Soluble Oligomers of Amyloid beta-protein Impair Hippocampal Synaptic Plasticity? Front. Cell. Neurosci. 2010, 4, 5. [Google Scholar]
- Sperling, R.A.; Dickerson, B.C.; Pihlajamaki, M.; Vannini, P.; LaViolette, P.S.; Vitolo, O.V.; Hedden, T.; Becker, J.A.; Rentz, D.M.; Selkoe, D.J.; et al. Functional alterations in memory networks in early Alzheimer’s disease. Neuromol. Med. 2010, 12, 27–43. [Google Scholar] [CrossRef] [Green Version]
- Olsen, I.; Singhrao, S.K. Inflammasome Involvement in Alzheimer’s Disease. J. Alzheimers Dis. 2016, 54, 45–53. [Google Scholar] [CrossRef] [Green Version]
- Hook, V.; Hook, G.; Kindy, M.S. Pharmacogenetic Features of Inhibitors to Cathepsin B that Improve Memory Deficit and Reduces Beta-Amyloid Related to Alzheimer’s Disease. Biol. Chem. 2010, 391, 861–872. [Google Scholar] [CrossRef]
- Nordengen, K.; Kirsebom, B.E.; Henjum, K.; Selnes, P.; Gísladóttir, B.; Wettergreen, M.; Torsetnes, S.B.; Grøntvedt, G.R.; Waterloo, K.K.; Aarsland, D.; et al. Glial activation and inflammation along the Alzheimer’s disease continuum. J. Neuroinflamm. 2019, 16, 46. [Google Scholar] [CrossRef] [PubMed]
- Su, C.; Zhao, K.; Xia, H.; Xu, Y. Peripheral inflammatory biomarkers in Alzheimer’s disease and mild cognitive impairment: A systematic review and meta-analysis. Psychogeriatrics 2019, 19, 300–309. [Google Scholar] [CrossRef] [PubMed]
- Akiyama, H.; Arai, T.; Kondo, H.; Tanno, E.; Haga, C.; Ikeda, K. Cell mediators of inflammation in the Alzheimer disease brain. Alzheimer Dis. Assoc. Disord. 2000, 14 (Suppl. 1), S47–S53. [Google Scholar] [CrossRef] [PubMed]
- Imbimbo, B.P. An update on the efficacy of non-steroidal anti-inflammatory drugs in Alzheimer’s disease. Expert Opin. Investig. Drugs. 2009, 18, 1147–1168. [Google Scholar] [CrossRef] [PubMed]

Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Yu, J.; Zhu, H.; Taheri, S.; Mondy, W.; Perry, S.; Kirstein, C.; Kindy, M.S. Effects of GrandFusion Diet on Cognitive Impairment in Transgenic Mouse Model of Alzheimer’s Disease. Nutrients 2021, 13, 117. https://doi.org/10.3390/nu13010117
Yu J, Zhu H, Taheri S, Mondy W, Perry S, Kirstein C, Kindy MS. Effects of GrandFusion Diet on Cognitive Impairment in Transgenic Mouse Model of Alzheimer’s Disease. Nutrients. 2021; 13(1):117. https://doi.org/10.3390/nu13010117
Chicago/Turabian StyleYu, Jin, Hong Zhu, Saeid Taheri, William Mondy, Stephen Perry, Cheryl Kirstein, and Mark S. Kindy. 2021. "Effects of GrandFusion Diet on Cognitive Impairment in Transgenic Mouse Model of Alzheimer’s Disease" Nutrients 13, no. 1: 117. https://doi.org/10.3390/nu13010117