Pathophysiological Basis for Nutraceutical Supplementation in Heart Failure: A Comprehensive Review

 ,
,  ,
, 

Abstract
:1. Introduction
2. Emerging Pathophysiological Mechanisms Involved in the Onset of HF
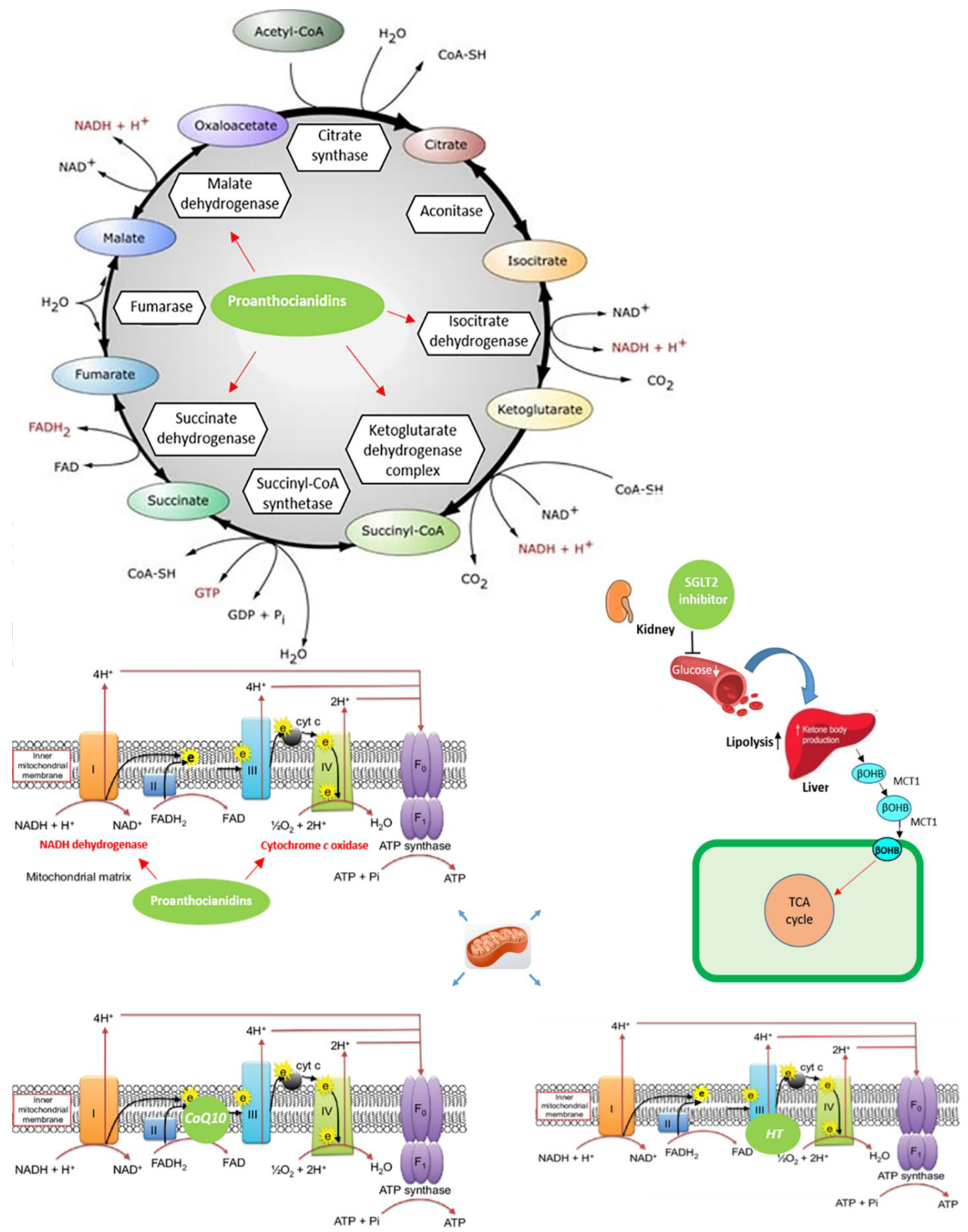
2.1. Energy Deficiency and Mitochondrial Impairment in HF
2.2. Endoplasmic Reticulum Stress in HF
2.3. Imbalanced Metalloproteinase Regulation in HF
2.4. The cGMO/NO Pathway in HF
2.5. HF and Sodium-Glucose Cotransporters
2.6. Impairment and Senescence of Cardiac Stem Cells in HF
3. Candidates for Nutraceutical Supplementation in the Failing Myocardium according to the Aforementioned Emerging Mechanisms
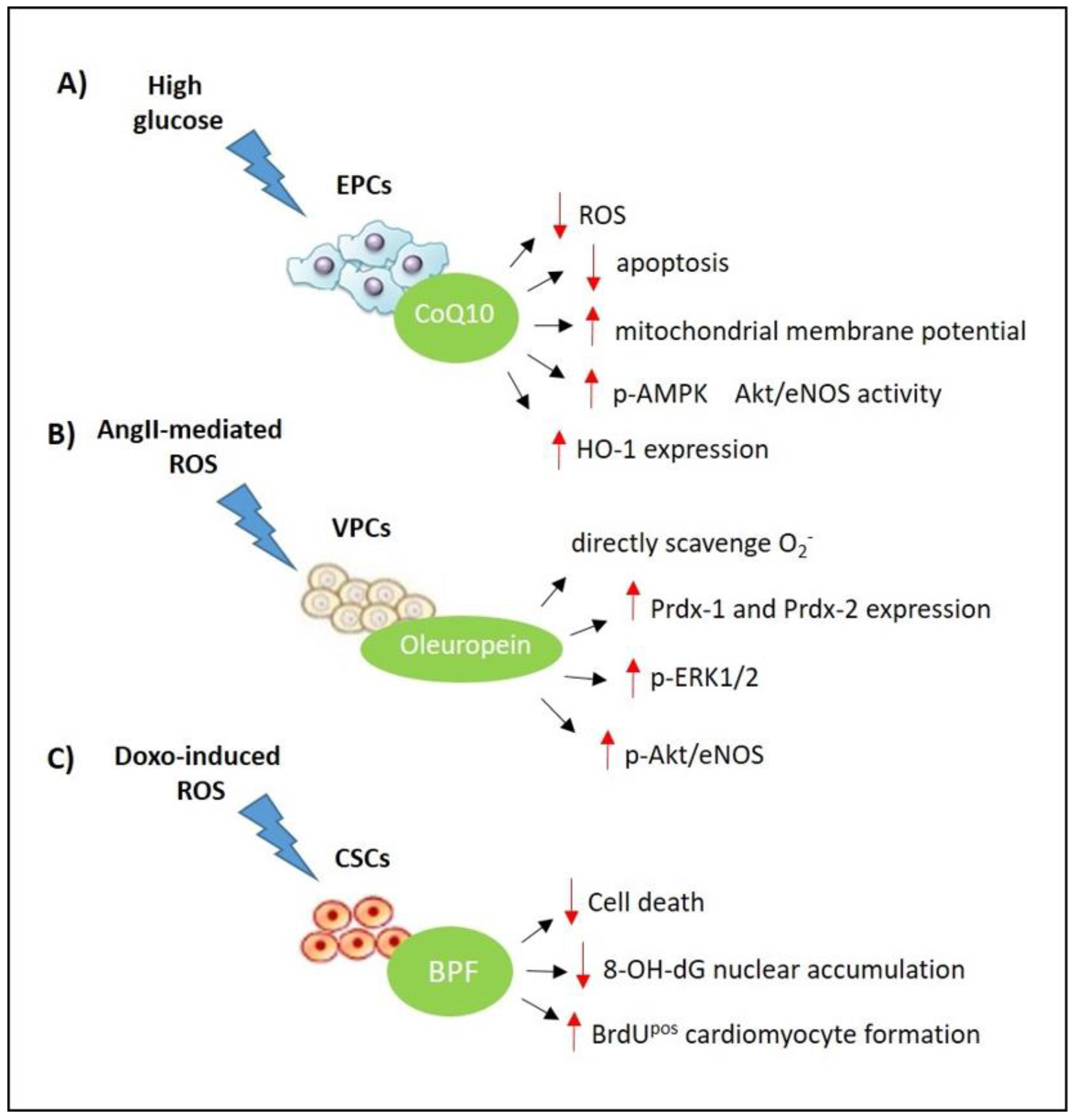
3.1. Coenzyme Q10
3.2. Bergamot Polyphenols
3.3. Olea Europea L. Extract
3.4. Apple-Derived Natural SGLT2-Inhibitors
3.5. Grape Seed Extract
4. Conclusions and Future Perspectives
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
References
- Ponikowski, P.; Anker, S.D.; AlHabib, K.F.; Cowie, M.R.; Force, T.L.; Hu, S.; Jaarsma, T.; Krum, H.; Rastogi, V.; Rohde, L.E.; et al. Heart failure: Preventing disease and death worldwide. ESC Heart Fail 2014, 1, 4–25. [Google Scholar] [CrossRef] [PubMed]
- Gomez-Soto, F.M.; Andrey, J.L.; Garcia-Egido, A.A.; Escobar, M.A.; Romero, S.P.; Garcia-Arjona, R.; Gutierrez, J.; Gomez, F. Incidence and mortality of heart failure: A community-based study. Int. J. Cardiol. 2011, 151, 40–45. [Google Scholar] [CrossRef] [PubMed]
- Kasznicki, J.; Drzewosk, J. Heart failure in the diabetic population—Pathophysiology, diagnosis and management. Med. Sci. 2014, 10, 546–556. [Google Scholar]
- Nieminen, M.S.; Brutsaert, D.; Dickstein, K.; Drexler, H.; Follath, F.; Harjola, V.P.; Hochadel, M.; Komajda, M.; Lassus, J.; Lopez-Sendon, J.L.; et al. EuroHeart Failure Survey II (EHFS II): A survey on hospitalized acute heart failure patients: Description of population. Eur. Heart J. 2006, 27, 2725–2736. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kurmani, S.; Squire, I. Acute Heart Failure: Definition, Classification and Epidemiology. Curr. Heart Fail. Rep. 2017, 14, 385–392. [Google Scholar] [CrossRef] [Green Version]
- Alissa, E.M.; Ferns, G.A. Functional foods and nutraceuticals in the primary prevention of cardiovascular diseases. J. Nutr. Metab. 2012, 2012, 569486. [Google Scholar] [CrossRef] [Green Version]
- Gliozzi, M.; Walker, R.; Mollace, V. Bergamot polyphenols: Pleiotropic players in the treatment of metabolic syndrome. J. Metabolic Synd. 2014, 2, 35–42. [Google Scholar]
- Tresserra-Rimbau, A.; Rimm, E.B.; Medina-Remón, A.; Martínez-González, M.A.; de la Torre, R.; Corella, D.; Salas-Salvadó, J.; Gómez-Gracia, E.; Lapetra, J.; Arós, F.; et al. Inverse association between habitual polyphenol intake and incidence of cardiovascular events in the PREDIMED study. Nutr. Metab. Cardiovasc. Dis. 2014, 24, 639–647. [Google Scholar] [CrossRef]
- Mollace, V.; Gliozzi, M.; Carresi, C.; Musolino, V.; Oppedisano, F. Re-assessing the mechanism of action of n-3 PUFAs. Int. J. Cardiol. 2013, 170, S8–S11. [Google Scholar] [CrossRef]
- Carresi, C.; Gliozzi, M.; Giancotta, C.; Scarcella, A.; Scarano, F.; Bosco, F.; Mollace, R.; Tavernese, A.; Vitale, C.; Musolino, V. Studies on the protective role of Bergamot polyphenols in doxorubicin-induced cardiotoxicity. PharmaNutrition 2016, 4, S19–S26. [Google Scholar] [CrossRef]
- Sciatti, E.; Lombardi, C.; Ravera, A.; Vizzardi, E.; Bonadei, I.; Carubelli Gorga, V.E.; Metra, M. Nutritional deficiency in patients with heart failure. Nutrients 2016, 8, 442. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Witte, K.K.; Clark, A.L.; Cleland, J.G. Chronic heart failure and micronutrients. J. Am. Coll. Cardiol. 2001, 37, 1765–1774. [Google Scholar] [CrossRef] [Green Version]
- Pinho, C.P.S.; da Silveira, A.C. Nutritional aspects in heart failure. J. Nutr. Health Sci. 2014, 1, 305. [Google Scholar]
- Witte, K.K.; Nikitin, N.P.; Parker, A.C.; von Haehling, S.; Volk, H.D.; Anker, S.D.; Clark, A.L.; Cleland, J.G. The effect of micronutrient supplementation on quality-of-life and left ventricular function in elderly patients with chronic heart failure. Eur. Heart J. 2005, 26, 2238–2244. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cvetinovic, N.; Loncar, G.; Isakovic, A.M.; von Haehling, S.; Doehner, W.; Lainscak, M.; Farkas, J. Micronutrient depletion in heart failure: Common, clinically relevant and treatable. Int. J. Mol. Sci. 2019, 20, 5627. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Witte, K.K.; Clark, A.L. Micronutrients and their supplementation in chronic cardiac failure. An update beyond theoretical perspectives. Heart Fail. Rev. 2006, 11, 65–74. [Google Scholar] [CrossRef]
- Shimon, I.; Almog, S.; Vered, Z.; Seligmann, H.; Shefi, M.; Peleg, E.; Rosenthal, T.; Motro, M.; Halkin, H.; Ezra, D. Improved left ventricular function after thiamine supplementation in patients with congestive heart failure receiving long-term furosemide therapy. Am. J. Med. 1995, 98, 485–490. [Google Scholar] [CrossRef]
- Deichmann, R.; Lavie, C.; Andrews, S. Coenzyme Q10 and statin-induced mitochondrial dysfunction. Ochsner J. 2010, 10, 16–21. [Google Scholar]
- Sharma, A.; Fonarow, G.C.; Butler, J.; Ezekowitz, J.A.; Felker, G.M. Coenzyme Q10 and heart failure: A state-of-the-art review. Circ. Heart Fail. 2016, 9, e002639. [Google Scholar] [CrossRef]
- Tsai, K.L.; Huang, Y.H.; Kao, C.L.; Yang, D.M.; Lee, H.C.; Chou, H.Y.; Chen, H.C.; Chiou, G.Y.; Chen, L.H.; Yang, Y.P.; et al. A novel mechanism of coenzyme Q10 protects against human endothelial cells from oxidative stress-induced injury by modulating NO-related pathways. J. Nutr. Biochem. 2012, 23, 458–468. [Google Scholar] [CrossRef]
- Fotino, D.; Thompson-Paul, A.M.; Bazzano, L.A. Effect of coenzyme Q10 supplementation on heart failure: A meta-analysis. Am. J. Clin. Nutr. 2013, 97, 268–275. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mortensen, S.A.; Rosenfeldt, F.; Kumar, A.; Dolliner, P.; Filipiak, K.J.; Pella, D.; Alehagen, U.; Steurer, G.; Littarru, G.P. Q-SYMBIO Study Investigators. The effect of coenzyme Q10 on morbidity and mortality in chronic heart failure: Results from Q-SYMBIO: A randomized double-blind trial. JACC Heart Fail. 2014, 2, 641–649. [Google Scholar] [CrossRef] [PubMed]
- Mollace, V.; Sacco, I.; Janda, E.; Malara, C.; Ventrice, D.; Colica, C.; Visalli, V.; Muscoli, S.; Ragusa, S.; Muscoli, C.; et al. Hypolipemic and hypoglycaemic activity of bergamot polyphenols: From animal models to human studies. Fitoterapia 2011, 82, 309–316. [Google Scholar] [CrossRef] [PubMed]
- Mollace, V.; Scicchitano, M.; Paone, S.; Casale, F.; Calandruccio, C.; Gliozzi, M.; Musolino, V.; Carresi, C.; Maiuolo, J.; Nucera, S.; et al. Hypoglycemic and Hypolipemic Effects of a new lecithin formulation of bergamot Polyphenolic Fraction: A double blind, randomized, placebo-controlled study. Endocr. Metab. Immune Disord. Drug Targets 2019, 19, 136–143. [Google Scholar] [CrossRef] [PubMed]
- Gliozzi, M.; Walker, R.; Muscoli, S.; Vitale, C.; Gratteri, S.; Carresi, C.; Musolino, V.; Russo, V.; Janda, E.; Ragusa, S.; et al. Bergamot polyphenolic fraction enhances rosuvastatin-induced effect on LDL-cholesterol, LOX-1 expression and protein kinase B phosphorylation in patients with hyperlipidemia. Int. J. Cardiol. 2013, 170, 140–145. [Google Scholar] [CrossRef] [PubMed]
- Perrinjaquet-Moccetti, T.; Busjahn, A.; Schmidlin, C.; Schmidt, A.; Bradl, B.; Aydogan, C. Food supplementation with an olive (Olea europaea L.) leaf extract reduces blood pressure in borderline hypertensive monozygotic twins. Phytother. Res. 2008, 22, 1239–1242. [Google Scholar] [CrossRef]
- Susalit, E.; Agus, N.; Effendi, I.; Tjandrawinata, R.R.; Nofiarny, D.; Perrinjaquet-Moccetti, T.; Verbruggen, M. Olive (Olea europaea) leaf extract effective in patients with stage-1 hypertension: Comparison with Captopril. Phytomedicine 2011, 18, 251–258. [Google Scholar] [CrossRef]
- Zinman, B.; Wanner, C.; Lachin, J.M.; Fitchett, D.; Bluhmki, E.; Hantel, S.; Mattheus, M.; Devins, T.; Johansen, O.E.; Woerle, H.J.; et al. Empagliflozin, cardiovascular outcomes, and mortality in Type 2 Diabetes. N. Engl. J. Med. 2015, 373, 2117. [Google Scholar] [CrossRef]
- Wiviott, S.D.; Raz, I.; Bonaca, M.P.; Mosenzon, O.; Kato, E.T.; Cahn, A.; Silverman, M.G.; Zelniker, T.A.; Kuder, J.F.; Murphy, S.A.; et al. Dapagliflozin and cardiovascular outcomes in Type 2 Diabetes. N. Engl. J. Med. 2019, 380, 347. [Google Scholar] [CrossRef]
- Sarewicz, M.; Osyczka, A. electronic connection between the quinone and cytochrome c redox pools and its role in regulation of mitochondrial electron transport and redox signalling. Physiol. Rev. 2015, 95, 219–243. [Google Scholar] [CrossRef] [Green Version]
- Tsai, H.Y.; Lin, C.P.; Huang, P.H.; Li, S.Y.; Chen, J.S.; Lin, F.Y.; Chen, J.W.; Lin, S.J. Coenzyme Q10 attenuates high glucose-induced endothelial progenitor cell dysfunction through AMP-Activated protein kinase pathways. J. Diabetes Res. 2016, 2016, 6384759. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mancuso, M.; Orsucci, D.; Volpi, L.; Calsolaro, V.; Siciliano, G. Coenzyme Q10 in neuromuscular and neurodegenerative disorders. Curr. Drug Targets 2010, 11, 111–121. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Garjani, A.; Andalib, S.; Biabani, S.; Soraya, H.; Doustar, Y.; Garjani, A.; Maleki-Dizaji, N. Combined atorvastatin and coenzyme Q10 improve the left ventricular function in isoproterenol-induced heart failure in rat. Eur. J. Pharmacol. 2011, 666, 135–141. [Google Scholar] [CrossRef] [PubMed]
- Zozina, V.I.; Covantev, S.; Goroshko, O.A.; Krasnykh, L.M.; Kukes, V.G. Coenzyme Q10 in cardiovascular and metabolic diseases: Current state of the problem. Curr. Cardiol. Rev. 2018, 14, 164–174. [Google Scholar] [CrossRef] [PubMed]
- Niklowitz, P.; Onur, S.; Fischer, A.; Laudes, M.; Palussen, M.; Menke, T.; Döring, F. Coenzyme Q10 serum concentration and redox status in European adults: Influence of age, sex, and lipoprotein concentration. J. Clin. Biochem. Nutr. 2016, 58, 240–245. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Molyneux, S.L.; Florkowski, C.M.; George, P.M.; Pilbrow, A.P.; Frampton, C.M.; Lever, M.; Richards, M. Coenzyme Q(10) an independent predictor of mortality in chronic heart failure. J. Am. Coll. Cardiol. 2008, 52, 1435–1441. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kjekshus, J.; Apetrei, E.; Barrios, V.; Böhm, M.; Cleland, J.G.F.; Cornel, J.H.; Dunselman, P.; Fonseca, C.; Goudev, A.; Grande, P.; et al. Rosuvastatin in older patients with systolic heart failure. Int. J. Clin. Pract. 2008, 62, 1. [Google Scholar] [CrossRef] [PubMed]
- Molyneux, S.L.; Young, J.M.; Florkowski, C.M.; Lever, M.; George, P.M. Coenzyme Q10: Is There a clinical role and a case for measurement? Clin. Biochem. Rev. 2008, 29, 71–82. [Google Scholar]
- Carresi, C.; Gliozzi, M.; Musolino, V.; Scicchitano, M.; Scarano, F.; Bosco, F.; Nucera, S.; Maiuolo, J.; Macrì, R.; Ruga, S.; et al. The effect of natural antioxidants in the development of metabolic syndrome: Focus on bergamot polyphenolic fraction. Nutrients 2020, 12, 1504. [Google Scholar] [CrossRef]
- Musolino, V.; Gliozzi, M.; Nucera, S.; Carresi, C.; Maiuolo, J.; Mollace, R.; Paone, S.; Bosco, F.; Scarano, F.; Scicchitano, M.; et al. The effect of bergamot polyphenolic fraction on lipid transfer protein system and vascular oxidative stress in a rat model of hyperlipemia. Lipids Health Dis. 2019, 18, 115. [Google Scholar] [CrossRef] [Green Version]
- Musolino, V.; Gliozzi, M.; Scarano, F.; Bosco, F.; Scicchitano, M.; Nucera, S.; Carresi, C.; Ruga, S.; Zito, M.C.; Maiuolo, J.; et al. Bergamot polyphenols improve dyslipidemia and pathophysiological features in a mouse model of Non-Alcoholic Fatty Liver Disease. Sci. Rep. 2020, 10, 2565. [Google Scholar] [CrossRef] [PubMed]
- Parafati, M.; Lascala, A.; Morittu, V.M.; Trimboli, F.; Rizzuto, A.; Brunelli, E.; Coscarelli, F.; Costa, N.; Britti, D.; Ehrlich, J.; et al. Bergamot polyphenol fraction prevents nonalcoholic fatty liver disease via stimulation of lipophagy in cafeteria diet-induced rat model of metabolic syndrome. Nutr. Biochem. 2015, 26, 938–948. [Google Scholar] [CrossRef] [PubMed]
- Miceli, N.; Mondello, M.R.; Monforte, M.T.; Sdrafkakis, V.; Dugo, P.; Crupi, M.L.; Taviano, M.F.; De Pasquale, R.; Trovato, A. Hypolipidemic effects of Citrus bergamia Risso et Poiteau juice in rats fed a hypercholesterolemic diet. J. Agric. Food Chem. 2007, 55, 10671–10677. [Google Scholar] [CrossRef] [PubMed]
- Musolino, V.; Gliozzi, M.; Carresi, C.; Maiuolo, J.; Mollace, R.; Bosco, F.; Scarano, F.; Scicchitano, M.; Maretta, A.; Palma, E.; et al. Lipid-lowering effect of bergamot polyphenolic fraction: Role of pancreatic cholesterol ester hydrolase. J. Biol. Regul. Homeost. Agents 2017, 31, 1087–1093. [Google Scholar] [PubMed]
- Di Donna, L.; De Luca, G.; Mazzotti, F.; Napoli, A.; Salerno, R.; Taverna, D.; Sindona, G. Statin-like principles of bergamot fruit (Citrus bergamia): Isolation of 3-hydroxymethylglutaryl flavonoid glycosides. J. Nat. Prod. 2009, 72, 1352–1354. [Google Scholar] [CrossRef]
- Leopoldini, M.; Malaj, N.; Toscano, M.; Sindona, G.; Russo, N. On the inhibitor effects of bergamot juice flavonoids binding to the 3-hydroxy-3-methylglutaryl-CoA reductase (HMGR) enzyme. J. Agric. Food Chem. 2010, 58, 10768–10773. [Google Scholar] [CrossRef]
- Di Donna, L.; Iacopetta, D.; Cappello, A.R.; Gallucci, G.; Martello, E.; Fiorillo, M.; Dolce, V.; Sindona, G. Hypocholesterolaemic activity of 3-hydroxy-3-methyl-glutaryl flavanones enriched fraction from bergamot fruit (Citrus bergamia): ‘In vivo’ studies. J. Funct. Foods 2014, 7, 558–568. [Google Scholar] [CrossRef]
- De Bock, M.; Thorstensen, E.B.; Derraik, J.G.; Henderson, H.V.; Hofman, P.L.; Cutfield, W.S. Human absorption and metabolism of oleuropein and hydroxytyrosol ingested as olive (Olea europaea L.) leaf extract. Mol. Nutr. Food Res. 2013, 57, 2079–2085. [Google Scholar] [CrossRef]
- Kouka, P.; Tsakiri, G.; Tzortzi, D.; Dimopoulou, S.; Sarikaki, G.; Stathopoulos, P.; Veskoukis, A.S.; Halabalaki, M.; Skaltsounis, A.L.; Kouretas, D. The polyphenolic composition of extracts derived from different greek extra virgin olive oils is correlated with their antioxidant potency. Oxid. Med. Cell. Longev. 2019, 2019, 1870965. [Google Scholar] [CrossRef]
- Talhaoui, N.; Gómez-Caravaca, A.M.; León, L.; De la Rosa, R.; Fernández-Gutiérrez, A.; Segura-Carretero, A. From olive fruits to olive oil: Phenolic compound transfer in six different olive cultivars grown under the same agronomical conditions. Int. J. Mol. Sci. 2016, 17, 337. [Google Scholar] [CrossRef] [Green Version]
- Gorzynik-Debicka, M.; Przychodzen, P.; Cappello, F.; Kuban-Jankowska, A.; Marino Gammazza, A.; Knap, N.; Wozniak, M.; Gorska-Ponikowska, M. Potential health benefits of olive oil and plant polyphenols. Int. J. Mol. Sci. 2018, 19, 686. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Žugčić, T.; Abdelkebir, R.; Alcantara, C.; Collado, M.C.; García-Pérez, J.V.; Meléndez-Martínez, A.J.; Režek Jambrak, A.; Lorenzo, J.M.; Barba, F.J. From extraction of valuable compounds to health promoting benefits of olive leaves through bioaccessibility, bioavailability and impact on gut microbiota. Trends Food Sci. Technol. 2019, 83, 63–77. [Google Scholar] [CrossRef]
- Bulotta, S.; Oliverio, M.; Russo, D.; Procopio, A. Biological Activity of Oleuropein and its Derivatives. In Natural Products; Springer: Heidelberg, Germany, 2013; Volume 156, pp. 3605–3638. [Google Scholar]
- Andrikopoulos, N.K.; Kaliora, A.C.; Assimopoulou, A.N.; Papageorgiou, V.P. Inhibitory activity of minor polyphenolic and nonpolyphenolic constituents of olive oil against in vitro low-density lipoprotein oxidation. J. Med. Food 2002, 5, 1–7. [Google Scholar] [CrossRef] [PubMed]
- Andreadou, I.; Mikros, E.; Ioannidis, K.; Sigala, F.; Naka, K.; Kostidis, S.; Farmakis, D.; Tenta, R.; Kavantzas, N.; Bibli, S.I.; et al. Oleuropein prevents doxorubicin-induced cardiomyopathy interfering with signaling molecules and cardiomyocyte metabolism. J. Mol. Cell. Cardiol. 2014, 69, 4–16. [Google Scholar] [CrossRef] [PubMed]
- Granados-Principal, S.; El-azem, N.; Pamplona, R.; Ramirez-Tortosa, C.; Pulido-Moran, M.; Vera-Ramirez, L.; Quiles, J.L.; Sanchez-Rovira, P.; Naudí, A.; Portero-Otin, M.; et al. Hydroxytyrosol ameliorates oxidative stress and mitochondrial dysfunction in doxorubicin-induced cardiotoxicity in rats with breast cancer. Biochem. Pharmacol. 2014, 90, 25–33. [Google Scholar] [CrossRef]
- Andreadou, I.; Iliodromitis, E.K.; Mikros, E.; Constantinou, M.; Agalias, A.; Magiatis, P.; Skaltsounis, A.L.; Kamber, E.; Tsantili-Kakoulidou, A.; Kremastinos, D.T. The olive constituent oleuropein exhibits anti-ischemic, antioxidative, and hypolipidemic effects in anesthetized rabbits. J. Nutr. 2006, 136, 2213–2219. [Google Scholar] [CrossRef] [Green Version]
- Mattera, R.; Benvenuto, M.; Giganti, M.G.; Tresoldi, I.; Pluchinotta, F.R.; Bergante, S.; Tettamanti, G.; Masuelli, L.; Manzari, V.; Modesti, A.; et al. Effects of Polyphenols on Oxidative Stress-Mediated Injury in Cardiomyocytes. Nutrients 2017, 9, 523. [Google Scholar] [CrossRef] [Green Version]
- Samuel, S.M.; Thirunavukkarasu, M.; Penumathsa, S.V.; Paul, D.; Maulik, N. Akt/FOXO3a/SIRT1-mediated cardioprotection by n-tyrosol against ischemic stress in rat in vivo model of myocardial infarction: Switching gears toward survival and longevity. J. Agric. Food Chem. 2008, 56, 9692–9698. [Google Scholar] [CrossRef] [Green Version]
- Jin, H.X.; Zhang, Y.H.; Guo, R.N.; Zhao, S.N. Inhibition of MEK/ERK/STAT3 signaling in oleuropein treatment inhibits myocardial ischemia/reperfusion. Int. J. Mol. Med. 2018, 2, 1034–1043. [Google Scholar] [CrossRef] [Green Version]
- Potočnjak, I.; Škoda, M.; Pernjak-Pugel, E.; Pavletić Peršić, M.; Domitrović, R. Oral administration of oleuropein attenuates cisplatin-induced acute renal injury in mice through inhibition of ERK signaling. Mol. Nutr. Food Res. 2016, 60, 530–541. [Google Scholar] [CrossRef]
- Wu, L.X.; Xu, Y.Y.; Yang, Z.J.; Feng, Q. Hydroxytyrosol and olive leaf extract exert cardioprotective effects by inhibiting GRP78 and CHOP expression. J. Biomed. Res. 2018, 32, 371–379. [Google Scholar] [PubMed]
- Choi, S.Y.; Joo, B.H.; Lee, S.J.; Choi, H.Y.; Park, J.H.; Baek, S.H.; Kwon, S.M. Oleuropein prevents angiotensin II-mediated human vascular progenitor cell depletion. Int. J. Cardiol. 2015, 181, 160–165. [Google Scholar] [CrossRef] [PubMed]
- Diedrich, D.F. Competitive inhibition of intestinal glucose transport by phlorizin analogs. Arch. Biochem. Biophys. 1966, 117, 248–256. [Google Scholar] [CrossRef]
- Pearson, D.A.; Tan, C.H.; German, J.B.; Davis, P.A.; Gershwin, M.E. Apple juice inhibits human low density lipoprotein oxidation. Life Sci. 1999, 64, 1913–1920. [Google Scholar] [CrossRef]
- Ehrenkranz, J.R.; Lewis, N.G.; Kahn, C.R.; Roth, J. Phlorizin: A review. Diabetes Metab. Res. Rev. 2005, 21, 31–38. [Google Scholar] [CrossRef]
- Figtree, G.A.; Griffiths, H.; Lu, Y.Q.; Webb, C.M.; MacLeod, K.; Collins, P. Plant-derived estrogens relax coronary arteries in vitro by a calcium antagonistic mechanism. Am. Coll. Cardiol. 2000, 35, 1977–1985. [Google Scholar] [CrossRef] [Green Version]
- Rossetti, L.; Smith, D.; Shculman, G.I.; Papachristou, D.; DeFronzo, R.A. Correction of hyperglycemia with phlorizin normalizes tissue sensitivity to insulin in diabetic rats. J. Clin. Investig. 1987, 79, 1510–1515. [Google Scholar] [CrossRef]
- Blondel, O.; Bailbe, D.; Portha, B. Insulin resistance in rats with non-insulin-dependent diabetes induced by neonatal (5 days) streptozotocin: Evidence for reversal following phlorizin treatment. Metabolism 1990, 39, 787–793. [Google Scholar] [CrossRef]
- Kahn, B.B.; Shulman, G.I.; DeFronzo, R.A.; Cushman, S.W.; Rossetti, L. Normalization of blood glucose in diabetic rats with phlorizin treatment reverses insulin-resistant glucose transport in adipose cells without restoring glucose transporter gene expression. J. Clin. Investig. 1991, 87, 561–570. [Google Scholar] [CrossRef] [Green Version]
- Takii, H.; Matsumoto, K.; Kometani, T.; Okada, S.; Fushiki, T. Lowering effect of phenolic glycosides on the rise in postprandial glucose in mice. Biosci. Biotechnol. Biochem. 1997, 61, 1531–1535. [Google Scholar] [CrossRef] [Green Version]
- Oku, A.; Ueta, K.; Arakawa, K.; Kano-Ishihara, T.; Matsumoto, T.; Adachi, T.; Yasuda, K.; Tsuda, K.; Ikezawa, K.; Saito, A. Correction of hyperglycemia and insulin sensitivity by T-1095, an inhibitor of renal Na+-glucose cotransporters, in streptozotocin-induced diabetic rats. Jpn. J. Pharmacol. 2000, 84, 351–354. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Adachi, T.; Yasuda, K.; Okamoto, Y.; Shihara, N.; Oku, A.; Ueta, K.; Kitamura, K.; Saito, A.; Iwakura, I.; Yamada, Y.; et al. T-1095, a renal Na+-glucose transporter inhibitor, improves hyperglycemia in streptozotocin-induced diabetic rats. Metabolism 2000, 49, 990–995. [Google Scholar] [CrossRef] [PubMed]
- Karthikeyan, K.; Sarala Bai, B.R.; Niranjali Devaraj, S. Grape seed proanthocyanidins ameliorates isoproterenol-induced myocardial injury in rats by stabilizing mitochondrial and lysosomal enzymes: An in vivo study. Life Sci. 2007, 81, 1615–1621. [Google Scholar] [CrossRef] [PubMed]
- Du, Y.; Guo, H.; Lou, H. Grape seed polyphenols protect cardiac cells from apoptosis via induction of endogenous antioxidant enzymes. J. Agric. Food Chem. 2007, 55, 1695–1701. [Google Scholar] [CrossRef]
- Bagchi, D.; Garg, A.; Krohn, R.L.; Bagchi, M.; Bagchi, D.J.; Balmoori, J.; Stohs, S.J. Protective effects of grape seed proanthocyanidins and selected antioxidants against TPA-induced hepatic and brain lipid peroxidation and DNA fragmentation, and peritoneal macrophage activation in mice. Gen. Pharmacol. 1998, 30, 771–776. [Google Scholar] [CrossRef]
- Bagchi, M.; Kuszynski, C.A.; Balmoori, J.; Joshi, S.S.; Stohs, S.J.; Bagchi, D. Protective effects of antioxidants against smokeless tobacco-induced oxidative stress and modulation of Bcl-2 and p53 genes in human oral keratinocytes. Free Radic. Res. 2001, 35, 181–194. [Google Scholar] [CrossRef]
- Joshi, S.S.; Kuszynski, C.A.; Benner, E.J.; Balmoori, J.C.A.; Bagchi, M.; Bagchi, D. Amelioration of cytotoxic effects of idarubicin and 4-hydroxyperoxycyclophosphamide on Chang liver cells by a novel grape seed proanthocyanidin extract. FASEB J. 1998, 12, A774. [Google Scholar]
- Ye, X.; Krohn, R.L.; Liu, W.; Joshi, S.S.; Kuszynski, C.A.; McGinn, T.R.; Bagchi, M.; Preuss, H.G.; Stohs, S.J.; Bagchi, D. The cytotoxic effects of a novel IH636 grape seed proanthocyanidin extract on cultured human cancer cells. Mol. Cell. Biochem. 1999, 196, 99–108. [Google Scholar] [CrossRef]
- Bagchi, D.; Ray, S.D.; Patel, D.; Bagchi, M. Protection against drug- and chemical-induced multiorgan toxicity by a novel grape seed proanthocyanidin extract. Drugs Exp. Clin. Res. 2001, 27, 3–15. [Google Scholar]
- Ray, S.D.; Kumar, M.A.; Bagchi, D. A novel proanthocyanidin IH636 grape seed extract increases in vivo bcl-XL expression and prevents acetaminophen-induced programmed and unprogrammed cell death in mouse liver. Arch. Biochem. Biophys. 1999, 369, 42–58. [Google Scholar] [CrossRef]
- Sato, M.; Bagchi, D.; Tosaki, A.; Das, D.K. Grape seed proanthocyanidin reduces cardiomyocyte apoptosis by inhibiting ischemia/reperfusion-induced activation of jnk-1 and c-jun. Free Radic. Biol. Med. 2001, 31, 729–737. [Google Scholar] [CrossRef]
- Yuan, Z.; Du, W.; He, X.; Zhang, D.; He, W. Tribulus terrestris ameliorates oxidative stress-induced ARPE-19 cell injury through the PI3K/Akt-Nrf2 Signaling Pathway. Oxid. Med. Cell. Longev. 2020, 28, 7962393. [Google Scholar]
- Raj, P.; Louis, X.L.; Thandapilly, S.J.; Movahed, A.; Zieroth, S.; Netticadan, T. Potential of resveratrol in the treatment of heart failure. Life Sci. 2014, 95, 63–71. [Google Scholar] [CrossRef] [PubMed]
- Dyck, G.J.B.; Raj, P.; Zieroth, S.; Dyck, J.R.B.; Ezekowitz, J.A. The effects of resveratrol in patients with cardiovascular disease and heart failure: A narrative review. Int. J. Mol. Sci. 2019, 20, 904. [Google Scholar] [CrossRef] [Green Version]
- Lagouge, M.; Argmann, C.; Gerhart-Hines, Z.; Meziane, H.; Lerin, C.; Daussin, F.; Messadeq, N.; Milne, J.; Lambert, P.; Elliott, P.; et al. Resveratrol improves mitochondrial function and protects against metabolic disease by activating SIRT1 and PGC-1a. Cell 2006, 127, 1109–1122. [Google Scholar] [CrossRef]
- Gu, X.S.; Wang, Z.B.; Ye, Z.; Lei, J.P.; Li, L.; Su, D.F.; Zheng, X. Resveratrol, an activator of SIRT1, upregulates AMPK and improves cardiac function in heart failure. Genet. Mol. Res. 2014, 13, 323–335. [Google Scholar] [CrossRef]
- Kanamori, H.; Takemura, Y.G.; Goto, K.; Tsujimoto, A.; Ogino, A.; Takeyama, T.; Kawaguchi, T.; Watanabe, T.; Morishita, K.; Kawasaki, M.; et al. Cardiovascular, pulmonary and renal pathology resveratrol reverses remodelling in hearts with large, old myocardial infarctions through enhanced autophagy-activating AMP Kinase pathway. Am. J. Pathol. 2013, 182, 3. [Google Scholar] [CrossRef]
- Marques, B.; Trindade, M.; Aquino, J.C.F.; Cunha, A.R.; Gismondi, R.O.; Neves, M.F.; Oigman, W. Beneficial effects of acute trans-resveratrol supplementation in treated hypertensive patients with endothelial dysfunction. Clin. Exp. Hypertens. 2018, 40, 218–223. [Google Scholar] [CrossRef]
- Tome-Carneiro, J.; Gonzalvez, M.; Larrosa, M.; Yanez-Gascon, M.J.; Garcia-Almagro, F.J.; Ruiz-Ros, J.A.; Tomas-Barberan, F.A.; Garcia-Conesa, M.T.; Espin, J.C. Grape resveratrol increases serum adiponectin and downregulates inflammatory genes in peripheral blood mononuclear cells: A triple-blind, placebo-controlled, one-year clinical trial in patients with stable coronary artery disease. Cardiovasc. Drugs Ther. 2013, 27, 37–48. [Google Scholar] [CrossRef] [Green Version]
- Tome-Carneiro, J.; Gonzalvez, M.; Larrosa, M.; Yanez-Gascon, M.J.; Garcia-Almagro, F.J.; Ruiz-Ros, J.A.; Garcia-Conesa, M.T.; Tomas-Barberan, F.A.; Espin, J.C. One-year consumption of a grape nutraceutical containing resveratrol improves the inflammatory and fibrinolytic status of patients in primary prevention of cardiovascular disease. Am. J. Cardiol. 2012, 110, 356–363. [Google Scholar] [CrossRef]
- Bhatt, J.K.; Thomas, S.; Nanjan, M.J. Resveratrol supplementation improves glycemic control in type 2 diabetes mellitus. Nutr. Res. 2012, 32, 537–541. [Google Scholar] [CrossRef] [PubMed]
- Magyar, K.; Halmosi, R.; Palfi, A.; Feher, G.; Czopf, L.; Fulop, A.; Battyany, I.; Sumegi, B.; Toth, K.; Szabados, E. Cardioprotection by resveratrol: A human clinical trial in patients with stable coronary artery disease. Clin. Hemorheol. Microcirc. 2012, 50, 179–187. [Google Scholar] [CrossRef] [PubMed]
- Neubauer, S. The Failing Heart—An Engine Out of Fuel. N. Engl. J. Med. 2007, 356, 1140–1151. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bottomley, P.A. MR spectroscopy of the human heart: The status and the challenges. Radiology 1994, 191, 593–612. [Google Scholar] [CrossRef]
- Ingwall, J.S.; Atkinson, D.E.; Clarke, K.; Fetters, J.K. Energetic correlates of cardiac failure: Changes in the creatine kinase system in the failing myocardium. Eur. Heart J. 1990, 11 (Suppl. B), 108–115. [Google Scholar] [CrossRef]
- Starling, R.C.; Hammer, D.F.; Altschuld, R.A. Human myocardial ATP content and in vivo contractile function. Mol. Cell. Biochem. 1998, 180, 171–177. [Google Scholar] [CrossRef]
- Zhou, B.; Tian, R. Mitochondrial dysfunction in pathophysiology of heart failure. J. Clin. Investig. 2018, 128, 3716–3726. [Google Scholar] [CrossRef] [Green Version]
- Gorski, P.A.; Ceholski, D.K.; Hajjar, R.J. Altered myocardial calcium cycling and energetics in heart failure—A rational approach for disease treatment. Cell Metab. 2015, 21, 183–194. [Google Scholar] [CrossRef] [Green Version]
- Montgomery, C.; Hamilton, N.; Ianuzzo, C.D. Energy status of the rapidly paced canine myocardium in congestive heart failure. J. Appl. Physiol. 1992, 73, 2363–2367. [Google Scholar] [CrossRef]
- Doenst, T.; Pytel, G.; Schrepper, A.; Amorim, P.; Färber, G.; Shingu, Y.; Mohr, F.W.; Schwarzer, M. Decreased rates of substrate oxidation ex vivo predict the onset of heart failure and contractile dysfunction in rats with pressure overload. Cardiovasc. Res. 2010, 86, 461–470. [Google Scholar] [CrossRef] [Green Version]
- Chidsey, C.A.; Weinbach, E.C.; Pool, P.E.; Morrow, A.G. Biochemical studies of energy production in the failing human heart. J. Clin. Investig. 1966, 45, 40–50. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sobel, B.E.; Spann, J.F.; Pool, P.E.; Sonnenblick, E.H.; Braunwald, E. Normal oxidative phosphorylation in mitochondria from the failing heart. Circ. Res. 1967, 21, 355–364. [Google Scholar] [CrossRef] [Green Version]
- Cordero-Reyes, A.M.; Gupte, A.A.; Youker, K.A.; Loebe, M.; Hsueh, W.A.; Torre-Amione, G.; Taegtmeyer, H.; Hamilton, D.J. Freshly isolated mitochondria from failing human hearts exhibit preserved respiratory function. J. Mol. Cell. Cardiol. 2014, 68, 98–105. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Holzem, K.M.; Vinnakota, K.C.; Ravikumar, V.K.; Madden, E.J.; Ewald, G.A.; Dikranian, K.; Beard, D.A.; Efimov, I.R. Mitochondrial structure and function are not different between non-failing donor and end-stage failing human hearts. FASEB J. 2016, 30, 2698–2707. [Google Scholar] [CrossRef] [Green Version]
- Sharov, V.G.; Todor, A.V.; Silverman, N.; Goldstein, S.; Sabbah, H.N. Abnormal mitochondrial respiration in failed human myocardium. J. Mol. Cell. Cardiol. 2000, 32, 2361–2367. [Google Scholar] [CrossRef]
- Nickel, A.; Loffler, J.; Maack, C. Myocardial energetics in heart failure. Basic Res. Cardiol. 2013, 108, 358. [Google Scholar] [CrossRef]
- Razeghi, P.; Young, M.E.; Alcorn, J.L.; Moravec, C.S.; Frazier, O.H.; Taegtmeyer, H. Metabolic gene expression in fetal and failing human heart. Circulation 2001, 104, 2923–2931. [Google Scholar] [CrossRef] [Green Version]
- Chen, Y.; Csordas, G.; Jowdy, C.; Schneider, T.G.; Csordas, N.; Wang, W.; Liu, Y.; Kohlhaas, M.; Meiser, M.; Bergem, S.; et al. Mitofusin 2-containing mitochondrial-reticular microdomains direct rapid cardiomyocyte bioenergetic responses via interorganelle Ca2+ crosstalk. Circ. Res. 2012, 111, 863–875. [Google Scholar] [CrossRef] [Green Version]
- Goh, K.Y.; Qu, J.; Hong, H.; Liu, T.; Dell’Italia, L.J.; Wu, Y.; O’Rourke, B.; Zhou, L. Impaired mitochondrial network excitability in failing guinea-pig cardiomyocytes. Cardiovasc. Res. 2016, 109, 79–89. [Google Scholar] [CrossRef]
- Maack, C. Orphaned mitochondria in heart failure. Cardiovasc. Res. 2016, 109, 6–8. [Google Scholar] [CrossRef]
- Yu, Z.; Chen, R.; Li, M.; Yu, Y.; Liang, Y.; Han, F.; Qin, S.; Chen, X.; Su, Y.; Ge, J. Mitochondrial calcium uniporter inhibition provides cardioprotection in pressure overload-induced heart failure through autophagy enhancement. Int. J. Cardiol. 2018, 271, 161–168. [Google Scholar] [CrossRef] [PubMed]
- Tahrir, F.G.; Langford, D.; Amini, S.; Mohseni Ahooyi, T.; Khalili, K. Mitochondrial quality control in cardiac cells: Mechanisms and role in cardiac cell injury and disease. J. Cell. Physiol. 2019, 234, 8122–8133. [Google Scholar] [CrossRef] [PubMed]
- Wang, K.; Xu, Y.; Sun, Q.; Long, J.; Liu, J.; Ding, J. Mitochondria regulate cardiac contraction through ATP-dependent and independent mechanisms. Free Radic. Res. 2018, 52, 1256–1265. [Google Scholar] [CrossRef] [PubMed]
- Octavia, Y.; Brunner-La Rocca, H.P.; Moens, A.L. NADPH oxidase-dependent oxidative stress in the failing heart: From pathogenic roles to therapeutic approach. Free Radic. Biol. Med. 2012, 15, 291–297. [Google Scholar] [CrossRef]
- Nickel, A.G.; von Hardenberg, A.; Hohl, M.; Löffler, J.R.; Kohlhaas, M.; Becker, J.; Reil, J.C.; Kazakov, A.; Bonnekoh, J.; Stadelmaier, M.; et al. Reversal of mitochondrial transhydrogenase causes oxidative stress in heart failure. Cell Metab. 2005, 22, 472–484. [Google Scholar] [CrossRef] [Green Version]
- Aon, M.A.; Cortassa, S.; O’Rourke, B. Redox-optimized ROS balance: A unifying hypothesis. Biochim. Biophys. Acta 2010, 1797, 865–877. [Google Scholar] [CrossRef] [Green Version]
- Huang, P.L. eNOS, metabolic syndrome and cardiovascular disease. Trends Endocrinol. Metab. 2009, 20, 295–302. [Google Scholar] [CrossRef] [Green Version]
- Zorov, D.B.; Juhaszova, M.; Sollott, S.J. Mitochondrial reactive oxygen species (ROS) and ROS-induced ROS release. Physiol. Rev. 2014, 94, 909–950. [Google Scholar] [CrossRef] [Green Version]
- Stehling, O.; Lill, R. The role of mitochondria in cellular iron–sulfur protein biogenesis: Mechanisms, connected processes, and diseases. Cold Spring Harb. Perspect. Biol. 2013, 5, a011312. [Google Scholar] [CrossRef] [Green Version]
- Paradies, G.; Paradies, V.; Ruggiero, F.M.; Petrosillo, G. Oxidative stress, cardiolipin and mitochondrial dysfunction in nonalcoholic fatty liver disease. World J. Gastroenterol. 2014, 20, 14205–14218. [Google Scholar] [CrossRef]
- Paradies, G.; Petrosillo, G.; Paradies, V.; Ruggiero, F.M. Role of cardiolipin peroxidation and Ca2+ in mitochondrial dysfunction and disease. Cell Calcium. 2009, 45, 643–650. [Google Scholar] [CrossRef] [PubMed]
- Paradies, G.; Paradies, V.; De Benedictis, V.; Ruggiero, F.M.; Petrosillo, G. Functional role of cardiolipin in mitochondrial bioenergetics. Biochimica et Biophysica Acta (BBA) 2014, 1837, 408–417. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cheng, J.; Nanayakkara, G.; Shao, Y.; Cueto, R.; Wang, L.; Yang, W.Y.; Tian, Y.; Wang, H.; Yang, X. mitochondrial proton leak plays a critical role in pathogenesis of cardiovascular diseases. Adv. Exp. Med. Biol. 2017, 982, 359–370. [Google Scholar] [PubMed] [Green Version]
- Karamanlidis, G.; Nascimben, L.; Couper, G.S.; Shekar, P.S.; del Monte, F.; Tian, R. Defective DNA replication impairs mitochondrial biogenesis in human failing hearts. Circ. Res. 2010, 106, 1541–1548. [Google Scholar] [CrossRef] [Green Version]
- Ide, T.; Tsutsui, H.; Hayashidani, S.; Kang, D.; Suematsu, N.; Nakamura, K.; Utsumi, H.; Hamasaki, N.; Takeshita, A. Mitochondrial DNA damage and dysfunction associated with oxidative stress in failing hearts after myocardial infarction. Circ. Res. 2001, 88, 529–535. [Google Scholar] [CrossRef] [Green Version]
- Dhalla, N.S.; Temsah, R.M.; Netticadan, T. Role of oxidative stress in cardiovascular diseases. J. Hypertens. 2000, 18, 655–673. [Google Scholar] [CrossRef]
- Arrieta, A.; Blackwood, E.A.; Stauffer, W.T.; Glembotski, C.C. Integrating ER and Mitochondrial Proteostasis in the Healthy and Diseased Heart. Front. Cardiovasc. Med. 2020, 6, 193. [Google Scholar] [CrossRef]
- Glembotski, C.C. Endoplasmic reticulum stress in the heart. Circ. Res. 2007, 101, 975–984. [Google Scholar] [CrossRef]
- Jin, J.K.; Blackwood, E.A.; Azizi, K.; Thuerauf, D.J.; Fahem, A.G.; Hofmann, C.; Kaufman, R.J.; Doroudgar, S.; Glembotski, C.C. ATF6 decreases myocardial ischemia/reperfusion damage and links ER stress and oxidative stress signaling pathways in the heart. Circ. Res. 2017, 120, 862–875. [Google Scholar] [CrossRef] [Green Version]
- Ochoa, C.D.; Wu, R.F.; Terada, L.S. ROS signaling and ER stress in cardiovascular disease. Mol. Asp. Med. 2018, 63, 18–29. [Google Scholar] [CrossRef]
- Chen, C.-Y.; Zhang, S.-L.; Liu, Z.-Y.; Tian, Y.; Sun, Q. Cadmium toxicity induces ER stress and apoptosis via impairing energy homoeostasis in cardiomyocytes. Biosci. Rep. 2015, 35, e00214. [Google Scholar] [CrossRef] [PubMed]
- Cao, S.S.; Kaufman, R.J. Endoplasmic Reticulum Stress and Oxidative Stress in Cell Fate Decision and Human Disease. Antioxid. Redox Signal. 2014, 21, 396–413. [Google Scholar] [CrossRef] [PubMed]
- Martucciello, S.; Masullo, M.; Cerulli, A.; Piacente, S. Natural Products Targeting ER Stress, and the Functional Link to Mitochondria. Int. J. Mol. Sci. 2020, 21, 1905. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, S.; Binder, P.; Fang, Q.; Wang, Z.; Xiao, W.; Liu, W.; Wang, X. Endoplasmic reticulum stress in the heart: Insights into mechanisms and drug targets. Br. J. Pharmacol. 2018, 175, 1293–1304. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Saito, A.; Ochiai, K.; Kondo, S.; Tsumagari, K.; Murakami, T.; Cavener, D.R.; Imaizumi, K. Endoplasmic reticulum stress response mediated by the PERK-eIF2(alpha)-ATF4 pathway is involved in osteoblast differentiation induced by BMP2. J. Biol. Chem. 2011, 286, 4809–4818. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ni, M.; Zhang, Y.; Lee, A.S. Beyond the endoplasmic reticulum: Atypical GRP78 in cell viability, signalling and therapeutic targeting. Biochem. J. 2011, 434, 181–188. [Google Scholar] [CrossRef] [PubMed]
- Ariyasinghe, N.R.; Lyra-Leite, D.M.; McCain, M.L. Engineering cardiac microphysiological systems to model pathological extracellular matrix remodeling. Am. J. Physiol. Heart Circ. Physiol. 2018, 315, H771–H789. [Google Scholar] [CrossRef]
- Frangogiannis, N.G. The extracellular matrix in ischemic and nonischemic heart failure. Circ. Res. 2019, 125, 117–146. [Google Scholar] [CrossRef]
- Takawale, A.; Sakamuri, S.S.; Kassiri, Z. Extracellular matrix communication and turnover in cardiac physiology and pathology. Compr. Physiol. 2015, 5, 687–719. [Google Scholar]
- Kelly, D.; Cockerill, G.; Ng, L.L.; Thompson, M.; Khan, S.; Samani, N.J.; Squire, I.B. Plasma matrix metalloproteinase-9 and left ventricular remodelling after acute myocardial infarction in man: A prospective cohort study. Eur. Heart J. 2007, 28, 711–718. [Google Scholar] [CrossRef] [Green Version]
- Hsiao, K.C.; Tsai, J.P.; Yang, S.F.; Lee, W.C.; Huang, J.Y.; Chang, S.C.; Hso, C.S.; Chang, H.R. MMP-2 serum concentrations predict mortality in hemodialysis patients: A 5-year cohort study. Clin. Chim. Acta 2016, 452, 161–166. [Google Scholar] [CrossRef] [PubMed]
- Hemdahl, A.L.; Gabrielsen, A.; Zhu, C.; Eriksson, P.; Hedin, U.; Kastrup, J.; Thorén, P.; Hansson, G.K. Expression of neutrophil gelatinase–associated lipocalin in atherosclerosis and myocardial infarction. Arterioscler. Thromb. Vasc. Biol. 2006, 26, 136–142. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sawicki, G.; Leon, H.; Sawicka, J.; Sariahmetoglu, M.; Schulze, C.J.; Scott, P.G.; Szczesna-Cordary, D.; Schulz, R. Degradation of myosin light chain in isolated rat hearts subjected to ischemia-reperfusion injury: A new intracellular target for matrix metalloproteinase-2. Circulation 2005, 112, 544–552. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Belo, V.A.; Guimarães, D.A.; Castro, M.M. Matrix metalloproteinase 2 as a potential mediator of vascular smooth muscle cell migration and chronic vascular remodelling in Hypertension. J. Vasc. Res. 2015, 52, 221–231. [Google Scholar] [CrossRef]
- Papazafiropoulou, A.; Tentolouris, N. Matrix metalloproteinases and cardiovascular diseases. Hippokratia 2009, 13, 76–82. [Google Scholar]
- Lalu, M.M.; Pasini, E.; Schulze, C.J.; Ferrari-Vivaldi, M.; Ferrari-Vivaldi, G.; Bachetti, T.; Schulz, R. Ischaemia-reperfusion injury activates matrix metalloproteinases in the human heart. Eur. Heart J. 2005, 26, 27–35. [Google Scholar] [CrossRef]
- DeCoux, A.; Lindsey, M.L.; Villarreal, F.; Garcia, R.A.; Schulz, R. Myocardial matrix metalloproteinase-2: Inside out and upside down. J. Mol. Cell. Cardiol. 2014, 77, 64–72. [Google Scholar] [CrossRef] [Green Version]
- Liu, P.; Sun, M.; Sader, S. Matrix metalloproteinases in cardiovascular disease. Can. J. Cardiol. 2006, 22 (Suppl. B), 25B–30B. [Google Scholar] [CrossRef] [Green Version]
- Wang, G.Y.; Bergman, M.R.; Nguyen, A.P.; Turcato, S.; Swigart, P.M.; Rodrigo, M.C.; Simpson, P.C.; Karliner, J.S.; Lovett, D.H.; Baker, A.J. Cardiac transgenic matrix metalloproteinase-2 expression directly induces impaired contractility. Cardiovasc. Res. 2006, 69, 688–696. [Google Scholar] [CrossRef] [Green Version]
- Azevedo, A.; Prado, A.F.; Antonio, R.C.; Issa, J.P.; Gerlach, R.F. Matrix metalloproteinases are involved in cardiovascular diseases. Basic Clin. Pharmacol. Toxicol. 2014, 115, 301–314. [Google Scholar] [CrossRef]
- DeLeon-Pennell, K.Y.; Meschiari, C.A.; Jung, M.; Lindsey, M.L. Matrix metalloproteinases in myocardial infarction and heart failure. Prog. Mol. Biol. Transl. Sci. 2017, 147, 75–100. [Google Scholar] [PubMed] [Green Version]
- Lovett, D.H.; Chu, C.; Wang, G.; Ratcliffe, M.B.; Baker, A.J. A N-terminal truncated intracellular isoform of matrix metalloproteinase-2 impairs contractility of mouse myocardium. Front. Physiol. 2014, 5, 363. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Giri, B.; Dey, S.; Das, T.; Sarkar, M.; Banerjee, J.; Dash, S.K. Chronic hyperglycemia mediated physiological alteration and metabolic distortion leads to organ dysfunction, infection, cancer progression and other pathophysiological consequences: An update on glucose toxicity. Biomed. Pharmacother. 2018, 107, 306–328. [Google Scholar] [CrossRef] [PubMed]
- Gliozzi, M.; Scarano, F.; Musolino, V.; Carresi, C.; Scarcella, A.; Nucera, S.; Scicchitano, M.; Ruga, S.; Bosco, F.; Maiuolo, J.; et al. Paradoxical effect of fat diet in matrix metalloproteinases induced mitochondrial dysfunction in diabetic cardiomyopathy. J. Cardiovasc. Med. (Hagerstown) 2020. [Google Scholar] [CrossRef]
- Ilkan, Z.; Akar, F.G. The Mitochondrial Translocator Protein and the Emerging Link between Oxidative Stress and Arrhythmias in the Diabetic Heart. Front. Physiol. 2018, 9, 1518. [Google Scholar] [CrossRef] [Green Version]
- Motloch, L.J.; Hu, J.; Akar, F.G. The mitochondrial translocator protein and arrhythmogenesis in ischemic heart disease. Oxid. Med. Cell. Longev. 2015, 2015, 234104. [Google Scholar] [CrossRef] [Green Version]
- Searles, C.D. The nitric oxide pathway and oxidative stress in heart failure. Congest. Heart Fail. 2002, 8, 142–155. [Google Scholar]
- Carnicer, R.; Crabtree, M.J.; Sivakumaran, V.; Casadei, B.; Kass, D.A. Nitric oxide synthases in heart failure. Antioxid. Redox Signal. 2013, 18, 1078–1099. [Google Scholar] [CrossRef] [Green Version]
- Ebong, I.A.; Goff, D.C.; Rodriguez, C.J.; Chen, H.; Bertoni, A.G. Mechanisms of heart failure in obesity. Obes. Res. Clin. Pract. 2014, 8, e540–e548. [Google Scholar] [CrossRef] [Green Version]
- Roger, V.L.; Go, A.S.; Lloyd-Jones, D.M.; Adams, R.J.; Berry, J.D.; Brown, T.M.; Carnethon, M.R.; Dai, S.; de Simone, G.; Ford, E.S.; et al. Heart disease and stroke statistics-2011 update: A report from the American Heart Association. Circulation 2011, 123, e18–e209. [Google Scholar] [CrossRef] [Green Version]
- Greene, S.J.; Gheorghiade, M.; Borlaug, B.A.; Pieske, B.; Vaduganathan, M.; Burnett, J.C.; Roessig, L.; Stasch, J.P.; Solomon, S.D.; Paulus, W.J.; et al. The cGMP signaling pathway as a therapeutic target in heart failure with preserved ejection fraction. J. Am. Heart Assoc. 2013, 2, e000536. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, Q.; Yon, J.Y.; Cai, H. Mechanisms and consequences of eNOS dysfunction in hypertension. J. Hypertens. 2015, 33, 1128–1136. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tsai, E.J.; Liu, Y.; Koitabashi, N.; Bedja, D.; Danner, T.; Jasmin, J.F.; Lisanti, M.; Friebe, A.; Takimoto, E.; Kass, D.A. Pressure-overload-induced subcellular relocalization/oxidation of soluble guanylyl cyclase in the heart modulates enzyme stimulation. Circ. Res. 2012, 110, 295–303. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, G.E.; Kass, D.A. Cardiac phosphodiesterases and their modulation for treating heart disease. Handb. Exp. Pharmacol. 2017, 243, 249–269. [Google Scholar]
- Nagendran, J.; Archer, S.L.; Soliman, D.; Gurtu, V.; Moudgil, R.; Haromy, A.; St Aubin, C.; Webster, L.; Rebeyka, I.M.; Ross, D.B.; et al. Phosphodiesterase type 5 is highly expressed in the hypertrophied human right ventricle, and acute inhibition of phosphodiesterase type 5 improves contractility. Circulation 2007, 116, 238–248. [Google Scholar] [CrossRef]
- Blanton, R.M. cGMP signaling and modulation in heart failure. J. Cardiovasc. Pharmacol. 2020, 75, 385–398. [Google Scholar] [CrossRef]
- Methawasin, M.; Strom, J.; Borkowski, T.; Hourani, Z.; Runyan, R.; Smith, J.E., III; Granzier, H. Phosphodiesterase 9a inhibition in mouse models of diastolic dysfunction. Circ. Heart Fail. 2020, 13, e006609. [Google Scholar] [CrossRef]
- Takimoto, E.; Koitabashi, N.; Hsu, S.; Ketner, E.A.; Zhang, M.; Nagayama, T.; Bedja, D.; Gabrielson, K.L.; Blanton, R.; Siderovski, D.P.; et al. Regulator of G protein signaling 2 mediates cardiac compensation to pressure overload and antihypertrophic effects of PDE5 inhibition in mice. J. Clin. Investig. 2009, 119, 408–420. [Google Scholar] [CrossRef] [Green Version]
- Lee, D.I.; Zhu, G.; Sasaki, T.; Cho, G.S.; Hamdani, N.; Holewinski, R.; Jo, S.H.; Danner, T.; Zhang, M.; Rainer, P.P. Phosphodiesterase 9A controls nitric-oxide-independent cGMP and hypertrophic heart disease. Nature 2015, 519, 472–476. [Google Scholar] [CrossRef]
- Van Heerebeek, L.; Paulus, W.J. Understanding heart failure with preserved ejection fraction: Where are we today? Neth. Heart J. 2016, 24, 227–236. [Google Scholar] [CrossRef] [Green Version]
- Verma, S.; McMurray, J.J.V. SGLT2 inhibitors and mechanisms of cardiovascular benefit: A state-of-the-art review. Diabetologia 2018, 61, 2108–2117. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gambardella, J.; Lombardi, A.; Santulli, G. Metabolic flexibility of mitochondria plays a key role in balancing glucose and fatty acid metabolism in the diabetic heart. Diabetes 2020, 69, 2054–2057. [Google Scholar] [CrossRef] [PubMed]
- Leichman, J.G.; Aguilar, D.; King, T.M.; Vlada, T.A.; Reyes, E.; Taegtmeyer, H. Association of plasma free fatty acids and left ventricular diastolic function in patients with clinically severe obesity. Am. J. Clin. Nutr. 2006, 84, 336–341. [Google Scholar] [CrossRef] [PubMed]
- Ekanayake, P.; Hupfeld, C.; Mudaliar, S. Sodium-glucose cotransporter Type 2 (SGLT-2) inhibitors and ketogenesis: The Good and the Bad. Curr. Diab. Rep. 2020, 20, 1–10. [Google Scholar] [CrossRef]
- Ferrannini, E.; Mark, M.; Mayoux, E. CV protection in the EMPA-REG OUTCOME Trial: A “Thrifty Substrate” hypothesis. Diabetes Care 2016, 39, 1108–1114. [Google Scholar] [CrossRef] [Green Version]
- Mizuno, Y.; Harada, E.; Nakagawa, H.; Morikawa, Y.; Shono, M.; Kugimiya, F.; Yoshimura, M.; Yasue, H. The diabetic heart utilizes ketone bodies as an energy source. Metabolism 2007, 77, 65–72. [Google Scholar] [CrossRef] [Green Version]
- Gormsen, L.C.; Svart, M.; Thomsen, H.H.; Søndergaard, E.; Vendelbo, M.H.; Christensen, N.; Tolbod, L.P.; Harms, H.J.; Nielsen, R.; Wiggers, H.; et al. Ketone body infusion with 3-hydroxybutyrate reduces myocardial glucose uptake and increases blood flow in humans: A positron emission tomography study. J. Am. Heart Assoc. 2017, 6, e005066. [Google Scholar] [CrossRef]
- Santos-Gallego, C.G.; Ibanez, J.A.R.; San Antonio, R.; Ishikawa, K.; Watanabe, S.; Botija, M.B.P.; Sanz Salvo, A.J.; Hajjar, R.; Fuster, V.; Badimon, J. Empagliflozin induces a myocardial metabolic shift from glucose consumption to ketone metabolism that mitigates adverse cardiac remodeling and improves myocardial contractility. J. Am. Coll. Cardiol. 2018, 71, A674. [Google Scholar] [CrossRef]
- Lopaschuk, G.D.; Verma, S. Empagliflozin’s fuel hypothesis: Not so soon. Cell Metab. 2016, 24, 200–202. [Google Scholar] [CrossRef] [Green Version]
- Kappel, B.A.; Lehrke, M.; Schutt, K.; Artati, A.; Adamski, J.; Lebherz, C.; Marz, N. Effect of empagliflozin on the metabolic signature of patients with type 2 diabetes mellitus and cardiovascular disease. Circulation 2017, 136, 969–972. [Google Scholar] [CrossRef]
- Packer, M.; Anker, S.D.; Butler, J.; Filippatos, G.; Zannad, F. Effects of sodium-glucose cotransporter 2 inhibitors for the treatment of patients with heart failure: Proposal of a novel mechanism of action. JAMA Cardiol. 2017, 2, 1025–1029. [Google Scholar] [CrossRef] [PubMed]
- Uthman, L.; Baartscheer, A.; Bleijlevens, B.; Schumacher, C.A.; Fiolet, J.W.T.; Koeman, A.; Jancev, M.; Hollmann, M.W.; Weber, N.C.; Coronel, R.; et al. Class effects of SGLT2 inhibitors in mouse cardiomyocytes and hearts: Inhibition of Na+/H+ exchanger, lowering of cytosolic Na+ and vasodilation. Diabetologia 2018, 61, 722–726. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gallo, L.A.; Wright, E.M.; Vallon, V. Probing SGLT2 as a therapeutic target for diabetes: Basic physiology and consequences. Diab. Vasc. Dis. Res. 2015, 12, 78–89. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fedak, P.W.; Verma, S.; Weisel, R.D.; Li, R.K. Cardiac remodeling and failure from molecules to man (part II). Cardiovasc. Pathol. 2005, 14, 49–60. [Google Scholar] [CrossRef]
- Lee, T.M.; Chang, N.C.; Lin, S.Z. Dapagliflozin, a selective SGLT2 inhibitor, attenuated cardiac fibrosis by regulating the macrophage polarization via STAT3 signaling in infarcted rat hearts. Free Radic. Biol. Med. 2017, 104, 298–310. [Google Scholar] [CrossRef]
- Kang, S.; Verma, S.; Hassanabad, A.F.; Teng, G.; Belke, D.D.; Dundas, J.A.; Guzzardi, D.G.; Svystonyuk, D.A.; Pattar, S.S.; Park, D.S.J.; et al. Direct effects of empagliflozin on extracellular matrix remodeling in human cardiac fibroblasts: Novel translational clues to EMPA-REG Outcome. Can. J. Cardiol. 2020, 36, 543–553. [Google Scholar] [CrossRef] [Green Version]
- Braunwald, E. The war against heart failure: The Lancet lecture. Lancet 2015, 385, 812–824. [Google Scholar] [CrossRef]
- Gerber, Y.; Weston, S.A.; Enriquez-Sarano, M.; Berardi, C.; Chamberlain, A.M.; Manemann, S.M.; Jiang, R.; Dunlay, S.M.; Roger, V.L. Mortality associated with heart failure after myocardial infarction: A contemporary community perspective. Circ. Heart Fail. 2016, 9, e002460. [Google Scholar] [CrossRef] [Green Version]
- Gerber, Y.; Weston, S.A.; Berardi, C.; McNallan, S.M.; Jiang, R.; Redfield, M.M.; Roger, V.L. Contemporary trends in heart failure with reduced and preserved ejection fraction after myocardial infarction: A community study. Am. J. Epidemiol. 2013, 178, 1272–1280. [Google Scholar] [CrossRef] [Green Version]
- Cruz, M.S.; da Silva, A.M.G.; da Souza, K.S.C.; Luchessi, A.D.; Silbiger, V.N. miRNAs emerge as circulating biomarkers of post-myocardial infarction heart failure. Heart Fail. Rev. 2020, 25, 321–329. [Google Scholar] [CrossRef]
- Nadal-Ginard, B.; Torella, D.; De Angelis, A.; Rossi, F. Monographic issue of pharmacological research on adult myocardial repair/regeneration. Pharmacol. Res. 2018, 127, 1–3. [Google Scholar] [CrossRef] [PubMed]
- Nadal-Ginard, B.; Ellison, G.M.; Torella, D. The cardiac stem cell compartment is indispensable for myocardial cell homeostasis, repair and regeneration in the adult. Stem. Cell. Res. 2014, 13 Pt B, 615–630. [Google Scholar] [CrossRef] [Green Version]
- Beltrami, A.P.; Barlucchi, L.; Torella, D.; Baker, M.; Limana, F.; Chimenti, S.; Kasahara, H.; Rota, M.; Musso, E.; Urbanek, K.; et al. Adult cardiac stem cells are multipotent and support myocardial regeneration. Cell 2003, 114, 763–776. [Google Scholar] [CrossRef] [Green Version]
- Chabot, B.; Stephenson, D.A.; Chapman, V.M.; Besmer, P.; Bernstein, A. The proto-oncogene c-kit encoding a transmembrane tyrosine kinase receptor maps to the mouse W locus. Nature 1988, 335, 88–89. [Google Scholar] [CrossRef] [PubMed]
- Morrison, S.J.; Wandycz, A.M.; Hemmati, H.D.; Wright, D.E.; Weissman, I.L. Identification of a lineage of multipotent hematopoietic progenitors. Development 1997, 124, 1929–1939. [Google Scholar] [PubMed]
- Sellers, S.E.; Tisdale, J.F.; Agricola, B.A.; Metzger, M.E.; Donahue, R.E.; Dunbar, C.E.; Sorrentino, B.P. The effect of multidrug-resistance 1 gene versus neo transduction on ex vivo and in vivo expansion of rhesus macaque hematopoietic repopulating cells. Blood 2001, 97, 1888–1891. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nocka, K.; Majumder, S.; Chabot, B.; Ray, P.; Cervone, M.; Bernstein, A.; Besmer, P. Expression of c-kit gene products in known cellular targets of W mutations in normal and W mutant mice--evidence for an impaired c-kit kinase in mutant mice. Genes Dev. 1989, 3, 816–826. [Google Scholar] [CrossRef] [Green Version]
- Vajravelu, B.N.; Hong, K.U.; Al-Maqtari, T.; Cao, P.; Keith, M.C.; Wysoczynski, M.; Zhao, J.; Moore, J.B., IV; Bolli, R. C-Kit promotes growth and migration of human cardiac progenitor cells via the PI3K-AKT and MEK-ERK Pathways. PLoS ONE 2015, 10, e0140798. [Google Scholar] [CrossRef] [Green Version]
- Cimini, M.; Fazel, S.; Zhuo, S.; Xaymardan, M.; Fujii, H.; Weisel, R.D.; Li, R.K. c-kit dysfunction impairs myocardial healing after infarction. Circulation 2007, 116 (Suppl. 11), I77–I82. [Google Scholar] [CrossRef] [Green Version]
- Li, M.; Naqvi, N.; Yahiro, E.; Liu, K.; Powell, P.C.; Bradley, W.E.; Martin, D.I.K.; Graham, R.M.; Dell’Italia, L.J.; Husain, A. c-kit is required for cardiomyocyte terminal differentiation. Circ. Res. 2008, 102, 677–685. [Google Scholar] [CrossRef] [Green Version]
- Ye, L.; Zhang, E.Y.; Xiong, Q.; Astle, C.M.; Zhang, P.; Li, Q.; From, A.H.; Harrison, D.E.; Zhang, J.J. Aging Kit mutant mice develop cardiomyopathy. PLoS ONE 2012, 7, e33407. [Google Scholar] [CrossRef] [PubMed]
- Lennartsson, J.; Rönnstrand, L. Stem cell factor receptor/c-Kit: From basic science to clinical implications. Physiol. Rev. 2012, 92, 1619–1649. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Van Berlo, J.H.; Kanisicak, O.; Maillet, M.; Vagnozzi, R.J.; Karch, J.; Lin, S.C.; Middleton, R.C.; Marbán, E.; Molkentin, J.D. c-kit+ cells minimally contribute cardiomyocytes to the heart. Nature 2014, 509, 337–341. [Google Scholar] [CrossRef] [PubMed]
- Vicinanza, C.; Aquila, I.; Cianflone, E.; Scalise, M.; Marino, F.; Mancuso, T.; Fumagalli, F.; Giovannone, E.D.; Cristiano, F.; Iaccino, E.; et al. Kitcre knock-in mice fail to fate-map cardiac stem cells. Nature 2018, 555, E1–E5. [Google Scholar] [CrossRef] [PubMed]
- Chimenti, C.; Kajstura, J.; Torella, D.; Urbanek, K.; Heleniak, H.; Colussi, C.; Di Meglio, F.; Nadal-Ginard, B.; Frustaci, A.; Leri, A.; et al. Senescence and death of primitive cells and myocytes lead to premature cardiac aging and heart failure. Circ. Res. 2003, 93, 604–613. [Google Scholar] [CrossRef] [PubMed]
- Torella, D.; Rota, M.; Nurzynska, D.; Musso, E.; Monsen, A.; Shiraishi, I.; Zias, E.; Walsh, K.; Rosenzweig, A.; Sussman, M.A.; et al. Cardiac stem cell and myocyte aging, heart failure, and insulin-like growth factor-1 overexpression. Circ. Res. 2004, 94, 514–524. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chua, C.C.; Gao, J.; Ho, Y.S.; Xu, X.; Kuo, I.C.; Chua, K.Y.; Wang, H.; Hamdy, R.C.; Reed, J.C.; Chua, B.H. Over-expression of a modified bifunctional apoptosis regulator protects against cardiac injury and doxorubicin-induced cardiotoxicity in transgenic mice. Cardiovasc. Res. 2009, 81, 20–27. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zakharova, L.; Nural-Guvener, H.; Nimlos, J.; Popovic, S.; Gaballa, M.A. Chronic heart failure is associated with transforming growth factor beta-dependent yield and functional decline in atrial explant-derived c-Kit+ cells. J. Am. Heart Assoc. 2013, 2, e000317. [Google Scholar] [CrossRef] [Green Version]
- Itzhaki-Alfia, A.; Leor, J.; Raanani, E.; Sternik, L.; Spiegelstein, D.; Netser, S.; Holbova, R.; Pevsner-Fischer, M.; Lavee, J.; Barbash, I.M. Patient characteristics and cell source determine the number of isolated human cardiac progenitor cells. Circulation 2009, 120, 2559–2566. [Google Scholar] [CrossRef]
- Huang, C.; Zhang, X.; Ramil, J.M.; Rikka, S.; Kim, L.; Lee, Y.; Gude, N.A.; Thistlethwaite, P.A.; Sussman, M.A.; Gottlieb, R.A.; et al. Juvenile exposure to anthracyclines impairs cardiac progenitor cell function and vascularization resulting in greater susceptibility to stress-induced myocardial injury in adult mice. Circulation 2010, 121, 675–683. [Google Scholar] [CrossRef]
- Carresi, C.; Musolino, V.; Gliozzi, M.; Maiuolo, J.; Mollace, R.; Nucera, S.; Maretta, A.; Sergi, D.; Muscoli, S.; Gratteri, S.; et al. Anti-oxidant effect of bergamot polyphenolic fraction counteracts doxorubicin-induced cardiomyopathy: Role of autophagy and ckitposCD45negCD31neg cardiac stem cell activation. J. Mol. Cell. Cardiol. 2018, 119, 10–18. [Google Scholar] [CrossRef] [PubMed]
- Enriqueza, J.A.; Lenazb, G. Coenzyme Q and the Respiratory Chain: Coenzyme Q Pool and Mitochondrial Supercomplexes. Mol. Syndromol. 2014, 5, 119–140. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ayer, A.M.P.; Stocker, R. CoQ10 function and role in heart failure and ischemic heart disease. Annu. Rev. Nutr. 2015, 2015, 175–213. [Google Scholar] [CrossRef] [PubMed]
- Crane, F.L. Discovery of ubiquinone (coenzyme Q) and an overview of function. Mitochondrion 2007, 7, S2–S7. [Google Scholar] [CrossRef]
- Burridge, P.W.; Li, Y.F.; Matsa, E.; Wu, H.; Ong, S.G.; Sharma, A.; Holmström, A.; Chang, A.C.; Coronado, M.J.; Ebert, A.D.; et al. Human induced pluripotent stem cell-derived cardiomyocytes recapitulate the predilection of breast cancer patients to doxorubicin-induced cardiotoxicity. Nat. Med. 2016, 22, 547–556. [Google Scholar] [CrossRef] [Green Version]
- Milano, G.; Biemmi, V.; Lazzarini, E.; Balbi, C.; Ciullo, A.; Bolis, S.; Ameri, P.; Di Silvestre, D.; Mauri, P.; Barile, L.; et al. Intravenous administration of cardiac progenitor cell-derived exosomes protects against doxorubicin/trastuzumab-induced cardiac toxicity. Cardiovasc. Res. 2020, 116, 383–392. [Google Scholar] [CrossRef]
- Barbaro, B.; Toietta, G.; Maggio, R.; Arciello, M.; Tarocchi, M.; Galli, A.; Balsano, C. Effects of the Olive-Derived Polyphenol Oleuropein on Human Health. Int. J. Mol. Sci. 2014, 15, 18508–18524. [Google Scholar] [CrossRef]
- Vogel, P.; Machado, I.K.; Garavaglia, J.; Zani, V.T.; de Souza, D.; Dal Bosco, S.M. Polyphenols benefits of olive leaf (Olea europaea L.) to human health. Nutr. Hosp. 2015, 31, 1427–1433. [Google Scholar]
- Rizzo, M.; Ventrice, D.; Giannetto, F.; Cirinnà, S.; Santagati, N.A.; Procopio, A.; Mollace, V.; Muscoli, C. Antioxidant activity of oleuropein and semisynthetic acetyl-derivatives determined by measuring malondialdehyde in rat brain. J. Pharm. Pharmacol. 2017, 69, 1502–1512. [Google Scholar] [CrossRef]
- Romani, A.; Ieri, F.; Urciuoli, S.; Noce, A.; Marrone, G.; Nediani, C.; Bernini, R. Effects of phenolic compounds found in extra-virgin olive oil, by-products, and leaf of Olea europaea L. Nutrients 2019, 11, 1776. [Google Scholar] [CrossRef] [Green Version]
- Taamalli, A.; Feriani, A.; Lozano-Sanchez, J.; Ghazouani, L.; El Mufti, A.; Allagui, M.S.; Segura-Carretero, A.; Mhamdi, R.; Arráez-Roman, D. Potential hepatoprotective activity of super critical carbon dioxide olive leaf extracts against CCl4-Induced Liver Damage. Foods 2020, 9, 804. [Google Scholar] [CrossRef] [PubMed]
- Okrouhlá, M.; Stupka, R.; Čítek, J.; Lebedová, N.; Zadinová, K. Effect of duration of dietary rapeseed and soybean oil feeding on physical characteristics, fatty acid profile, and oxidative stability of pig backfat. Animals 2018, 8, 193. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Flori, L.; Donnini, S.; Calderone, V.; Zinnai, A.; Taglieri, I.; Venturi, F.; Testai, L. The nutraceutical value of olive oil and its bioactive constituents on the cardiovascular system. Focusing on main strategies to slow down its quality decay during production and storage. Nutrients 2019, 11, 1962. [Google Scholar]
- Beltran, G.; Aguilera, M.P.; Del-Rio, C.; Sanchez, S.; Martinez, L. Influence of fruit ripening on the natural antioxidant content of Hojiblanca virgin olive oils. Food Chem. 2005, 89, 207–215. [Google Scholar] [CrossRef]
- Bulotta, S.; Celano, M.; Lepore, S.M.; Montalcini, T.; Pujia, A.; Russo, D. Beneficial effects of the olive oil phenolic components oleuropein and hydroxytyrosol: Focus on protection against cardiovascular and metabolic diseases. J. Transl. Med. 2014, 12, 219. [Google Scholar] [CrossRef] [Green Version]
- Bendini, A.; Cerretani, L.; Carrasco-Pancorbo, A.; Gómez-Caravaca, A.M.; Segura-Carretero, A.; Fernández-Gutiérrez, A.; Lercker, G. Phenolic molecules in virgin olive oils: A survey of their sensory properties, health effects, antioxidant activity and analytical methods. An overview of the last decade. Molecules 2007, 12, 1679–1719. [Google Scholar] [CrossRef]
- Servili, M.; Montedoro, G. Contribution of phenolic compound to virgin olive oil quality. Eur. J. Lipid Sci. Technol. 2002, 104, 602–613. [Google Scholar] [CrossRef]
- Tripoli, E.; Giammanco, M.; Tabacchi, G.; Di Majo, D.; Giammanco, S.; La Guardia, M. The phenolic compounds of olive oil: Structure, biological activity and beneficial effects on human health. Nutr. Res. Rev. 2005, 18, 98–112. [Google Scholar] [CrossRef]
- Nediani, C.; Ruzzolini, J.; Romani, A.; Calorini, L. Oleuropein, a bioactive compound from Olea europaea L., as a potential preventive and therapeutic agent in non-communicable diseases. Antioxidants 2019, 8, 578. [Google Scholar] [CrossRef] [Green Version]
- Zhang, X.Z.; Zhao, Y.B.; Li, C.M.; Chen, D.M.; Wang, G.P.; Chang, R.F.; Shu, H. Potential polyphenol markers of phase change in apple (Malus domestica). J. Plant. Physiol. 2007, 164, 574–580. [Google Scholar] [CrossRef]
- Rodríguez-Pérez, C.; García-Villanova, B.; Guerra-Hernández, E.; Verardo, V. Grape Seeds proanthocyanidins: An overview of in vivo bioactivity in animal models. Nutrients 2019, 11, 2435. [Google Scholar] [CrossRef] [PubMed] [Green Version]



{kind=link}
{kind=link}
| Bioactive Component | Clinical Trials | Study Duration | Dosage Supplement | Properties | References |
|---|---|---|---|---|---|
| Coenzyme Q10 | - Meta-analysis of 13 randomised controlled trials - Includes 395 participants (49.8 to 68.0 years) At baseline: - Blood CoQ10 concentration 0.61–1.01 μg/mL. - EF 22–46%. - NYHA functional class 2.3–3.4 | 4–28 weeks | 60–300 mg/day | - Pooled mean net increase in blood CoQ10 concentration: 1.4 μg/mL (95% CI: 1.1, 1.7 μg/mL) - Pooled mean net increase in EF: 3.67% (95% CI: 1.60%, 5.74%) - Study-specific changes in EF CoQ10 supplementation vs. placebo: −3.0% (95% CI: −10.7%, 4.7%) to 17.8% (95% CI: 7.2%, 28.4%) - Pooled mean net change in NYHA classification: −0.30 (95% CI: −0.66, 0.06) | [21] |
| - A randomised controlled multicenter trial Q-SYMBIO - Includes 420 patients with moderate to severe HF At baseline: - Chronic HF in NYHA functional class III or IV - Typical symptoms and signs of HF - No specific cut-point related to EF | Primary short-term endpoints (16 weeks): - NYHA functional class and functional status - VAS for symptoms - 6MWT - echocardiography - Serum samples for CoQ10 and NT-proBNP. Primary long-term endpoint (106 weeks): - Morbidity and mortality - VAS for symptoms - MACE | 100 mg 3 times/day | After 16weeks: - The level of serum CoQ10-treated group increased to about 3 times the baseline value - NT-proBNP reduction (mean value of 384 pg/mL (20%) vs. 199 pg/mL (12%) After 106 weeks: - Fewer MACE (N = 30, 15% vs. N = 57, 26%) - Improved NYHA functional classification (N = 86, 58% vs. N = 68, 45%) - At least 1 grade of improvement in NYHA functional class - Reduced serum NT-proBNP (mean value of 1137 pg/mL; 60% vs. mean value 881 pg/mL; 52%) - Lower total number of cardiovascular deaths (N = 18, 9% vs. N = 34, 16%), corresponding to a 43% relative reduction. - Lower number of hospital stays for HF (N = 17, 8% vs. N = 31, 14%) - Lower number of adverse events (mean value 26 (13%) vs. 41 (19%) | [22] | |
| BPF | - A randomised, double-blind, placebo-controlled study - Includes 237 patients: group A (104 patients) with isolated HC (LDL-C levels ≥130 mg/dL) group B (42 patients) with HC/HT group C (59 patients) with mixed HC/HT/HG over 110 mg/dL group D (32 patients) who stopped simvastatin therapy | 30 consecutive days | 500, 1000 or 1500 mg/day | - In group A, B and C reduction in: - tChol (mean value from 278 mg/mL to 199 mg/mL) - LDL-C (mean value from 188 mg/mL to 126 mg/mL) - TG (mean value from 267 mg/mL to 158 mg/mL) - Increase in HDL-C was observed with the best response in the 10% subjects (−64.6%) - Reduction in blood glucose levels (mean value of −18.9% under 500 mg BPF treatment and −22.4% in 1000 mg BPF treatment) - Group D, 30/32 patients showed a reduction in tChol (mean −25% and LDL-C −27.6%) - Increasing flow-mediated vasodilation | [23] |
| - A randomised double-blind, placebo-controlled study - Includes 60 patients suffering from T2DM with: Serum glucose >110 mg/dL)) Mixed hyperlipemia (LDL-C >120 mg/dL and TG >175 mg/dL) | 30 consecutive days | 650 mg BPF twice/day | - Fasting plasma glucose (from 120 ± 1.6 mg/mL to 98 ± 1.3 mg/mL) - tChol (262 ± 14 mg/dL to 196 ± 12 mg/dL) - LDL-C (175 ± 5.8 mg/mL to 116 ± 3.2 mg/mL) - HDL-C (44 ± 4.1 mg/mL to 48 + 3.8 mg/mL) - Triglycerides (252 ± 9 mg/mL to 170 + 7 mg/mL) - Relevant changes in mean particle diameters for VLDL, LDL, and HDL - Decrease the mean concentration of IDL particles to Increase large LDL and to decrease small LDL. - Increase in total HDL particles | [24] | |
| - A prospective, open-label, parallel group, placebo- controlled study - Includes 77 patients mixed hypercholesterolemia (LDL-C = 160 mg/dL, TG = 225 mg/dL) | 30 consecutive days | - Rosuvastatin (10 and 20 mg/day) - BPF (1000 mg/day) - BPF+rosuvastatin (10 mg/day) | - tChol (mean value from 278 ± 4 mg/dL to 191 ± 5 mg/dL) - LDL-C (mean value from 191 ± 3 mg/dL to 191 ± 3 mg/dL) - TG (mean value from 238 ± 5 mg/dL to 165 ± 3 mg/dL) - HDL-C (mean value from 38 ± 2 mg/dL to 45 ± 4 mg/dL) - Association of BPF+rosuvastatin enhance the hypolipidemic effect of rosuvastatin: - tChol (mean value from 172 ± 3 mg/dL to 195 ± 3 mg/dL) - LDL-C (mean value from 90 ± 4 mg/dL to 115 ± 4 mg/dL), - TG (mean value from 152 ± 5 mg/dL to 200 ± 4 mg/dL) - HDL-C (mean value from 52 ± 4 mg/dL to 42 ± 3 mg/dL) - Reduced urinary MVA excretion - Decreased both MDA levels and LOX-1 expression in PMNs of patients - Enhanced phosphorylation of PKB1 expression in PMNs of patients - The effect of BPF produced a further enhancement of rosuvastatin antioxidant and vasoprotective effects | [25] | |
| Oleuropein, hydroxytyrosol | - An open, controlled, parallel-group, co-twin study - Including 40 borderline hypertensive monozygotic patients (18 and 60 years) with: an untreated SBP >120 mmHg or DBP >80 mmHg at rest | 8 weeks | 500 mg and 1000 mg/day | - 1000 mg significantly reduces: SBP (mean value 126 ± 9 vs. 137 ± 10) DBP (76 ± 6 vs. 80 ± 10) - Reduction in LDL-C | [26] |
| - A double-blind, randomised, parallel and active- controlled clinical study - Includes 232 patients (25 and 60 years) in stage-1 hypertension with SBP of 140–159 mmHg, DBP <90 mmHg or in between 90 and 99 mmHg | - 4-weeks single-blind placebo (is diet-alone) - Run-in period and followed by 8-weeks double-blind treatment period | 500 mg twice/day vs. captopril treatment (12.5 mg twice/day) | - significant reduction in: SBP (−11.5 ± 8.6 mmHg vs−13.7 ± 7.6 mmHg in captopril) DBP (−4.8 ± 5.5 mmHg vs. −6.4 ± 5.2 mmHg captopril) - LDL-C (−3.89 ± 19.40 mg/dL vs. 2.14 ± 14.20 mg/dL in captopril) - TG (−11.90 ± 46.17 mg/dL vs. −1.26 ± 43.31 mg/dL in captopril) | [27] | |
| SGLT2-inhibitors | - EMPA-REGOUTCOMEtrial, a cardiovascular outcome trial - Includes 7020 patients (≥18 years) with T2DM at high risk of CV disease with: - BMI ≤ 45 kg/m2; - No glucose-lowering therapy in previous 12 weeks -HbA1c 7.0–9.0%, or stable glucose-lowering therapy and HbA1c 7.0–10.0% - Glomerular filtration rate (eGFR) of at least 30 mL/min × 1.73 m2 of body-surface area | 3 years Primary long-term endpoint: 3P-MACE | 10 mg or 25 mg | Primary outcome: - All-cause mortality: HR 0.68 (95% CI 0.57, 0.82; p < 0.001) - Incident or worsening nephropathy: HR 0.61 (95% CI 0.53, 0.70; p < 0.001) - Lower rates of death from cardiovascular causes (3.7%, vs. 5.9% in te placebo; 38% relative risk reduction; HR 0.62 (95% CI 0.49, 0.77; p < 0.001) - Hospitalization for heart failure (2.7% and 4.1%, respectively; 35% relative risk reduction; HR 0.65 (95% CI 0.50, 0.85; p = 0.002) - Death from any cause (5.7% and 8.3%, respectively; 32% relative risk reduction) | [28] |
| - Declare-TIMI 58 trial - includes 17,160 patients (40 years) with T2DM and risk of atherosclerotic CV disease T2DM; HbA1c ≥6.5% | 4, 5 years Primary long-term end-point: - Non-inferiority for 3P-MACE - Composite kidney outcome | 10 mg | Co-primary efficacy outcomes−3P-MACE: - lower rate of cardiovascular death or hospitalization for heart failure (4.9% vs. 5.8%; HR, 0.83; 95% CI, 0.73 to 0.95; p = 0.005) - Lower rate of hospitalization for heart failure (HR, 0.73; 95% CI, 0.61 to 0.88) kidney composite outcome: - A renal event occurred in 4.3% vs. 5.6% in placebo (HR, 0.76; 95% CI, 0.67 to 0.87) - Death from any cause occurred in 6.2% and 6.6%, respectively (HR, 0.93; 95% CI, 0.82 to 1.04) | [29] |
| Plant | Bioactive Component | Properties | In Vitro/In Vivo Models | Clinical Trials | References |
|---|---|---|---|---|---|
| Brassicaceae family Gramineae family | Coenzyme Q10 | Antioxidant and anti-inflammatory activity Key component of METC and in ATP production Bioenergetic effect ↑ p-AMPK ↑ Akt/eNOS activity ↑ HO-1 expression ↑ hemodynamic parameters ↑ LV function ↓ 3-NT ↓ MDA ↓ Nox2 gene expression ↑ Vasodilation ↓ Aldosterone levels ↑ Fatty acid oxidation ↑ VLDL ↓ LDLc/HDLc ↓TC/HDLc ↓ Fibrinogen ↓ SBP ↓ DBP | - EPCs - Isoproterenol- induced HF in rats - Diabetic cardiomyopathy in mice | -HFrEF - Hypertension -T2DM - MetS - Hyperlipideima - MI | [18,19,20,21,22,30,31,32,33,34,35,36,37,38] |
| Citrus Bergamia Risso et Poiteau | BPF | ↓ Serum glucose, TG, TC, LDL-C, VLDL-C ↑ HDL-C ↑ fecal sterol excretion Re-arrangement of lipoprotein particles ↑ Lipid transfer protein system ↓ pCEH ↑ SOD, catalase ↓ SMC proliferation, LOX-1, p-PKB ↓ ROS, TBARS, MDA, Nitrotyrosine ↑ LV function ↓ pathologic cardiac remodelling ↓ detrimental autophagy ↓ apoptosis ↓ 8OHdG ↑ newly formed myocytes | - eCSCs - Rat neointimal hyperplasia - Hypercholesterolemic diet fed rats - Doxo-induced cardiotoxicity in rats | - Hyperlipemia - MetS - T2DM | [23,24,25,39,40,41,42,43,44,45,46,47] |
| Oleaceae family (Olea europaea Linn.) | Oleuropein, hydroxytyrosol | ↓ CK-MB, GSSG, TBARS, LDH ↓ MDA, 3-NT, ET-1, IL-1 β, IL-6, TNFα ↑ eNOS ↓ PCs, iNOS ↑ p-Akt, p-AMPK ↑Prdx-1 and Prdx-2 ↓ TC, TG ↑ SOD and GSH activity ↑ integrity of complex III of the METC ↓ infarct size ↓ myocyte apoptosis ↑ LV fuction ↑ pGS3K-β/GS3K-β ↑ Sirt-1, pFOXO3a ↓ myocardial fibrosis ↓ pMEK, pERK1/2, p53, p-IκBα ↑ pSTAT-3 ↓ CYP2E1, OH-1, NF-κB, COX-2 ↓ GRP78, CHOP | - VPCs - CoCl2-induced hypoxia in H9C2 cells - ISO-induced MI in rats - Myocardial I/R in rats - Doxo-treated rats - Myocardial I/R in hypercholesterolemic rabbits - Myocardial infarction in rats - T2DM and renal hypertension in rats - Cisplatin-induced kidney injury in mice | [48,49,50,51,52,53,54,55,56,57,58,59,60,61,62,63] | |
| Malus Malus sieversii | Phlorizin | ↑ endothelium-indipendent relaxation ↓ human ox-LDL ↓ postprandial blood glucose rise, HbA1c ↑ urinary glucose excretion ↓ urinary albumin excretion ↓ glycohemoglobin, insulin, TG ↑ muscle GLUT4 ↓ renal GLUT2 ↓ kidney epithelial vacuolization ↓ hepatic glucose production ↑ ketone bodies amount | - Isolated rabbit coronary artery - High-glucose diet in Std ddY mice - Neonatally STZ-induced diabetic rats - STZ-induced diabetic rats | [64,65,66,67,68,69,70,71,72,73] | |
| Vitis vinifera | Proanthocyanidins | free radical scavenging activity ↓ TBARS levels ↓ DNA fragmentation ↓ p53 ↑ Bcl-2, Bcl-XL ↓ liver toxicity and DNA fragmentation ↓ hepatocyte apoptosis and necrosis ↓ JNK-1, c-Jun ↓cardiomyocyte apoptosis ↑ post-ischemic cardiac function ↑ mitochondrial IDH, SDH, MDH, α-KGDH ↑ respiratory chain NADH dehydrogenase and CCO | - TPA-induced ROS/RNS in mice - Human oral keratinocytes - AAP-induced liver injury in mice - Myocardial I/R in rats | [74,75,76,77,78,79,80,81,82] | |
| Zygophyllaceae family (Tribulus terrestris L.) | ferulic acid, diosgenin, saponins | ↑Total antioxidant activity ↑ SOD, GPx, CAT activity ↓ MDA ↓ LDH, CK-MB, SGOT, SGPT, Calcium ↑ cell viability ↑ Integrity of mitochondrial PTP ↑ Activity of mitochondrial respiratory complexes ↑ oxygen consumption rate ↑ ATP level ↓ HIF-1α ↑ Mitochondrial OPA1, Mfn1, Mfn2 ↓ Drp1 and Fis1 ↓ Bax, Bad ↑ Bcl-2, p-Akt ↓p-P38, p-JNK ↓ Heart rate ↓ cardiac fibrosis ↓ IL-6, TNFα, IL-1β, MCP-1 ↑ IL-10 ↓ Nuclear traslocation of NF-κB ↑ coronary artery dilation ↑ ECG | - Myocardial ischemia in H9C2 - ISO-induced ischemia in rats | - Angina pectoris | [83] |
| Vitis vinifera | Resveratrol | ↑ oxygen consumption rate ↑ SIRT1 and p-AMPK ↓ PCG1α acetylation ↑ PCG1α activity ↑ insulin sensitivity ↑ endothelial function ↑SBP ↑ adiponectin, IL-10 ↓ PAI-1, hsCRP, TNF α, IL-6 ↓ HbA1c, TC, TG ↓ number of angina episodes ↑ LV diastolic function ↓ ANP ↑ LC3II/LC3I ↓p-mTOR, p-p70S6K ↑myocardial ATP content ↓ cleaved-caspase-3 | - C2C12 mouse myoblast cells - MEFs (mouse embrionic fibroblasts) - H9C2 cells - Isolated gastrocnemius muscle - HFD-treated mice - KKAy mice - MI in mice and rats - SHRs rats | - Hypertension - CAD - T2DM - Stable angina pectoris - MI | [84,85,86,87,88,89,90,91,92,93] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Mollace, V.; Rosano, G.M.C.; Anker, S.D.; Coats, A.J.S.; Seferovic, P.; Mollace, R.; Tavernese, A.; Gliozzi, M.; Musolino, V.; Carresi, C.; et al. Pathophysiological Basis for Nutraceutical Supplementation in Heart Failure: A Comprehensive Review. Nutrients 2021, 13, 257. https://doi.org/10.3390/nu13010257
Mollace V, Rosano GMC, Anker SD, Coats AJS, Seferovic P, Mollace R, Tavernese A, Gliozzi M, Musolino V, Carresi C, et al. Pathophysiological Basis for Nutraceutical Supplementation in Heart Failure: A Comprehensive Review. Nutrients. 2021; 13(1):257. https://doi.org/10.3390/nu13010257
Chicago/Turabian StyleMollace, Vincenzo, Giuseppe M. C. Rosano, Stefan D. Anker, Andrew J. S. Coats, Petar Seferovic, Rocco Mollace, Annamaria Tavernese, Micaela Gliozzi, Vincenzo Musolino, Cristina Carresi, and et al. 2021. "Pathophysiological Basis for Nutraceutical Supplementation in Heart Failure: A Comprehensive Review" Nutrients 13, no. 1: 257. https://doi.org/10.3390/nu13010257