
Nicotinamide Riboside for the Prevention and Treatment of Doxorubicin Cardiomyopathy. Opportunities and Prospects


Abstract
:1. Introduction
2. Article Search and Selection Strategy
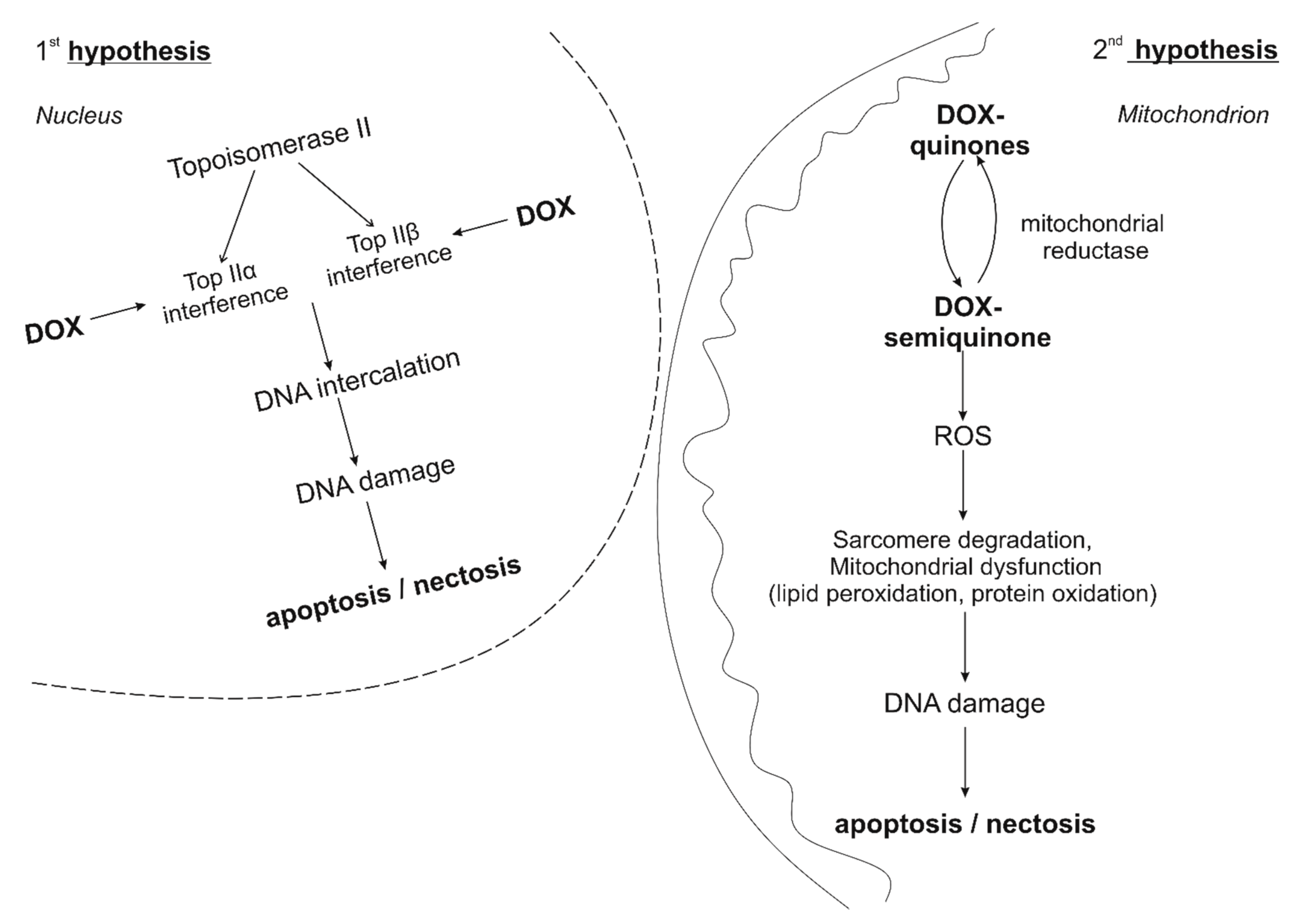
3. Features of the Development of Doxorubicin Cardiomypathy
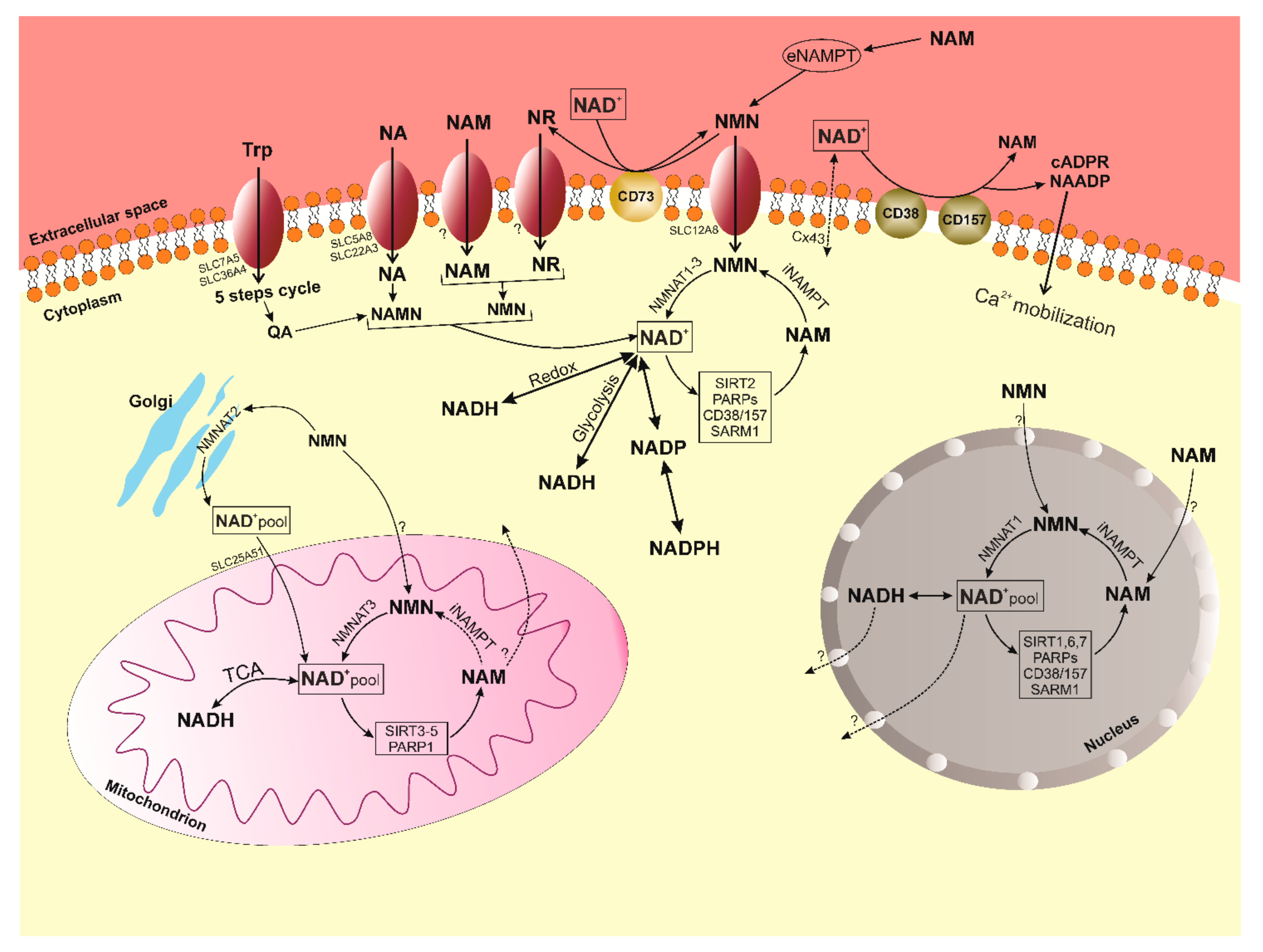
4. Known Pathways of the Synthesis of NAD+ Feature of NR
5. Metabolism of NAD+
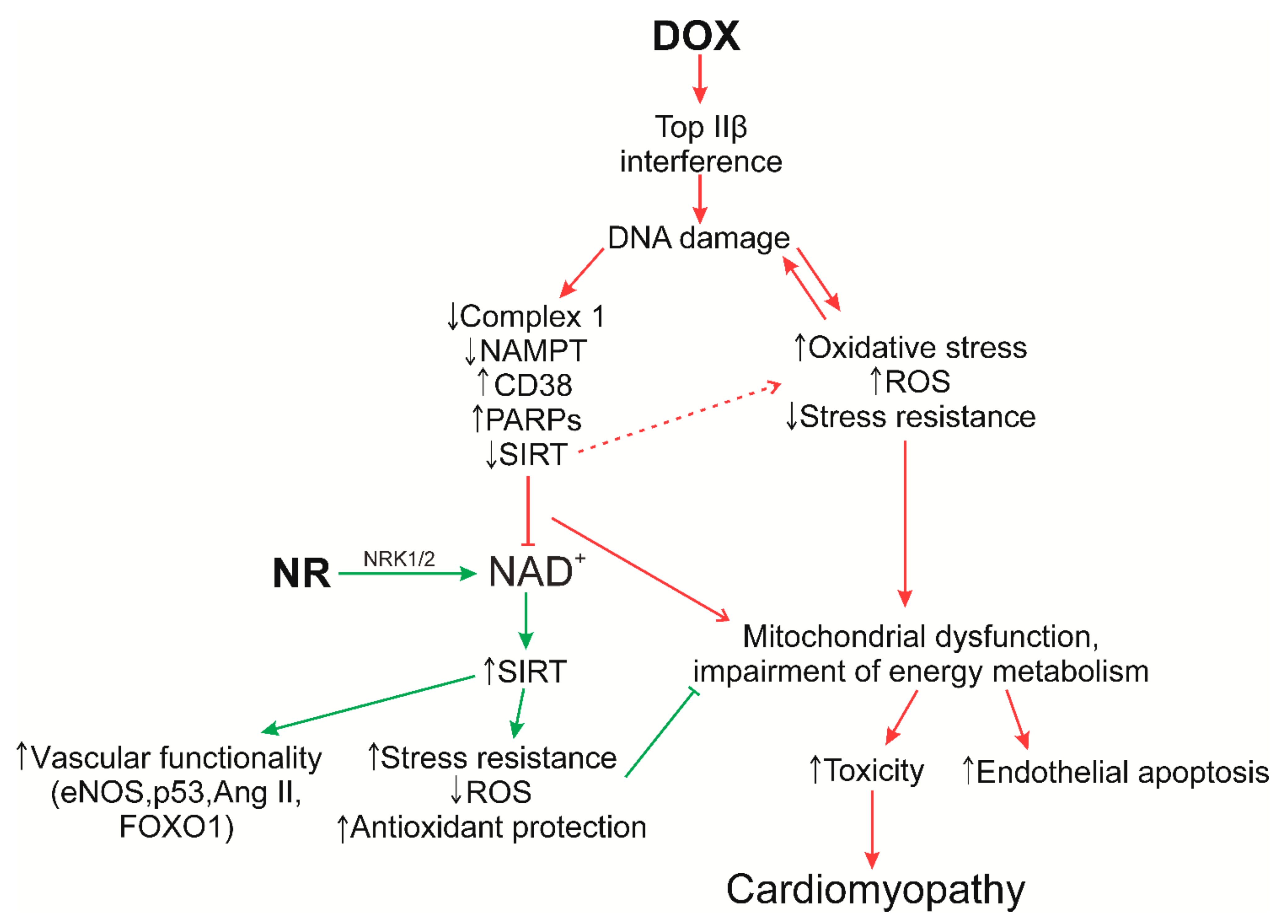
6. Role of NAD+/SIRT in the Regulation of Oxidative Stress
7. Effects of NR in Small Laboratory Animal Models
8. Supposed Role of NR in the Prevention of Doxorubicin Cardiomyopathy
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Markham, M.J.; Wachter, K.; Agarwal, N.; Bertagnolli, M.M.; Chang, S.M.; Dale, W.; Diefenbach, C.S.M.; Rodriguez-Galindo, C.; George, D.J.; Gilligan, T.D.; et al. Clinical Cancer Advances 2020: Annual report on progress against cancer from the American Society of Clinical oncology. J. Clin. Oncol. 2020, 38, 1081–1101. [Google Scholar] [CrossRef]
- Bray, F.; Ferlay, J.; Soerjomataram, I.; Siegel, R.L.; Torre, L.A.; Jemal, A. Global cancer statistics 2018: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J. Clin. 2018, 68, 394–424. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kleinerman, E.S. Current Advances in Osteosarcoma; Springer: Berlin/Heidelberg, Germany, 2014; Volume 804, ISBN 9783030430313. [Google Scholar]
- Gharagozloo, M.; Kalantari, H.; Rezaei, A.; Maracy, M.R.; Salehi, M.; Bahador, A.; Hassannejad, N.; Narimani, M.; Sanei, M.H.; Bayat, B.; et al. Cardioprotective effect of melatonin and agomelatine on doxorubicin-induced cardiotoxicity in a rat model: An electrocardiographic, scintigraphic and biochemical study. Bratisl. Lek. Listy. 2015, 116, 296–301. [Google Scholar] [CrossRef] [PubMed]
- Podyacheva, E.Y.; Kushnareva, E.A.; Karpov, A.A.; Toropova, Y.G. Analysis of Models of Doxorubicin-Induced Cardiomyopathy in Rats and Mice. A Modern View from the Perspective of the Pathophysiologist and the Clinician. Front. Pharmacol. 2021, 12, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Yoshino, J.; Baur, J.A.; Imai, S. ichiro NAD+ Intermediates: The Biology and Therapeutic Potential of NMN and NR. Cell Metab. 2018, 27, 513–528. [Google Scholar] [CrossRef] [Green Version]
- Xu, W.; Barrientos, T.; Mao, L.; Rockman, H.A.; Sauve, A.A.; Andrews, N.C. Lethal Cardiomyopathy in Mice Lacking Transferrin Receptor in the Heart. Cell Rep. 2015, 13, 533–545. [Google Scholar] [CrossRef] [Green Version]
- Yang, Y.; Sauve, A.A. NAD(+) metabolism: Bioenergetics, signaling and manipulation for therapy. Biochim. Biophys. Acta 2017, 1864, 1787–1800. [Google Scholar] [CrossRef] [Green Version]
- Menna, P.; Gonzalez Paz, O.; Chello, M.; Covino, E.; Salvatorelli, E.; Minotti, G. Anthracycline cardiotoxicity. Expert Opin. Drug Saf. 2012, 11. [Google Scholar] [CrossRef]
- Wang, J.C. Cellular roles of DNA topoisomerases: A molecular perspective. Nat. Rev. Mol. Cell Biol. 2002, 3, 430–440. [Google Scholar] [CrossRef]
- McGowan, J.V.; Chung, R.; Maulik, A.; Piotrowska, I.; Walker, J.M.; Yellon, D.M. Anthracycline Chemotherapy and Cardiotoxicity. Cardiovasc. Drugs Ther. 2017, 31, 63–75. [Google Scholar] [CrossRef] [Green Version]
- Tewey, K.; Rowe, T.; Yang, L.; Halligan, B.; Liu, L. Adriamycin-induced DNA damage mediated by mammalian DNA topoisomerase II. Science 1984, 226, 466–468. [Google Scholar] [CrossRef]
- dos Santos, D.S.; Goldenberg, R.C. Doxorubicin-Induced Cardiotoxicity: From Mechanisms to Development of Efficient Therapy. Cardiotoxicity 2018, 3–24. [Google Scholar] [CrossRef] [Green Version]
- Zhang, S.; Liu, X.; Bawa-Khalfe, T.; Lu, L.S.; Lyu, Y.L.; Liu, L.F.; Yeh, E.T.H. Identification of the molecular basis of doxorubicin-induced cardiotoxicity. Nat. Med. 2012, 18, 1639–1642. [Google Scholar] [CrossRef] [PubMed]
- Minotti, G.; Menna, P.; Salvatorelli, E.; Cairo, G.; Gianni, L. Anthracyclines: Molecular advances and pharmacologie developments in antitumor activity and cardiotoxicity. Pharmacol. Rev. 2004, 56, 185–229. [Google Scholar] [CrossRef] [Green Version]
- Deng, S.; Wojnowski, L. Genotyping the risk of anthracycline-induced cardiotoxicity. Cardiovasc. Toxicol. 2007, 7, 129–134. [Google Scholar] [CrossRef] [PubMed]
- Renu, K.; Abilash, V.G.; Tirupathi, T.P.; Arunachalam, S. Molecular Mechanism of Doxorubicin-Induced Cardiomyopathy—An Update. Eur. J. Pharmacol. 2018, 818, 241–253. [Google Scholar] [CrossRef]
- Octavia, Y.; Tocchetti, C.G.; Gabrielson, K.L.; Janssens, S.; Crijns, H.J.; Moens, A.L. Doxorubicin-induced cardiomyopathy: From molecular mechanisms to therapeutic strategies. J. Mol. Cell. Cardiol. 2012, 52, 1213–1225. [Google Scholar] [CrossRef] [Green Version]
- Yi, L.L.; Kerrigan, J.E.; Lin, C.P.; Azarova, A.M.; Tsai, Y.C.; Ban, Y.; Liu, L.F. Topoisomerase IIβ-mediated DNA double-strand breaks: Implications in doxorubicin cardiotoxicity and prevention by dexrazoxane. Cancer Res. 2007, 67, 8839–8846. [Google Scholar] [CrossRef] [Green Version]
- James Kang, Y.; Chen, Y.; Epstein, P.N. Suppression of doxorubicin cardiotoxicity by overexpression of catalase in the heart of transgenic mice. J. Biol. Chem. 1996, 271, 12610–12616. [Google Scholar] [CrossRef] [Green Version]
- Yen, H.C.; Oberley, T.D.; Vichitbandha, S.; Ho, Y.S.; St. Clair, D.K. The protective role of manganese superoxide dismutase against adriamycin-induced acute cardiac toxicity in transgenic mice. J. Clin. Invest. 1996, 98, 1253–1260. [Google Scholar] [CrossRef] [Green Version]
- Bieganowski, P.; Brenner, C. Discoveries of nicotinamide riboside as a nutrient and conserved NRK genes establish a preiss-handler independent route to NAD+ in fungi and humans. Cell 2004, 117, 495–502. [Google Scholar] [CrossRef] [Green Version]
- Dietrich, L.S.; Muniz, O.; Powanda, M. NAD synthesis in animal tissues. J. Vitaminol. 1968, 14, 123–129. [Google Scholar] [CrossRef] [Green Version]
- Croft, T.; Venkatakrishnan, P.; Lin, S.J. Nad+ metabolism and regulation: Lessons from yeast. Biomolecules 2020, 10, 330. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xiao, W.; Wang, R.S.; Handy, D.E.; Loscalzo, J. NAD(H) and NADP(H) Redox Couples and Cellular Energy Metabolism. Antioxid. Redox Signal. 2018, 28, 251–272. [Google Scholar] [CrossRef] [PubMed]
- Hwang, E.S.; Song, S.B. Possible adverse effects of high-dose nicotinamide: Mechanisms and safety assessment. Biomolecules 2020, 10, 687. [Google Scholar] [CrossRef]
- Imai, S. Nicotinamide Phosphoribosyltransferase (Nampt): A Link Between NAD Biology, Metabolism, and Diseases. Curr. Pharm. Des. 2009, 15, 20–28. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yoshino, J.; Mills, K.F.; Yoon, M.J.; Imai, S.I. Nicotinamide mononucleotide, a key NAD+ intermediate, treats the pathophysiology of diet- and age-induced diabetes in mice. Cell Metab. 2011, 14, 528–536. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Braidy, N.; Poljak, A.; Grant, R.; Jayasena, T.; Mansour, H.; Chan-Ling, T.; Guillemin, G.J.; Smythe, G.; Sachdev, P. Mapping NAD+ metabolism in the brain of ageing Wistar rats: Potential targets for influencing brain senescence. Biogerontology 2014, 15, 177–198. [Google Scholar] [CrossRef] [PubMed]
- Mehmel, M.; Jovanović, N.; Spitz, U. Nicotinamide riboside—the current state of research and therapeutic uses. Nutrients 2020, 12, 1616. [Google Scholar] [CrossRef] [PubMed]
- Hopp, A.K.; Grüter, P.; Hottiger, M.O. Regulation of Glucose Metabolism by NAD+ and ADP-Ribosylation. Cells 2019, 8, 890. [Google Scholar] [CrossRef] [Green Version]
- Ratajczak, J.; Joffraud, M.; Trammell, S.A.J.; Ras, R.; Canela, N.; Boutant, M.; Kulkarni, S.S.; Rodrigues, M.; Redpath, P.; Migaud, M.E.; et al. NRK1 controls nicotinamide mononucleotide and nicotinamide riboside metabolism in mammalian cells. Nat. Commun. 2016, 7, 1–12. [Google Scholar] [CrossRef]
- Tempel, W.; Rabeh, W.M.; Bogan, K.L.; Belenky, P.; Wojcik, M.; Seidle, H.F.; Nedyalkova, L.; Yang, T.; Sauve, A.A.; Park, H.W.; et al. Nicotinamide riboside kinase structures reveal new pathways to NAD+. PLoS Biol. 2007, 5, 2220–2230. [Google Scholar] [CrossRef]
- Fletcher, R.S.; Lavery, G.G. The Emergence of the Nicotinamide Riboside Kinases in the regulation of NAD+ Metabolism. J. Mol. Endocrinol. 2018, 44, 1–18. [Google Scholar] [CrossRef] [Green Version]
- Trammell, S.A.J.; Schmidt, M.S.; Weidemann, B.J.; Redpath, P.; Jaksch, F.; Dellinger, R.W.; Li, Z.; Abel, E.D.; Migaud, M.E.; Brenner, C. Nicotinamide riboside is uniquely and orally bioavailable in mice and humans. Nat. Commun. 2016, 7, 1–14. [Google Scholar] [CrossRef]
- Bogan, K.L.; Brenner, C. Nicotinic acid, nicotinamide, and nicotinamide riboside: A molecular evaluation of NAD+ precursor vitamins in human nutrition. Annu. Rev. Nutr. 2008, 28, 115–130. [Google Scholar] [CrossRef] [Green Version]
- Yang, T.; Chan, N.Y.K.; Sauve, A.A. Syntheses of nicotinamide riboside and derivatives: Effective agents for increasing nicotinamide adenine dinucleotide concentrations in mammalian cells. J. Med. Chem. 2007, 50, 6458–6461. [Google Scholar] [CrossRef] [PubMed]
- Kulikova, V.; Shabalin, K.; Nerinovski, K.; Dölle, C.; Niere, M.; Yakimov, A.; Redpath, P.; Khodorkovskiy, M.; Migaud, M.E.; Ziegler, M.; et al. Generation, release, and uptake of the NAD precursor nicotinic acid riboside by human cells. J. Biol. Chem. 2015, 290, 27124–27137. [Google Scholar] [CrossRef] [Green Version]
- Covarrubias, A.J.; Perrone, R.; Grozio, A.; Verdin, E. NAD+ metabolism and its roles in cellular processes during ageing. Nat. Rev. Mol. Cell Biol. 2021, 22, 119–141. [Google Scholar] [CrossRef]
- Cantó, C.; Sauve, A.A.; Bai, P. Crosstalk between poly(ADP-ribose) polymerase and sirtuin enzymes. Mol. Aspects Med. 2013, 34, 1168–1201. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Masri, S.; Rigor, P.; Cervantes, M.; Ceglia, N.; Sebastian, C.; Xiao, C.; Roqueta-Rivera, M.; Deng, C.; Osborne, T.F.; Mostoslavsky, R.; et al. Partitioning circadian transcription by SIRT6 leads to segregated control of cellular metabolism. Cell 2014, 158, 659–672. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Carrico, C.; Meyer, J.G.; He, W.; Gibson, B.W.; Verdin, E. The Mitochondrial Acylome Emerges: Proteomics, Regulation by Sirtuins, and Metabolic and Disease Implications. Cell Metab. 2018, 27, 497–512. [Google Scholar] [CrossRef] [Green Version]
- Jang, S.Y.; Kang, H.T.; Hwang, E.S. Nicotinamide-induced mitophagy: Event mediated by high NAD+/NADH ratio and SIRT1 protein activation. J. Biol. Chem. 2012, 287, 19304–19314. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cantó, C.; Jiang, L.Q.; Deshmukh, A.S.; Mataki, C.; Coste, A.; Lagouge, M.; Zierath, J.R.; Auwerx, J. Interdependence of AMPK and SIRT1 for Metabolic Adaptation to Fasting and Exercise in Skeletal Muscle. Cell Metab. 2010, 11, 213–219. [Google Scholar] [CrossRef] [Green Version]
- Cantó, C.; Menzies, K.J.; Auwerx, J. NAD+ Metabolism and the Control of Energy Homeostasis: A Balancing Act between Mitochondria and the Nucleus. Cell Metab. 2015, 22, 31–53. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cantó, C.; Auwerx, J. Targeting sirtuin 1 to improve metabolism: All you need is NAD+? Pharmacol. Rev. 2012, 64, 166–187. [Google Scholar] [CrossRef] [Green Version]
- Jing, E.; Gesta, S.; Kahn, C.R. SIRT2 Regulates Adipocyte Differentiation through FoxO1 Acetylation/Deacetylation. Cell Metab. 2007, 6, 105–114. [Google Scholar] [CrossRef] [Green Version]
- Rothgiesser, K.M.; Erener, S.; Waibel, S.; Lüscher, B.; Hottiger, M.O. SIRT2 regulates NF-κB-dependent gene expression through deacetylation of p65 Lys310. J. Cell Sci. 2010, 123, 4251–4258. [Google Scholar] [CrossRef] [Green Version]
- Giralt, A.; Villarroya, F. SIRT3, a pivotal actor in mitochondrial functions: Metabolism, cell death and aging. Biochem. J. 2012, 444, 1–10. [Google Scholar] [CrossRef] [Green Version]
- Bordone, L.; Motta, M.C.; Picard, F.; Robinson, A.; Jhala, U.S.; Apfeld, J.; McDonagh, T.; Lemieux, M.; McBurney, M.; Szilvasi, A.; et al. Sirt1 regulates insulin secretion by repressing UCP2 in pancreatic β cells. PLoS Biol. 2006, 4, 210–220. [Google Scholar] [CrossRef] [Green Version]
- Hirschey, M.D.; Shimazu, T.; Goetzman, E.; Jing, E.; Schwer, B.; Lombard, D.B.; Grueter, C.A.; Harris, C.; Biddinger, S.; Ilkayeva, O.R.; et al. SIRT3 regulates mitochondrial fatty-acid oxidation by reversible enzyme deacetylation. Nature 2010, 464, 121–125. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Du, J.; Zhou, Y.; Su, X.; Yu, J.J.; Khan, S.; Jiang, H.; Kim, J.; Woo, J.; Kim, J.H.; Choi, B.H.; et al. Sirt5 is a NAD-dependent protein lysine demalonylase and desuccinylase. Science 2011, 334, 806–809. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhong, L.; D’Urso, A.; Toiber, D.; Sebastian, C.; Henry, R.E.; Vadysirisack, D.D.; Guimaraes, A.; Marinelli, B.; Wikstrom, J.D.; Nir, T.; et al. The Histone Deacetylase Sirt6 Regulates Glucose Homeostasis via Hif1α. Cell 2010, 140, 280–293. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ford, E.; Voit, R.; Liszt, G.; Magin, C.; Grummt, I.; Guarente, L. Mammalian Sir2 homolog SIRT7 is an activator of RNA polymerase I transcription. Genes Dev. 2006, 20, 1075–1080. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Barber, M.F.; Michishita-Kioi, E.; Xi, Y.; Tasselli, L.; Kioi, M.; Moqtaderi, Z.; Tennen, R.I.; Paredes, S.; Young, N.L.; Chen, K.; et al. SIRT7 links H3K18 deacetylation to maintenance of oncogenic transformation. Nature 2012, 487, 114–118. [Google Scholar] [CrossRef] [PubMed]
- Bai, P.; Cantó, C. The role of PARP-1 and PARP-2 enzymes in metabolic regulation and disease. Cell Metab. 2012, 16, 290–295. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Módis, K.; Gerö, D.; Erdélyi, K.; Szoleczky, P.; Dewitt, D.; Szabo, C. Cellular bioenergetics is regulated by PARP1 under resting conditions and during oxidative stress. Biochem. Pharmacol. 2012, 83, 633–643. [Google Scholar] [CrossRef] [Green Version]
- Scheibye-Knudsen, M.; Mitchell, S.J.; Fang, E.F.; Iyama, T.; Ward, T.; Wang, J.; Dunn, C.A.; Singh, N.; Veith, S.; Hasan-Olive, M.M.; et al. A high-fat diet and NAD+ activate sirt1 to rescue premature aging in cockayne syndrome. Cell Metab. 2014, 20, 840–855. [Google Scholar] [CrossRef] [Green Version]
- Bai, P.; Canto, C.; Brunyánszki, A.; Huber, A.; Szántó, M.; Cen, Y.; Yamamoto, H.; Houten, S.M.; Kiss, B.; Oudart, H.; et al. PARP-2 regulates SIRT1 expression and whole-body energy expenditure. Cell Metab. 2011, 13, 450–460. [Google Scholar] [CrossRef] [Green Version]
- Boehler, C.; Gauthier, L.R.; Mortusewicz, O.; Biard, D.S.; Saliou, J.M.; Bresson, A.; Sanglier-Cianferani, S.; Smith, S.; Schreiber, V.; Boussin, F.; et al. Poly(ADP-ribose) polymerase 3 (PARP3), a newcomer in cellular response to DNA damage and mitotic progression. Proc. Natl. Acad. Sci. USA 2011, 108, 2783–2788. [Google Scholar] [CrossRef] [Green Version]
- Graeff, R.; Liu, Q.; Kriksunov, I.A.; Hao, Q.; Hon, C.L. Acidic residues at the active sites of CD38 and ADP-ribosylt cyclase determine nicotinic acid adenine dinucleotide phosphate (NAADP) synthesis and hydrolysis activities. J. Biol. Chem. 2006, 281, 28951–28957. [Google Scholar] [CrossRef] [Green Version]
- Ortolan, E.; Augeri, S.; Fissolo, G.; Musso, I.; Funaro, A. CD157: From immunoregulatory protein to potential therapeutic target. Immunol. Lett. 2019, 205, 59–64. [Google Scholar] [CrossRef] [Green Version]
- Aksoy, P.; White, T.A.; Thompson, M.; Chini, E.N. Regulation of intracellular levels of NAD: A novel role for CD38. Biochem. Biophys. Res. Commun. 2006, 345, 1386–1392. [Google Scholar] [CrossRef]
- Escande, C.; Nin, V.; Price, N.L.; Capellini, V.; Gomes, A.P.; Barbosa, M.T.; O’Neil, L.; White, T.A.; Sinclair, D.A.; Chini, E.N. Flavonoid apigenin is an inhibitor of the NAD+ase CD38: Implications for cellular NAD+ metabolism, protein acetylation, and treatment of metabolic syndrome. Diabetes 2013, 62, 1084–1093. [Google Scholar] [CrossRef] [Green Version]
- Shubinsky, G.; Schlesinger, M. The CD38 lymphocyte differentiation marker: New insight into its ectoenzymatic activity and its role as a signal transducer. Immunity 1997, 7, 315–324. [Google Scholar] [CrossRef] [Green Version]
- Quarona, V.; Zaccarello, G.; Chillemi, A.; Brunetti, E.; Singh, V.K.; Ferrero, E.; Funaro, A.; Horenstein, A.L.; Malavasi, F. CD38 and CD157: A long journey from activation markers to multifunctional molecules. Cytom. Part B—Clin. Cytom. 2013, 84, 207–217. [Google Scholar] [CrossRef]
- Deaglio, S.; Morra, M.; Mallone, R.; Ausiello, C.M.; Prager, E.; Garbarino, G.; Dianzani, U.; Stockinger, H.; Malavasi, F. Human CD38 (ADP-ribosyl cyclase) is a counter-receptor of CD31, an Ig superfamily member. J. Immunol. 1998, 160, 395–402. [Google Scholar]
- Deaglio, S.; Aydin, S.; Grand, M.M.; Vaisitti, T.; Bergui, L.; D’Arena, G.; Chiorino, G.; Malavasi, F. CD38/CD31 interactions activate genetic pathways leading to proliferation and migration in chronic lymphocytic leukemia cells. Mol. Med. 2010, 16, 87–91. [Google Scholar] [CrossRef] [PubMed]
- Wang, Q.; Zhang, S.; Liu, T.; Wang, H.; Liu, K.; Wang, Q.; Zeng, W. Sarm1/Myd88-5 Regulates Neuronal Intrinsic Immune Response to Traumatic Axonal Injuries. Cell Rep. 2018, 23, 716–724. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, H.C.; Zhao, Y.J. Resolving the topological enigma in Ca2+ signaling by cyclic ADP-ribose and NAADP. J. Biol. Chem. 2019, 294, 19831–19843. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Carty, M.; Bowie, A.G. SARM: From immune regulator to cell executioner. Biochem. Pharmacol. 2019, 161, 52–62. [Google Scholar] [CrossRef] [PubMed]
- Braidy, N.; Guillemin, G.J.; Mansour, H.; Chan-Ling, T.; Poljak, A.; Grant, R. Age related changes in NAD+ metabolism oxidative stress and sirt1 activity in wistar rats. PLoS ONE 2011, 6, 1–18. [Google Scholar] [CrossRef] [PubMed]
- Gomes, A.P.; Price, N.L.; Ling, A.J.Y.; Moslehi, J.J.; Montgomery, M.K.; Rajman, L.; White, J.P.; Teodoro, J.S.; Wrann, C.D.; Hubbard, B.P.; et al. Declining NAD+ induces a pseudohypoxic state disrupting nuclear-mitochondrial communication during aging. Cell 2013, 155, 1624–1638. [Google Scholar] [CrossRef] [Green Version]
- Ramsey, K.M.; Mills, K.F.; Satoh, A.; Imai, S.I. Age-associated loss of Sirt1-mediated enhancement of glucose-stimulated insulin secretion in beta cell-specific Sirt1-overexpressing (BESTO) mice. Aging Cell 2008, 7, 78–88. [Google Scholar] [CrossRef] [Green Version]
- Cantó, C.; Gerhart-Hines, Z.; Feige, J.N.; Lagouge, M.; Noriega, L.; Milne, J.C.; Elliott, P.J.; Puigserver, P.; Auwerx, J. AMPK regulates energy expenditure by modulating NAD+ metabolism and SIRT1 activity. Nature 2009, 458, 1056–1060. [Google Scholar] [CrossRef]
- Chen, D.; Bruno, J.; Easlon, E.; Lin, S.J.; Cheng, H.L.; Alt, F.W.; Guarente, L. Tissue-specific regulation of SIRT1 by calorie restriction. Genes Dev. 2008, 22, 1753–1757. [Google Scholar] [CrossRef] [Green Version]
- Cerutti, R.; Pirinen, E.; Lamperti, C.; Marchet, S.; Sauve, A.A.; Li, W.; Leoni, V.; Schon, E.A.; Dantzer, F.; Auwerx, J.; et al. NAD+-dependent activation of Sirt1 corrects the phenotype in a mouse model of mitochondrial disease. Cell Metab. 2014, 19, 1042–1049. [Google Scholar] [CrossRef] [Green Version]
- Khan, N.A.; Auranen, M.; Paetau, I.; Pirinen, E.; Euro, L.; Forsström, S.; Pasila, L.; Velagapudi, V.; Carroll, C.J.; Auwerx, J.; et al. Effective treatment of mitochondrial myopathy by nicotinamide riboside, a vitamin B3. EMBO Mol. Med. 2014, 6, 721–731. [Google Scholar] [CrossRef]
- Imai, S.-i.; Guarente, L. NAD+ and sirtuins in aging and disease. Trends Cell Biol. 2014, 24, 464–471. [Google Scholar] [CrossRef]
- Kaelin, W.G.; McKnight, S.L. Influence of metabolism on epigenetics and disease. Cell 2013, 153, 56–69. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bei, Y. NAD+ metabolism and the control of energy homeostasis—A balancing act between mitochondria and the nucleus. Physiol. Behav. 2017, 176, 139–148. [Google Scholar] [CrossRef]
- Belenky, P.; Bogan, K.L.; Brenner, C. NAD+ metabolism in health and disease. Trends Biochem. Sci. 2007, 32, 12–19. [Google Scholar] [CrossRef] [PubMed]
- Zhang, W.; Huang, Q.; Zeng, Z.; Wu, J.; Zhang, Y.; Chen, Z. Sirt1 Inhibits Oxidative Stress in Vascular Endothelial Cells. Oxid. Med. Cell. Longev. 2017, 2017. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yu, J.; Auwerx, J. Protein deacetylation by SIRT1: An emerging key post-translational modification in metabolic regulation. Pharmacol. Res. 2010, 62, 35–41. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Van Der Horst, A.; Tertoolen, L.G.J.; De Vries-Smits, L.M.M.; Frye, R.A.; Medema, R.H.; Burgering, B.M.T. FOXO4 is acetylated upon peroxide stress and deacetylated by the longevity protein hSir2SIRT1. J. Biol. Chem. 2004, 279, 28873–28879. [Google Scholar] [CrossRef] [Green Version]
- Menghini, R.; Casagrande, V.; Cardellini, M.; Martelli, E.; Terrinoni, A.; Amati, F.; Vasa-Nicotera, M.; Ippoliti, A.; Novelli, G.; Melino, G.; et al. MicroRNA 217 modulates endothelial cell senescence via silent information regulator 1. Circulation 2009, 120, 1524–1532. [Google Scholar] [CrossRef] [Green Version]
- Liu, J.; Bi, X.; Chen, T.; Zhang, Q.; Wang, S.X.; Chiu, J.J.; Liu, G.S.; Zhang, Y.; Bu, P.; Jiang, F. Shear stress regulates endothelial cell autophagy via redox regulation and Sirt1 expression. Cell Death Dis. 2015, 6, e1827. [Google Scholar] [CrossRef] [Green Version]
- Kauppinen, A.; Suuronen, T.; Ojala, J.; Kaarniranta, K.; Salminen, A. Antagonistic crosstalk between NF-κB and SIRT1 in the regulation of inflammation and metabolic disorders. Cell. Signal. 2013, 25, 1939–1948. [Google Scholar] [CrossRef]
- Johnson, F.; Giulivi, C. Superoxide dismutases and their impact upon human health. Mol. Aspects Med. 2005, 26, 340–352. [Google Scholar] [CrossRef]
- Shimada, T.; Furuta, H.; Doi, A.; Ariyasu, H.; Kawashima, H.; Wakasaki, H.; Nishi, M.; Sasaki, H.; Akamizu, T. Des-acyl ghrelin protects microvascular endothelial cells from oxidative stress-induced apoptosis through sirtuin 1 signaling pathway. Metabolism 2014, 63, 469–474. [Google Scholar] [CrossRef]
- de Picciotto, N.E.; Gano, L.B.; Johnson, L.C.; Martens, C.R.; Sindler, A.L.; Mills, K.F.; Imai, S.I.; Seals, D.R. Nicotinamide mononucleotide supplementation reverses vascular dysfunction and oxidative stress with aging in mice. Aging Cell 2016, 15, 522–530. [Google Scholar] [CrossRef]
- Chen, Z.; Peng, I.C.; Cui, X.; Li, Y.S.; Chien, S.; Shyy, J.Y.J. Shear stress, SIRT1, and vascular homeostasis. Proc. Natl. Acad. Sci. USA 2010, 107, 10268–10273. [Google Scholar] [CrossRef] [Green Version]
- Cantó, C.; Houtkooper, R.H.; Pirinen, E.; Youn, D.Y.; Oosterveer, M.H.; Cen, Y.; Fernandez-Marcos, P.J.; Yamamoto, H.; Andreux, P.A.; Cettour-Rose, P.; et al. The NAD+ precursor nicotinamide riboside enhances oxidative metabolism and protects against high-fat diet-induced obesity. Cell Metab. 2012, 15, 838–847. [Google Scholar] [CrossRef] [Green Version]
- Brown, K.D.; Maqsood, S.; Huang, J.; Pan, Y.; Harkcom, W.; Li, W.; Sauve, A.; Verdin, E.; Jaffrey, S.R.; Surgery, N.; et al. Activation of SIRT3 by the NAD+ precursor nicotinamide riboside protects from noise-induced hearing loss. Cell Metab. 2016, 20, 1059–1068. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Frederick, D.W.; Loro, E.; Liu, L.; Davila, A.; Chellappa, K.; Silverman, I.M.; Quinn, W.J.; Gosai, S.J.; Tichy, E.D.; Davis, J.G.; et al. Loss of NAD Homeostasis Leads to Progressive and Reversible Degeneration of Skeletal Muscle. Cell Metab. 2016, 24, 269–282. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, H.; Ryu, D.; Wu, Y.; Gariani, K.; Wang, X.; Luan, P.; D’Amico, D.; Ropelle, E.R.; Lutolf, M.P.; Aebersold, R.; et al. NAD+ repletion improves mitochondrial and stem cell function and enhances life span in mice. Science 2016, 352, 1436–1443. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gong, B.; Pan, Y.; Vempati, P.; Zhao, W.; Knable, L.; Ho, L.; Wang, J.; Sastre, M.; Ono, K.; Sauve, A.A.; et al. Nicotinamide riboside restores cognition through an upregulation of proliferator-activated receptor-γ coactivator 1α regulated β-secretase 1 degradation and mitochondrial gene expression in Alzheimer’s mouse models. Neurobiol. Aging 2013, 34, 1581–1588. [Google Scholar] [CrossRef] [Green Version]
- Trammell, S.A.J.; Brenner, C. Targeted, LCMS-based metabolomics for quantitative measurement of NAD+ metabolites. Comput. Struct. Biotechnol. J. 2013, 4, e201301012. [Google Scholar] [CrossRef] [Green Version]
- Trammell, S.A.J.; Weidemann, B.J.; Chadda, A.; Yorek, M.S.; Holmes, A.; Coppey, L.J.; Obrosov, A.; Kardon, R.H.; Yorek, M.A.; Brenner, C. Nicotinamide riboside opposes type 2 diabetes and neuropathy in mice. Sci. Rep. 2016, 6, 1–7. [Google Scholar] [CrossRef] [Green Version]
- Trammell, S.A.J.; Yu, L.; Redpath, P.; Migaud, M.E.; Brenner, C. Nicotinamide riboside is a major NAD+ precursor vitamin in cow milk. J. Nutr. 2016, 146, 957–963. [Google Scholar] [CrossRef] [Green Version]
- Hong, G.; Zheng, D.; Zhang, L.; Ni, R.; Wang, G.; Fan, G.C.; Lu, Z.; Peng, T. Administration of nicotinamide riboside prevents oxidative stress and organ injury in sepsis. Free Radic. Biol. Med. 2018, 123, 125–137. [Google Scholar] [CrossRef]
- Zheng, D.; Zhang, Y.; Zheng, M.; Cao, T.; Wang, G.; Zhang, L.; Brockman, J.; Zhong, H.; Fan, G.; Peng, T.; et al. Nicotinamide riboside promotes autolysosome clearance in preventing doxorubicin-induced cardiotoxicity. Clin. Sci. 2019, 133, 1505–1521. [Google Scholar] [CrossRef]
- Zhang, X.; Henneman, N.F.; Girardot, P.E.; Sellers, J.T.; Chrenek, M.A.; Li, Y.; Wang, J.; Brenner, C.; Nickerson, J.M.; Boatright, J.H. Systemic Treatment with Nicotinamide Riboside Is Protective in a Mouse Model of Light-Induced Retinal Degeneration. Investig. Ophthalmol. Vis. Sci. 2020, 61. [Google Scholar] [CrossRef]
- Conze, D.B.; Crespo-Barreto, J.; Kruger, C.L. Safety assessment of nicotinamide riboside, a form of Vitamin B3. Hum. Exp. Toxicol. 2016, 35, 1149–1160. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kourtzidis, I.A.; Stoupas, A.T.; Gioris, I.S.; Veskoukis, A.S.; Margaritelis, N.V.; Tsantarliotou, M.; Taitzoglou, I.; Vrabas, I.S.; Paschalis, V.; Kyparos, A.; et al. The NAD+ precursor nicotinamide riboside decreases exercise performance in rats. J. Int. Soc. Sports Nutr. 2016, 13, 1–4. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kourtzidis, I.A.; Dolopikou, C.F.; Tsiftsis, A.N.; Margaritelis, N.V.; Theodorou, A.A.; Zervos, I.A.; Tsantarliotou, M.P.; Veskoukis, A.S.; Vrabas, I.S.; Paschalis, V.; et al. Nicotinamide riboside supplementation dysregulates redox and energy metabolism in rats: Implications for exercise performance. Exp. Physiol. 2018, 103, 1357–1366. [Google Scholar] [CrossRef] [Green Version]
- Hamity, M.V.; White, S.R.; Walder, R.Y.; Schmidt, M.S.; Brenner, C.; Hammond, D.L. Nicotinamide riboside, a form of vitamin B3 and NAD+ precursor, relieves the nociceptive and aversive dimensions of paclitaxel-induced peripheral neuropathy in female rats. Pain 2017, 158, 962–972. [Google Scholar] [CrossRef]
- Toropova, Y.G.; Pechnikova, N.A.; Zelinskaya, I.A.; Zhuravsky, S.G.; Kornyushin, O.V.; Gonchar, A.I.; Ivkin, D.Y.; Leonova, Y.V.; Karev, V.E.; Karabak, I.A. Nicotinamide riboside has protective effects in a rat model of mesenteric ischaemia-reperfusion. Int. J. Exp. Pathol. 2018, 99, 304–311. [Google Scholar] [CrossRef] [PubMed]
- Brunet, A.; Sweeney, L.B.; Sturgill, J.F.; Chua, K.F.; Greer, P.L.; Lin, Y.; Tran, H.; Ross, S.E.; Mostoslavsy, R.; Cohen, H.Y.; et al. Stress-Dependent Regulation of FOXO Transcription Factors by the SIRT1 Deacetylase. Science 2004, 303, 2011–2015. [Google Scholar] [CrossRef] [Green Version]
- Han, M.K.; Song, E.K.; Guo, Y.; Ou, X.; Mantel, C.; Broxmeyer, H.E. SIRT1 Regulates Apoptosis and Nanog Expression in Mouse Embryonic Stem Cells by Controlling p53 Subcellular Localization. Cell Stem Cell 2008, 2, 241–251. [Google Scholar] [CrossRef] [Green Version]
- Brookins Danz, E.D.; Skramsted, J.; Henry, N.; Bennett, J.A.; Keller, R.S. Resveratrol prevents doxorubicin cardiotoxicity through mitochondrial stabilization and the Sirt1 pathway. Free Radic. Biol. Med. 2009, 46, 1589–1597. [Google Scholar] [CrossRef]
- Alcendor, R.R.; Gao, S.; Zhai, P.; Zablocki, D.; Holle, E.; Yu, X.; Tian, B.; Wagner, T.; Vatner, S.F.; Sadoshima, J. Sirt1 regulates aging and resistance to oxidative stress in the heart. Circ. Res. 2007, 100, 1512–1521. [Google Scholar] [CrossRef]
- Hsu, C.P.; Yamamoto, T.; Oka, S.; Sadoshima, J. The function of nicotinamide phosphoribosyltransferase in the heart. DNA Repair 2014, 23, 64–68. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Luo, M.; Anderson, M.E. Mechanisms of altered Ca2+ handling in heart failure. Circ. Res. 2013, 113, 690–708. [Google Scholar] [CrossRef] [Green Version]
- Luu, A.Z.; Chowdhury, B.; Al-Omran, M.; Teoh, H.; Hess, D.A.; Verma, S. Role of Endothelium in Doxorubicin-Induced Cardiomyopathy. JACC Basic Transl. Sci. 2018, 3, 861–870. [Google Scholar] [CrossRef] [PubMed]
- Mateuszuk, Ł.; Campagna, R.; Kutryb-Zając, B.; Kuś, K.; Słominska, E.M.; Smolenski, R.T.; Chlopicki, S. Reversal of endothelial dysfunction by nicotinamide mononucleotide via extracellular conversion to nicotinamide riboside. Biochem. Pharmacol. 2020, 178, 114019. [Google Scholar] [CrossRef] [PubMed]
- Westman, E.L.; Canova, M.J.; Radhi, I.J.; Koteva, K.; Kireeva, I.; Waglechner, N.; Wright, G.D. Bacterial inactivation of the anticancer drug doxorubicin. Chem. Biol. 2012, 19, 1255–1264. [Google Scholar] [CrossRef] [Green Version]
- Ma, W.; Mao, Q.; Xia, W.; Dong, G.; Yu, C.; Jiang, F. Gut microbiota shapes the efficiency of cancer therapy. Front. Microbiol. 2019, 10. [Google Scholar] [CrossRef] [Green Version]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
| No. Reference | Sex, Age | Dose of NR | Duration/Administration Frequency | Route of Administration | Effect |
|---|---|---|---|---|---|
| [93] | C57Bl/6J mice, male, 8 weeks | 400 mg/kg | 12 weeks | diet | Enhanced oxidative metabolism; protection against high fat diet-induced metabolic abnormalities; improved insulin sensitivity |
| [97] | Tg2576 mice | 250 mg/kg | 3 months | diet | Benefited cognitive function and synaptic plasticity |
| [94] | C57Bl/6J mice, male, 8–10 weeks | 1000 mg/kg | twice daily for 5 days | IP | Activated a NAD+-SIRT3 pathway; reduces neurite degeneration |
| [77] | unspecified | 400 mg/kg | 4 weeks | diet | Improved mitochondrial respiratory capacity in muscle |
| [99] | C57BL/6J mice, male | 500 mg/kg | 6 days | IP | Improved glucose tolerance; reduced weight gain, liver damage and the development of hepatic steatosis in prediabetic mice; protect against sensory neuropathy |
| [35] | C57Bl/6J mice, 12-week-old male; 6–8-week-old | 185 mg/kg; 500 mg/kg | 1 week; 6 days | diet; IP | The increase in NAAD is a highly sensitive biomarker of effective NAD+ repletion. |
| [96] | C57BL/10ScSn-Dmdmdx/J mice, male | 400 mg/kg | 6–8 weeks | diet | Induced the mitochondrial unfolded protein response; delayed senescence of neural SCs and melanocyte SCs; increased mouse life span |
| [101] | C57BL/6 mice, male, 2 months | 100, 300, 500 mg/kg | single dose | IP | prevented lung and heart injury; improved the survival in sepsis |
| [102] | C57BL/6 mice, male, 2 months | 100, 300, 500 mg/kg | single dose | IP | Elevated NAD+ levels, reduced cardiac injury and myocardial dysfunction |
| [103] | BALB/c mice, male, 3 months | 1000 mg/kg | single dose | IP | Protective effects of NR treatment in a mouse model of retinal degeneration |
| [104] | Sprague-Dawley rats, male and female | 5000 mg/kg; 750, 1500, 2500, 5000 mg/kg;, 300, 1000, 3000 mg/kg | Single; 14 days; 90 days | gavage | Toxicity profile similar to nicotinamide, target organs of toxicity were liver, kidney, ovaries, and testes; the lowest observed adverse effect level for NR was 1000 mg/kg/day; the no observed adverse effect level was 300 mg/kg/day |
| [105] | Wistar rats, male, 4 months | 300 mg/kg | 21 days | gavage | Negative effect of NR administration on physical performance |
| [106] | Wistar rats, male, 4 months | 300 mg/kg | 21 days | gavage | Increased NADPH levels in liver, but not in muscle, decreased the activity of major antioxidant enzymes in muscle; excessively increased glycogen in liver, but not in muscle; decreased glucose concentrations in blood; decrease maximal lactate production during exercise |
| [107] | Sprague-Dawley rats, female | 200 mg/kg | 7 days prior to and 24 days post-paclitaxe; 21 days beginning 14 days post-paclitaxel | gavage | Reversed the well-established tactile hypersensitivity in a subset of rats and blunte escape–avoidance behavior |
| [108] | Wistar rats, male | 50 mg/kg | single dose | IV | Protect the intestinal wall from ischaemia-reperfusion injury; improving the relaxation function of mesenteric vessels |
| [57] | Wistar rats, male | 400 mg/kg | 28 days | IP | Reduced adiposity (visceral and subcutaneous); improved insulin resistance; increased the antioxidant capacity via glutathione peroxidase and catalase enzymes (in rats under calorie restriction) |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Podyacheva, E.; Toropova, Y. Nicotinamide Riboside for the Prevention and Treatment of Doxorubicin Cardiomyopathy. Opportunities and Prospects. Nutrients 2021, 13, 3435. https://doi.org/10.3390/nu13103435
Podyacheva E, Toropova Y. Nicotinamide Riboside for the Prevention and Treatment of Doxorubicin Cardiomyopathy. Opportunities and Prospects. Nutrients. 2021; 13(10):3435. https://doi.org/10.3390/nu13103435
Chicago/Turabian StylePodyacheva, Ekaterina, and Yana Toropova. 2021. "Nicotinamide Riboside for the Prevention and Treatment of Doxorubicin Cardiomyopathy. Opportunities and Prospects" Nutrients 13, no. 10: 3435. https://doi.org/10.3390/nu13103435