
Astaxanthin, a Marine Carotenoid, Maintains the Tolerance and Integrity of Adipose Tissue and Contributes to Its Healthy Functions

 ,
,  , , , and
, , , and
Abstract
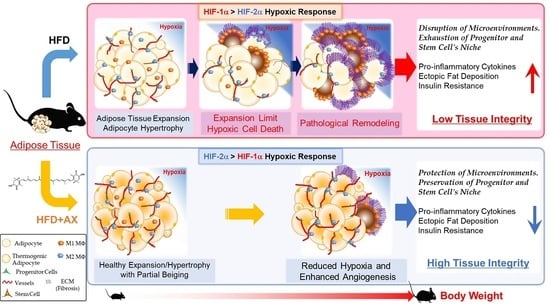
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Materials and Methods
2.1. Reagents
2.2. Animals
2.3. Body Composition Analysis
2.4. Realtime Reverse Transcription-Polymerase Chain Reaction (Realtime RT-PCR)
2.5. Glucose Tolerance Test and Insulin Tolerance Test
2.6. Flow Cytometry Analysis
2.7. Immunohistochemistry
2.8. Oxidative Stress Analysis
2.9. Statistical Analysis
3. Results
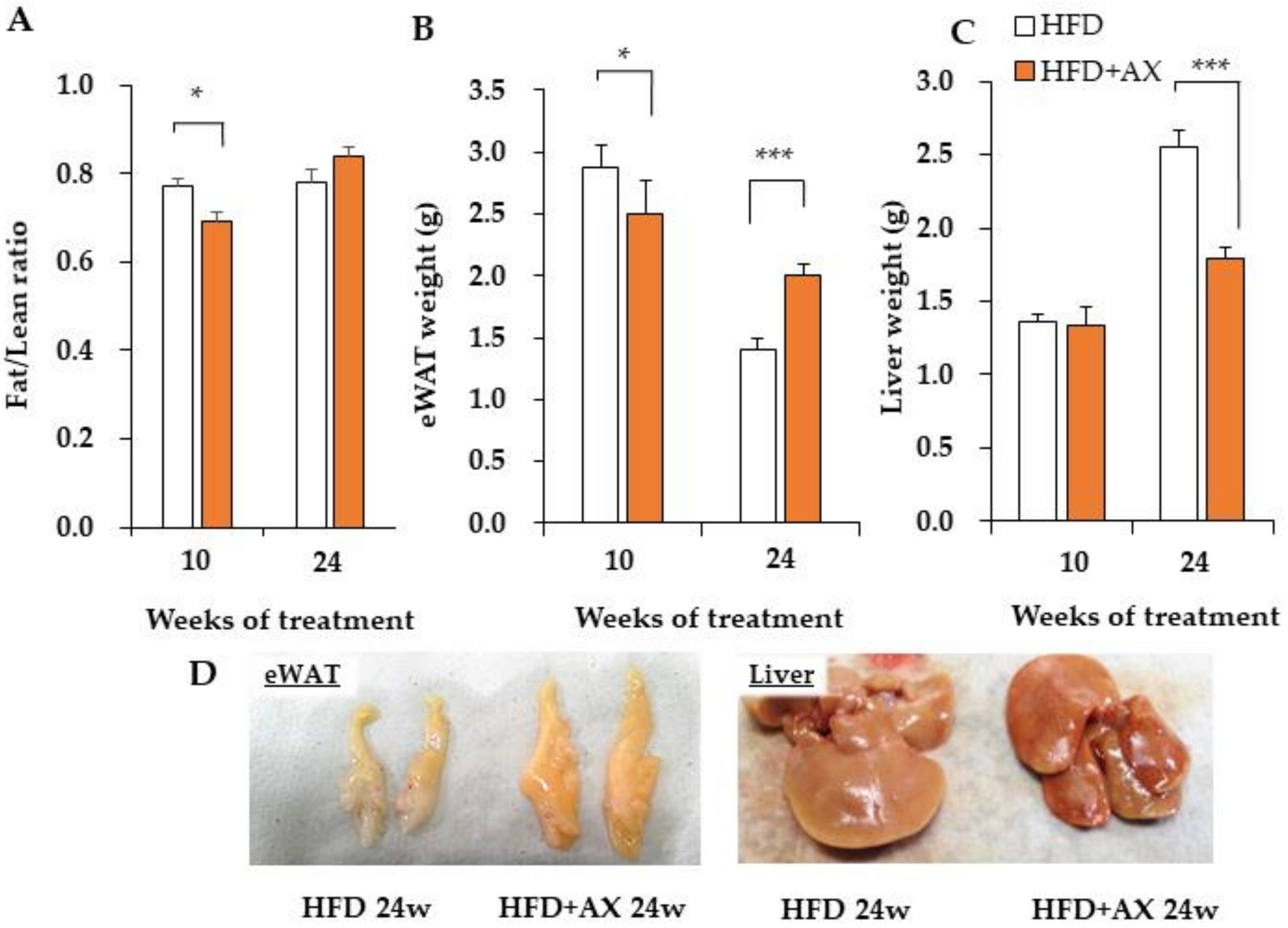
3.1. AX Inhibits Increases in Liver Weight, but Increases the Weight of Adipose Tissue upon High-Fat Diet Stimuli
3.2. AX Decreases Oxidative Stress in Adipose Tissue and Suppresses the Expression of Pro-Inflammatory Cytokine Genes
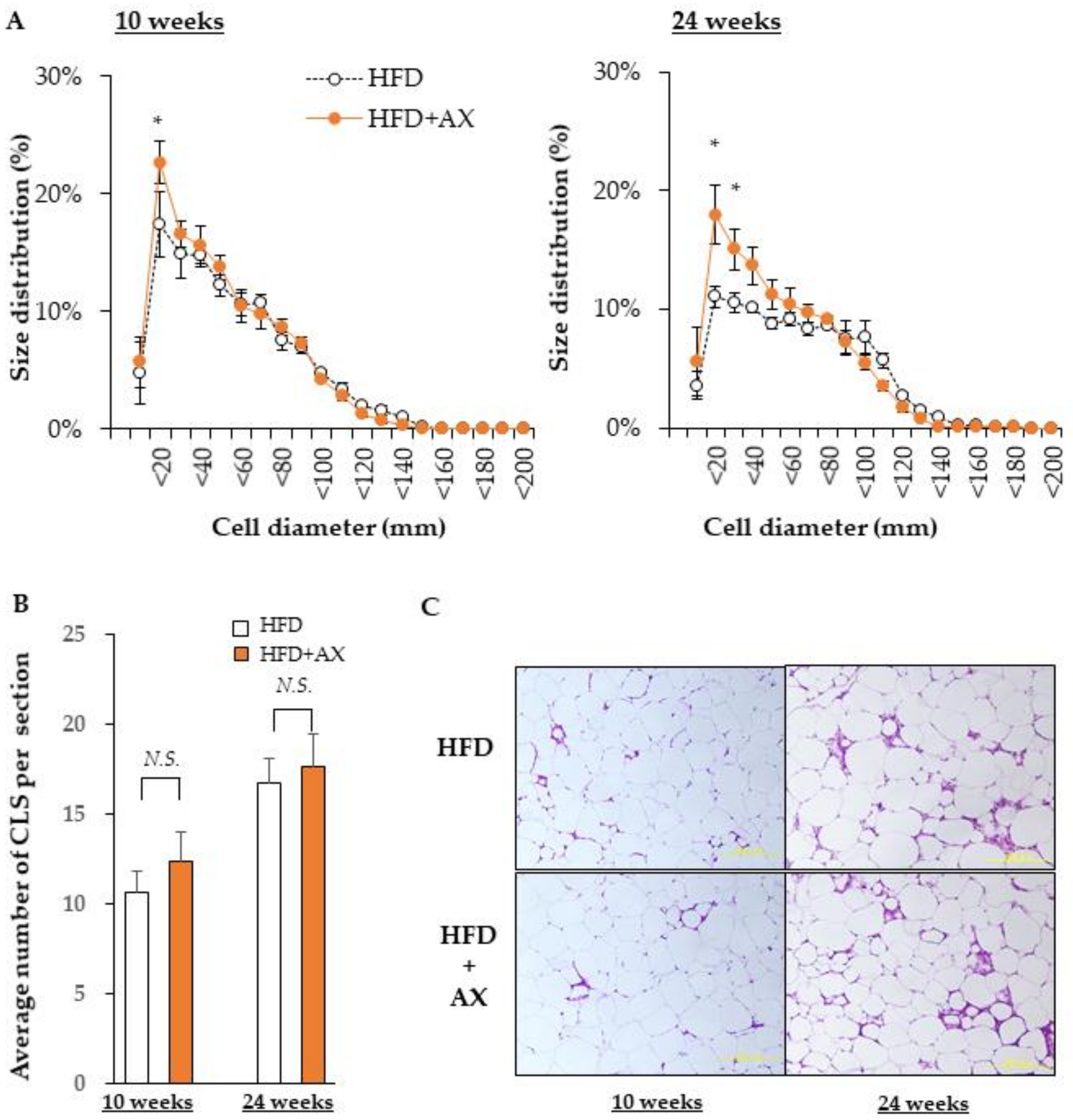
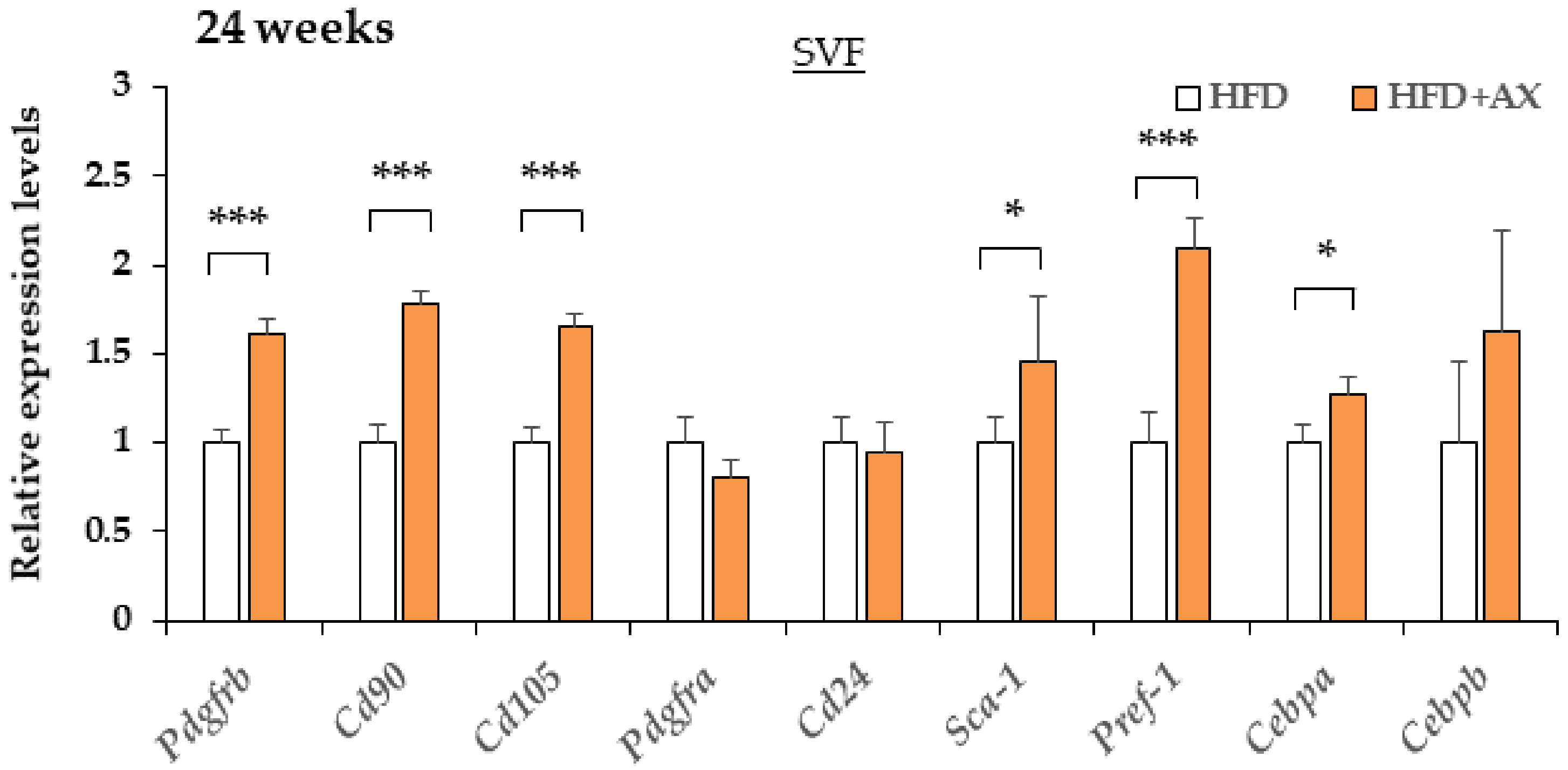
3.3. AX Reduces Inflammation of SVF in eWAT and Maintain Pool Size of Adipocyte Progenitors
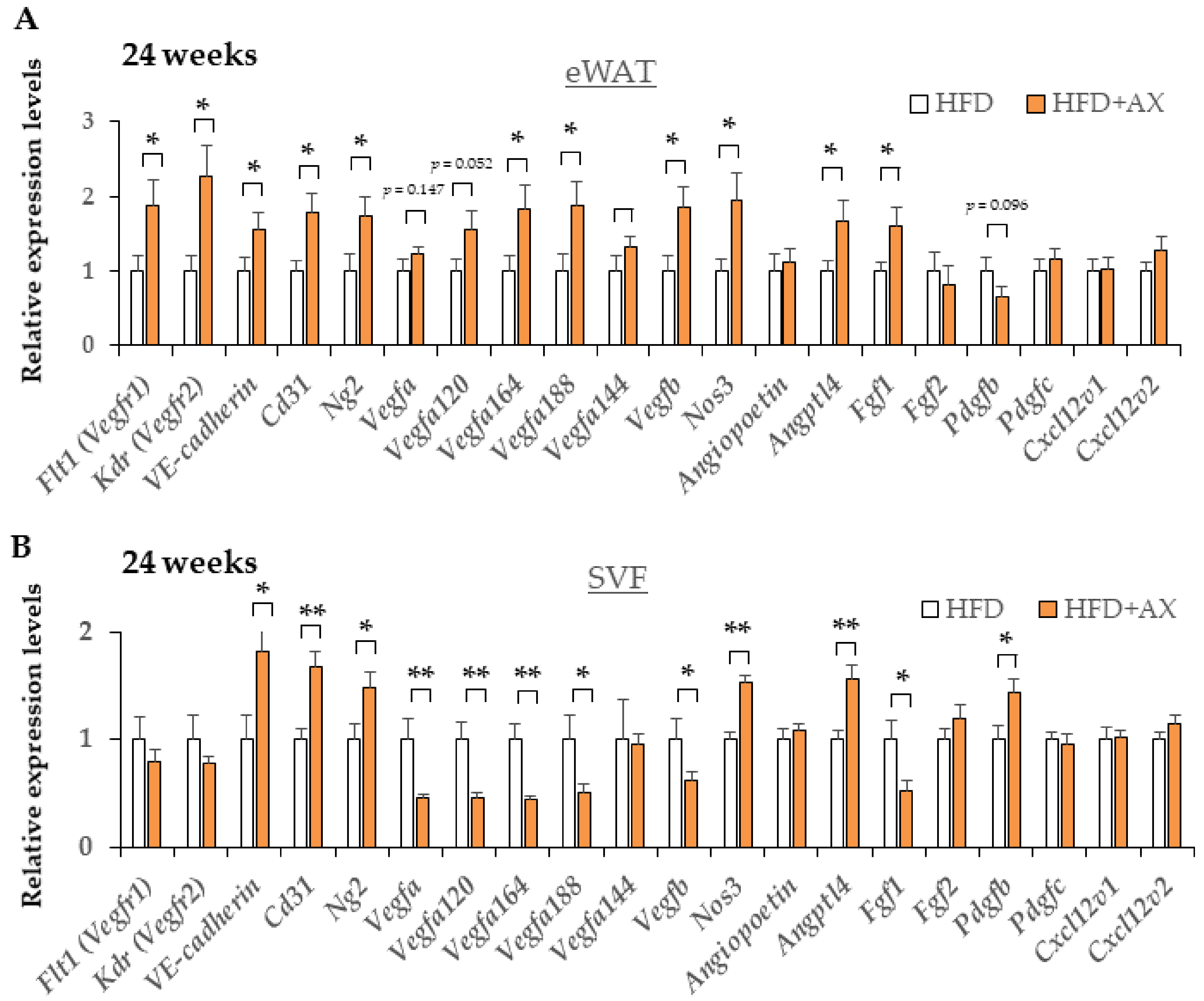
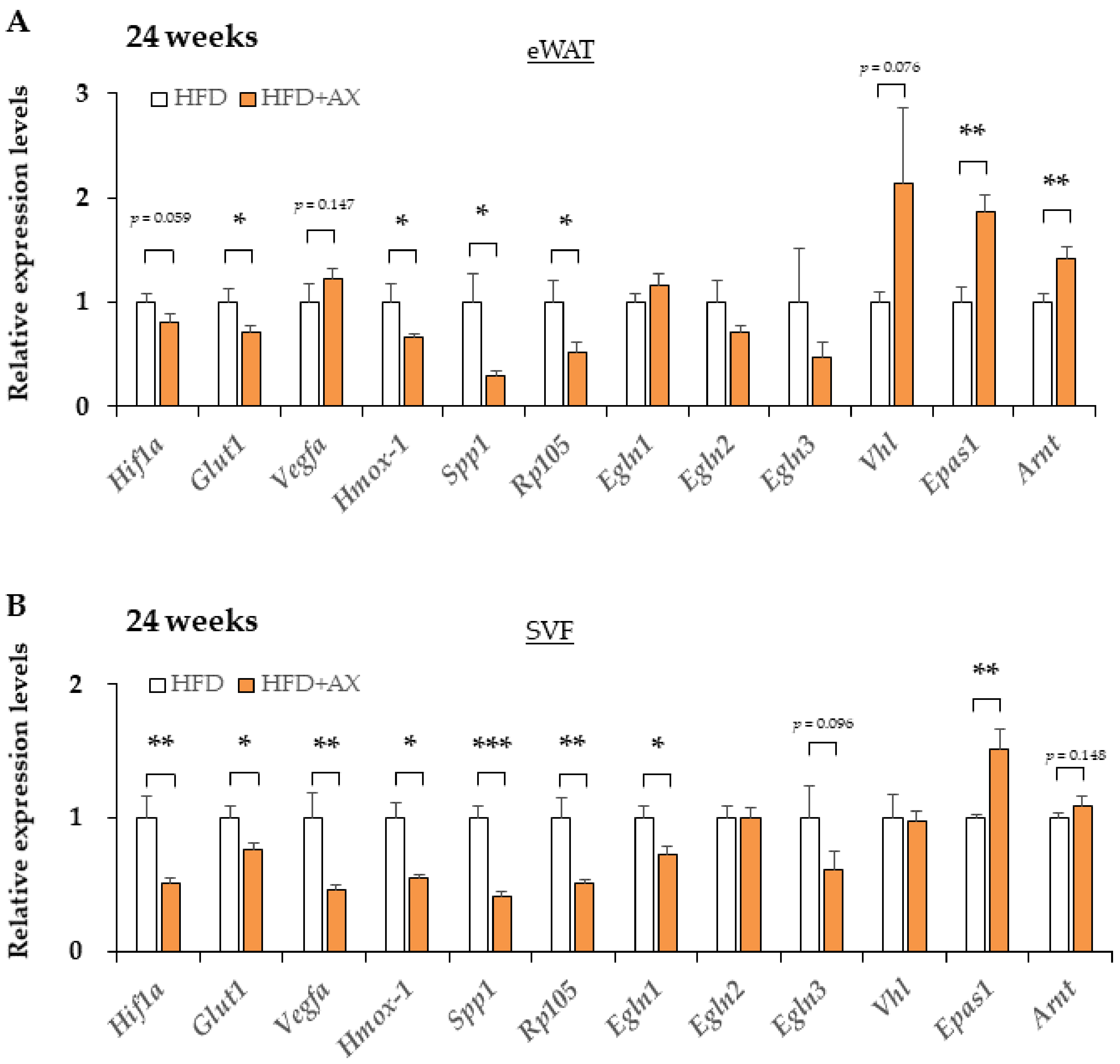
3.4. AX Induces a Predominantly HIF-2α, but Not HIF-1α, Hypoxic Response in eWAT
3.5. Intervention with AX in Mice after the Completion of Pathological Obesity Improved Insulin Resistance and Glucose Intolerance
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Friedman, J.M. Obesity in the new millennium. Nature 2000, 404, 632–634. [Google Scholar] [CrossRef]
- Cho, N.H.; Shaw, J.E.; Karuranga, S.; Huang, Y.; da Rocha Fernandes, J.D.; Ohlrogge, A.W.; Malanda, B. IDF Diabetes Atlas: Global estimates of diabetes prevalence for 2017 and projections for 2045. Diabetes Res. Clin. Pract. 2018, 138, 271–281. [Google Scholar] [CrossRef]
- Reinehr, T. Type 2 diabetes mellitus in children and adolescents. World J. Diabetes 2013, 4, 270–281. [Google Scholar] [CrossRef]
- Chait, A.; den Hartigh, L.J. Adipose Tissue Distribution, Inflammation and Its Metabolic Consequences, Including Diabetes and Cardiovascular Disease. Front. Cardiovasc. Med. 2020, 7, 22. [Google Scholar] [CrossRef] [Green Version]
- Balistreri, C.R.; Caruso, C.; Candore, G. The role of adipose tissue and adipokines in obesity-related inflammatory diseases. Mediat. Inflamm. 2010, 2010, 802078. [Google Scholar] [CrossRef] [PubMed]
- Nawaz, A.; Aminuddin, A.; Kado, T.; Takikawa, A.; Yamamoto, S.; Tsuneyama, K.; Igarashi, Y.; Ikutani, M.; Nishida, Y.; Nagai, Y.; et al. CD206+ M2-like macrophages regulate systemic glucose metabolism by inhibiting proliferation of adipocyte progenitors. Nat. Commun. 2017, 8, 286. [Google Scholar] [CrossRef] [PubMed]
- Heenan, K.A.; Carrillo, A.E.; Fulton, J.L.; Ryan, E.J.; Edsall, J.R.; Rigopoulos, D.; Markofski, M.M.; Flouris, A.D.; Dinas, P.C. Effects of Nutrition/Diet on Brown Adipose Tissue in Humans: A Systematic Review and Meta-Analysis. Nutrients 2020, 12, 2752. [Google Scholar] [CrossRef] [PubMed]
- Landrier, J.F.; Marcotorchino, J.; Tourniaire, F. Lipophilic micronutrients and adipose tissue biology. Nutrients 2012, 4, 1622–1649. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ishiki, M.; Nishida, Y.; Ishibashi, H.; Wada, T.; Fujisaka, S.; Takikawa, A.; Urakaze, M.; Sasaoka, T.; Usui, I.; Tobe, K. Impact of divergent effects of astaxanthin on insulin signaling in L6 cells. Endocrinology 2013, 154, 2600–2612. [Google Scholar] [CrossRef] [Green Version]
- Nishida, Y.; Nawaz, A.; Kado, T.; Takikawa, A.; Igarashi, Y.; Onogi, Y.; Wada, T.; Sasaoka, T.; Yamamoto, S.; Sasahara, M.; et al. Astaxanthin stimulates mitochondrial biogenesis in insulin resistant muscle via activation of AMPK pathway. J. Cachexia Sarcopenia Muscle 2020, 11, 241–258. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Uchiyama, K.; Naito, Y.; Hasegawa, G.; Nakamura, N.; Takahashi, J.; Yoshikawa, T. Astaxanthin protects beta-cells against glucose toxicity in diabetic db/db mice. Redox Rep. Commun. Free Radic. Res. 2002, 7, 290–293. [Google Scholar] [CrossRef]
- Hussein, G.; Nakagawa, T.; Goto, H.; Shimada, Y.; Matsumoto, K.; Sankawa, U.; Watanabe, H. Astaxanthin ameliorates features of metabolic syndrome in SHR/NDmcr-cp. Life Sci. 2007, 80, 522–529. [Google Scholar] [CrossRef]
- Preuss, H.G.; Echard, B.; Yamashita, E.; Perricone, N.V. High dose astaxanthin lowers blood pressure and increases insulin sensitivity in rats: Are these effects interdependent? Int. J. Med. Sci. 2011, 8, 126–138. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ni, Y.; Nagashimada, M.; Zhuge, F.; Zhan, L.; Nagata, N.; Tsutsui, A.; Nakanuma, Y.; Kaneko, S.; Ota, T. Astaxanthin prevents and reverses diet-induced insulin resistance and steatohepatitis in mice: A comparison with vitamin E. Sci. Rep. 2015, 5, 17192. [Google Scholar] [CrossRef]
- Choi, H.D.; Youn, Y.K.; Shin, W.G. Positive effects of astaxanthin on lipid profiles and oxidative stress in overweight subjects. Plant Foods Hum. Nutr. 2011, 66, 363–369. [Google Scholar] [CrossRef] [PubMed]
- Uchiyama, A.O.Y. Clinical efficacy of astaxanthin-containing Haematococcus pluvialis extract for the volunteers at risk of metabolic syndrome. J. Clin. Biochem. Nutr. 2008, 43, 38–43. [Google Scholar]
- Yoshida, H.; Yanai, H.; Ito, K.; Tomono, Y.; Koikeda, T.; Tsukahara, H.; Tada, N. Administration of natural astaxanthin increases serum HDL-cholesterol and adiponectin in subjects with mild hyperlipidemia. Atherosclerosis 2010, 209, 520–523. [Google Scholar] [CrossRef]
- Mashhadi, N.S.; Zakerkish, M.; Mohammadiasl, J.; Zarei, M.; Mohammadshahi, M.; Haghighizadeh, M.H. Astaxanthin improves glucose metabolism and reduces blood pressure in patients with type 2 diabetes mellitus. Asia Pac. J. Clin. Nutr. 2018, 27, 341–346. [Google Scholar] [CrossRef]
- Igarashi, Y.; Nawaz, A.; Kado, T.; Bilal, M.; Kuwano, T.; Yamamoto, S.; Sasahara, M.; Jiuxiang, X.; Inujima, A.; Koizumi, K.; et al. Partial depletion of CD206-positive M2-like macrophages induces proliferation of beige progenitors and enhances browning after cold stimulation. Sci. Rep. 2018, 8, 14567. [Google Scholar] [CrossRef]
- Takikawa, A.; Mahmood, A.; Nawaz, A.; Kado, T.; Okabe, K.; Yamamoto, S.; Aminuddin, A.; Senda, S.; Tsuneyama, K.; Ikutani, M.; et al. HIF-1alpha in Myeloid Cells Promotes Adipose Tissue Remodeling Toward Insulin Resistance. Diabetes 2016, 65, 3649–3659. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kadowaki, T.; Yamauchi, T.; Kubota, N.; Hara, K.; Ueki, K.; Tobe, K. Adiponectin and adiponectin receptors in insulin resistance, diabetes, and the metabolic syndrome. J. Clin. Investig. 2006, 116, 1784–1792. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Inoue, M.; Tanabe, H.; Matsumoto, A.; Takagi, M.; Umegaki, K.; Amagaya, S.; Takahashi, J. Astaxanthin functions differently as a selective peroxisome proliferator-activated receptor gamma modulator in adipocytes and macrophages. Biochem. Pharm. 2012, 84, 692–700. [Google Scholar] [CrossRef] [PubMed]
- Hernandez-Quiles, M.; Broekema, M.F.; Kalkhoven, E. PPARgamma in Metabolism, Immunity, and Cancer: Unified and Diverse Mechanisms of Action. Front. Endocrinol. 2021, 12, 624112. [Google Scholar] [CrossRef]
- Maurer, S.; Harms, M.; Boucher, J. The colorful versatility of adipocytes: White-to-brown transdifferentiation and its therapeutic potential in humans. FEBS J. 2021, 288, 3628–3646. [Google Scholar] [CrossRef] [PubMed]
- Nawaz, A.; Tobe, K. M2-like macrophages serve as a niche for adipocyte progenitors in adipose tissue. J. Diabetes Investig. 2019, 10, 1394–1400. [Google Scholar] [CrossRef]
- Nawaz, A.; Mehmood, A.; Kanatani, Y.; Kado, T.; Igarashi, Y.; Takikawa, A.; Yamamoto, S.; Okabe, K.; Nakagawa, T.; Yagi, K.; et al. Sirt1 activator induces proangiogenic genes in preadipocytes to rescue insulin resistance in diet-induced obese mice. Sci. Rep. 2018, 8, 11370. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Koh, M.Y.; Powis, G. Passing the baton: The HIF switch. Trends Biochem. Sci. 2012, 37, 364–372. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Poyton, R.; Hendrickson, M. Crosstalk between nitric oxide and hypoxia-inducible factor signaling pathways: An update. Res. Rep. Biochem. 2015, 5, 147–161. [Google Scholar] [CrossRef] [Green Version]
- Michailidou, Z.; Morton, N.M.; Moreno Navarrete, J.M.; West, C.C.; Stewart, K.J.; Fernandez-Real, J.M.; Schofield, C.J.; Seckl, J.R.; Ratcliffe, P.J. Adipocyte pseudohypoxia suppresses lipolysis and facilitates benign adipose tissue expansion. Diabetes 2015, 64, 733–745. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Choe, S.S.; Shin, K.C.; Ka, S.; Lee, Y.K.; Chun, J.S.; Kim, J.B. Macrophage HIF-2alpha ameliorates adipose tissue inflammation and insulin resistance in obesity. Diabetes 2014, 63, 3359–3371. [Google Scholar] [CrossRef] [Green Version]
- Obradovic, M.; Sudar-Milovanovic, E.; Soskic, S.; Essack, M.; Arya, S.; Stewart, A.J.; Gojobori, T.; Isenovic, E.R. Leptin and Obesity: Role and Clinical Implication. Front. Endocrinol. 2021, 12, 585887. [Google Scholar] [CrossRef] [PubMed]
- Funcke, J.B.; Scherer, P.E. Beyond adiponectin and leptin: Adipose tissue-derived mediators of inter-organ communication. J. Lipid Res. 2019, 60, 1648–1684. [Google Scholar] [CrossRef]
- Bilal, M.; Nawaz, A.; Kado, T.; Aslam, M.R.; Igarashi, Y.; Nishimura, A.; Watanabe, Y.; Kuwano, T.; Liu, J.; Miwa, H.; et al. Fate of adipocyte progenitors during adipogenesis in mice fed a high-fat diet. Mol. Metab. 2021, 54, 101328. [Google Scholar] [CrossRef]
- Sung, H.K.; Doh, K.O.; Son, J.E.; Park, J.G.; Bae, Y.; Choi, S.; Nelson, S.M.; Cowling, R.; Nagy, K.; Michael, I.P.; et al. Adipose vascular endothelial growth factor regulates metabolic homeostasis through angiogenesis. Cell Metab. 2013, 17, 61–72. [Google Scholar] [CrossRef] [Green Version]
- Fujisaka, S.; Usui, I.; Ikutani, M.; Aminuddin, A.; Takikawa, A.; Tsuneyama, K.; Mahmood, A.; Goda, N.; Nagai, Y.; Takatsu, K.; et al. Adipose tissue hypoxia induces inflammatory M1 polarity of macrophages in an HIF-1alpha-dependent and HIF-1alpha-independent manner in obese mice. Diabetologia 2013, 56, 1403–1412. [Google Scholar] [CrossRef] [Green Version]
- Zhang, X.; Zhang, Y.; Wang, P.; Zhang, S.Y.; Dong, Y.; Zeng, G.; Yan, Y.; Sun, L.; Wu, Q.; Liu, H.; et al. Adipocyte Hypoxia-Inducible Factor 2alpha Suppresses Atherosclerosis by Promoting Adipose Ceramide Catabolism. Cell Metab. 2019, 30, 937–951.e935. [Google Scholar] [CrossRef] [PubMed]
- Tabatabaie, T.; Potts, J.D.; Floyd, R.A. Reactive oxygen species-mediated inactivation of pyruvate dehydrogenase. Arch. Biochem. Biophys. 1996, 336, 290–296. [Google Scholar] [CrossRef]
- Kowshik, J.; Baba, A.B.; Giri, H.; Deepak Reddy, G.; Dixit, M.; Nagini, S. Astaxanthin inhibits JAK/STAT-3 signaling to abrogate cell proliferation, invasion and angiogenesis in a hamster model of oral cancer. PLoS ONE 2014, 9, e109114. [Google Scholar] [CrossRef]
- Onyango, A.N. The Contribution of Singlet Oxygen to Insulin Resistance. Oxidative Med. Cell. Longev. 2017, 2017, 8765972. [Google Scholar] [CrossRef] [Green Version]
- Murotomi, K.; Umeno, A.; Yasunaga, M.; Shichiri, M.; Ishida, N.; Abe, H.; Yoshida, Y.; Nakajima, Y. Switching from singlet-oxygen-mediated oxidation to free-radical-mediated oxidation in the pathogenesis of type 2 diabetes in model mouse. Free Radic. Res. 2015, 49, 133–138. [Google Scholar] [CrossRef]
- Umeno, A.; Shichiri, M.; Ishida, N.; Hashimoto, Y.; Abe, K.; Kataoka, M.; Yoshino, K.; Hagihara, Y.; Aki, N.; Funaki, M.; et al. Singlet oxygen induced products of linoleates, 10- and 12-(Z,E)-hydroxyoctadecadienoic acids (HODE), can be potential biomarkers for early detection of type 2 diabetes. PLoS ONE 2013, 8, e63542. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Miki, W. Biological functions and activities of animal carotenoids. Pure Appl. Chem. 1991, 63, 141–146. [Google Scholar] [CrossRef]
- Nishida, Y.Y.E.; Miki, W. Quenching Activities of Common Hydrophilic and Lipophilic Antioxidants against Singlet Oxygen Using Chemiluminescence Detection System. Carotenoid Sci. 2007, 11, 16–20. [Google Scholar]
- Lee, S.J.; Bai, S.K.; Lee, K.S.; Namkoong, S.; Na, H.J.; Ha, K.S.; Han, J.A.; Yim, S.V.; Chang, K.; Kwon, Y.G.; et al. Astaxanthin inhibits nitric oxide production and inflammatory gene expression by suppressing I(kappa)B kinase-dependent NF-kappaB activation. Mol. Cells 2003, 16, 97–105. [Google Scholar]
- Koivunen, P.; Hirsila, M.; Gunzler, V.; Kivirikko, K.I.; Myllyharju, J. Catalytic properties of the asparaginyl hydroxylase (FIH) in the oxygen sensing pathway are distinct from those of its prolyl 4-hydroxylases. J. Biol. Chem. 2004, 279, 9899–9904. [Google Scholar] [CrossRef] [Green Version]
- Rasbach, K.A.; Gupta, R.K.; Ruas, J.L.; Wu, J.; Naseri, E.; Estall, J.L.; Spiegelman, B.M. PGC-1alpha regulates a HIF2alpha-dependent switch in skeletal muscle fiber types. Proc. Natl. Acad. Sci. USA 2010, 107, 21866–21871. [Google Scholar] [CrossRef] [Green Version]
- Kanazashi, M.; Tanaka, M.; Murakami, S.; Kondo, H.; Nagatomo, F.; Ishihara, A.; Roy, R.R.; Fujino, H. Amelioration of capillary regression and atrophy of the soleus muscle in hindlimb-unloaded rats by astaxanthin supplementation and intermittent loading. Exp. Physiol. 2014, 99, 1065–1077. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, X.; Matsumoto, T.; Takuwa, M.; Saeed Ebrahim Shaiku Ali, M.; Hirabashi, T.; Kondo, H.; Fujino, H. Protective Effects of Astaxanthin Supplementation against Ultraviolet-Induced Photoaging in Hairless Mice. Biomedicines 2020, 8, 18. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hu, X.; Xu, X.; Lu, Z.; Zhang, P.; Fassett, J.; Zhang, Y.; Xin, Y.; Hall, J.L.; Viollet, B.; Bache, R.J.; et al. AMP activated protein kinase-alpha2 regulates expression of estrogen-related receptor-alpha, a metabolic transcription factor related to heart failure development. Hypertension 2011, 58, 696–703. [Google Scholar] [CrossRef] [Green Version]
- Matsakas, A.; Yadav, V.; Lorca, S.; Evans, R.M.; Narkar, V.A. Revascularization of ischemic skeletal muscle by estrogen-related receptor-gamma. Circ. Res. 2012, 110, 1087–1096. [Google Scholar] [CrossRef]
- LaBarge, S.; McDonald, M.; Smith-Powell, L.; Auwerx, J.; Huss, J.M. Estrogen-related receptor-alpha (ERRalpha) deficiency in skeletal muscle impairs regeneration in response to injury. FASEB J. 2014, 28, 1082–1097. [Google Scholar] [CrossRef] [PubMed] [Green Version]



Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Nawaz, A.; Nishida, Y.; Takikawa, A.; Fujisaka, S.; Kado, T.; Aminuddin, A.; Bilal, M.; Jeelani, I.; Aslam, M.R.; Nishimura, A.; et al. Astaxanthin, a Marine Carotenoid, Maintains the Tolerance and Integrity of Adipose Tissue and Contributes to Its Healthy Functions. Nutrients 2021, 13, 4374. https://doi.org/10.3390/nu13124374
Nawaz A, Nishida Y, Takikawa A, Fujisaka S, Kado T, Aminuddin A, Bilal M, Jeelani I, Aslam MR, Nishimura A, et al. Astaxanthin, a Marine Carotenoid, Maintains the Tolerance and Integrity of Adipose Tissue and Contributes to Its Healthy Functions. Nutrients. 2021; 13(12):4374. https://doi.org/10.3390/nu13124374
Chicago/Turabian StyleNawaz, Allah, Yasuhiro Nishida, Akiko Takikawa, Shiho Fujisaka, Tomonobu Kado, Aminuddin Aminuddin, Muhammad Bilal, Ishtiaq Jeelani, Muhammad Rahil Aslam, Ayumi Nishimura, and et al. 2021. "Astaxanthin, a Marine Carotenoid, Maintains the Tolerance and Integrity of Adipose Tissue and Contributes to Its Healthy Functions" Nutrients 13, no. 12: 4374. https://doi.org/10.3390/nu13124374