
Hypothalamic Inflammation as a Potential Pathophysiologic Basis for the Heterogeneity of Clinical, Hormonal, and Metabolic Presentation in PCOS

 ,
, 
Abstract
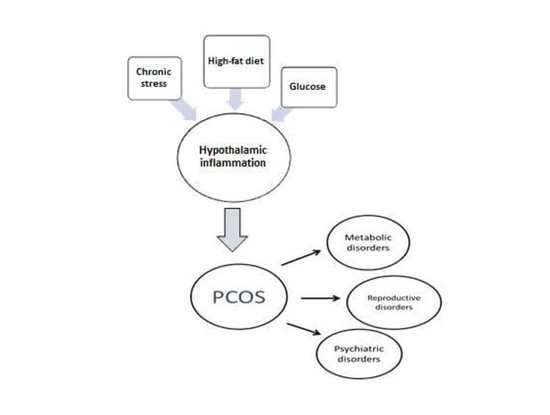
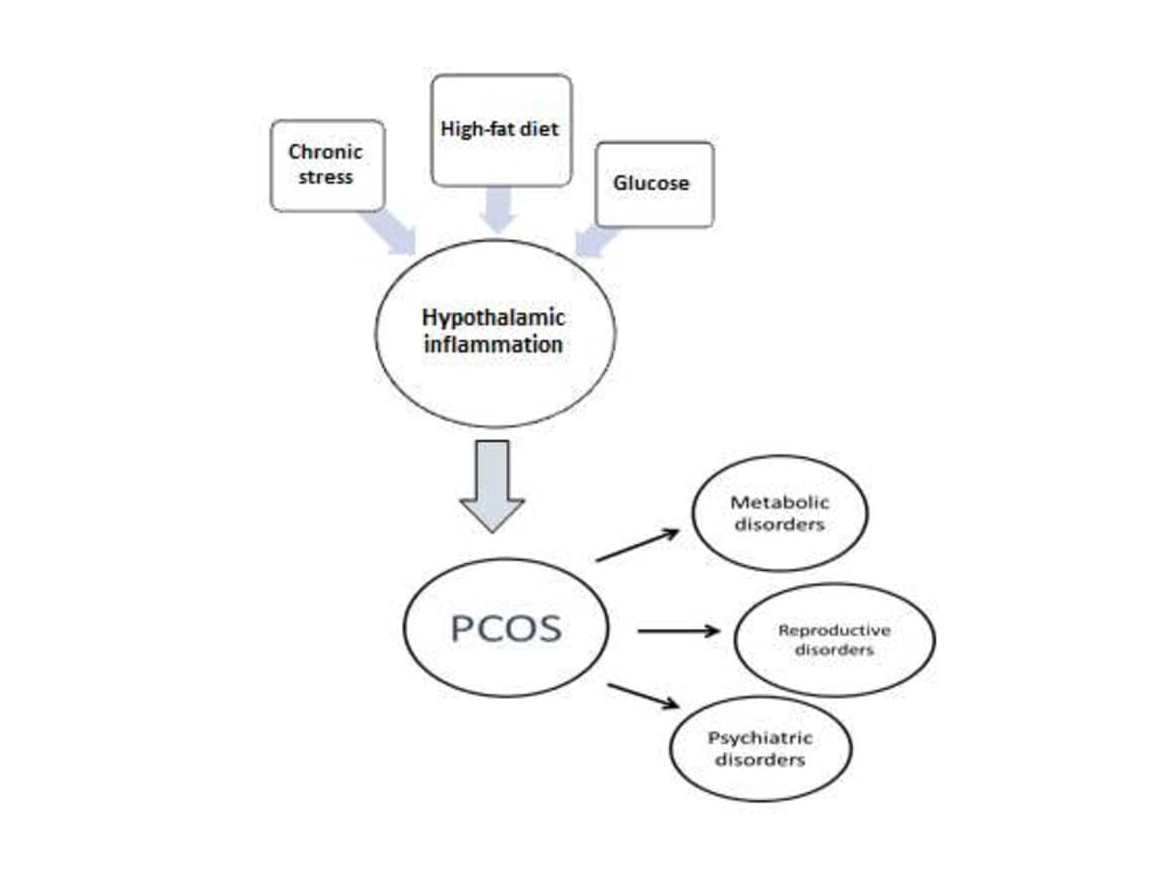{kind=link}
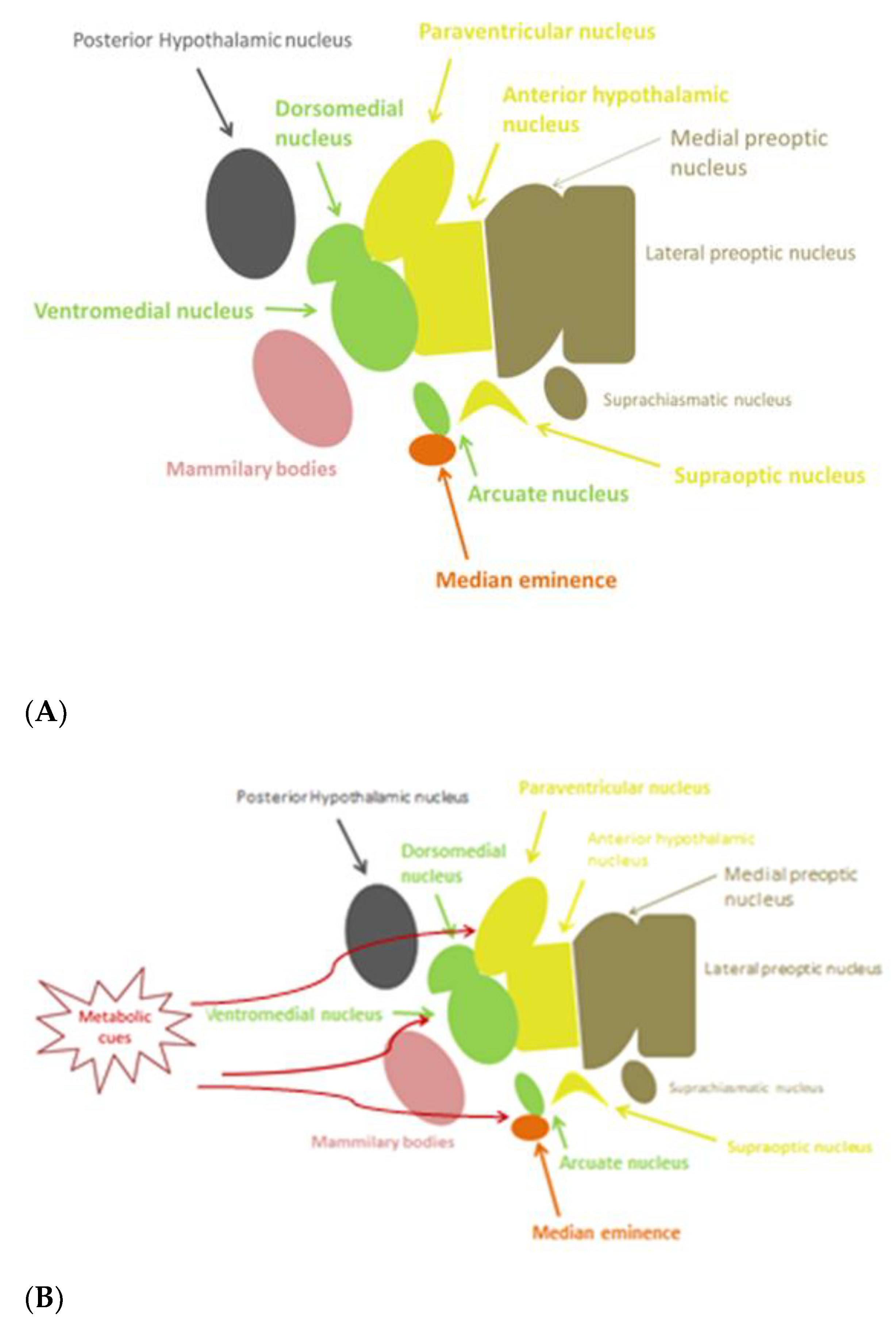{kind=link}
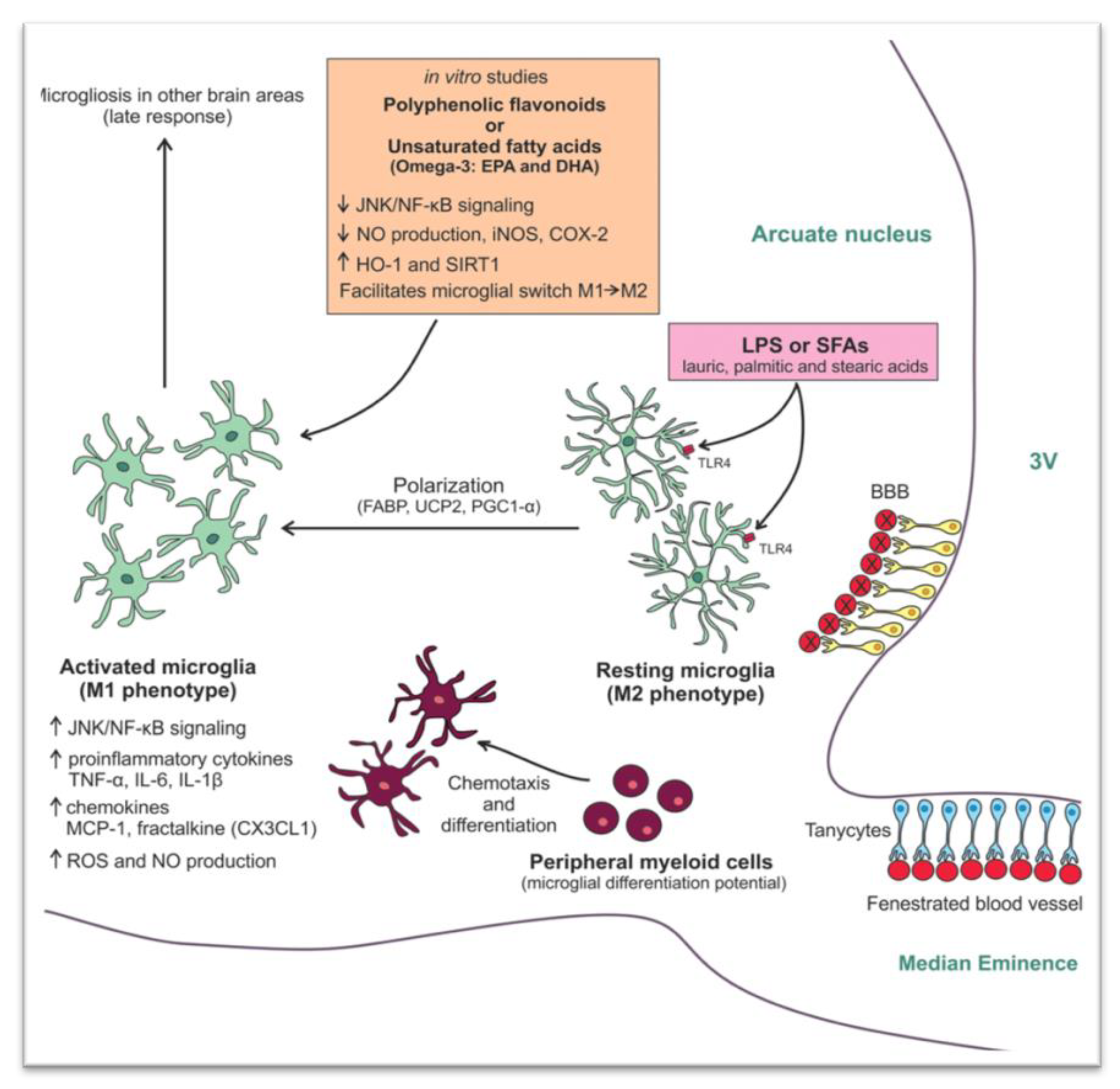{kind=link}
1. Introduction
2. Methodology
3. Potential Mechanisms Triggering Inflammation in the Hypothalamus
3.1. Glucose
3.2. Saturated Fatty Acids
3.3. Uric Acid
3.4. Stress
4. Heterogeneity of Clinical, Hormonal, and Metabolic Presentation in PCOS Physiopathologic Mechanisms
4.1. Heterogeneity of Clinical, Hormonal, and Metabolic Presentation in PCOS
4.2. Physiopathologic Mechanisms in PCOS
5. Hypothalamic Inflammation as a Potential Pathophysiological Mechanism of the Heterogeneity of PCOS
5.1. Hypothalamic Inflammation-Induced Metabolic Disorders
5.2. Potential Hypothalamic Inflammation-Associated Mechanisms in Reproductive Disorders
5.3. Potential Mechanisms of Hypothalamic Inflammation-Associated Psychiatric Disorders
6. Discussion
7. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Financial Disclosure Statement
Conflicts of Interest
References
- Markakis, E.A. Development of the neuroendocrine hypothalamus. Front. Neuroendocrinol. 2002, 23, 257–291. [Google Scholar] [CrossRef]
- Lechan, R.M.; Toni, R. Functional Anatomy of the Hypothalamus and Pituitary; MDText.com, Inc.: South Dartmouth, MA, USA, 2000; Available online: https://www.ncbi.nlm.nih.gov/books/NBK279126/ (accessed on 17 December 2020).
- Marshall, J.C.; Dalkin, A.C.; Haisenleder, D.J.; Griffin, M.L.; Kelch, R.P. GnRH pulses—The regulators of human reproduction. Trans. Am. Clin. Climatol. Assoc. 1992, 104, 31–46. [Google Scholar]
- Hotamisligil, G.S. Inflammation and metabolic disorders. Nature 2006, 444, 860–867. [Google Scholar] [CrossRef]
- De Souza, C.T.; Araujo, E.P.; Bordin, S.; Ashimine, R.; Zollner, R.L.; Boschero, A.C.; Saad, M.J.A.; Velloso, L.A. Consumption of a fat-rich diet activates a proinflammatory response and induces insulin resistance in the hypothalamus. Endocrinology 2005, 146, 4192–4199. [Google Scholar] [CrossRef]
- Mraz, M.; Haluzik, M. The role of adipose tissue immune cells in obesity and low-grade inflammation. J. Endocrinol. 2014, 222, R113–R127. [Google Scholar] [CrossRef]
- Thaler, J.P.; Yi, C.-X.; Schur, E.A.; Guyenet, S.J.; Hwang, B.H.; Dietrich, M.O.; Zhao, X.; Sarruf, D.A.; Izgur, V.; Maravilla, K.R.; et al. Obesity is associated with hypothalamic injury in rodents and humans. J. Clin. Investig. 2012, 122, 153–162. [Google Scholar] [CrossRef] [PubMed]
- Spalding, K.L.; Arner, E.; Westermark, P.O.; Bernard, S.; Buchholz, B.A.; Bergmann, O.; Blomqvist, L.; Hoffstedt, J.; Näslund, E.; Britton, T.; et al. Dynamics of fat cell turnover in humans. Nat. Cell Biol. 2008, 453, 783–787. [Google Scholar] [CrossRef]
- Boden, G. Obesity and Free Fatty Acids. Endocrinol. Metab. Clin. N. Am. 2008, 37, 635–646. [Google Scholar] [CrossRef]
- Ehses, J.A.; Perren, A.; Eppler, E.; Ribaux, P.; Pospisilik, J.A.; Maor-Cahn, R.; Gueripel, X.; Ellingsgaard, H.; Schneider, M.K.; Biollaz, G.; et al. Increased Number of Islet-Associated Macrophages in Type 2 Diabetes. Diabetes 2007, 56, 2356–2370. [Google Scholar] [CrossRef]
- Eguchi, K.; Manabe, I.; Oishi-Tanaka, Y.; Ohsugi, M.; Kono, N.; Ogata, F.; Yagi, N.; Ohto, U.; Kimoto, M.; Miyake, K.; et al. Saturated Fatty Acid and TLR Signaling Link β Cell Dysfunction and Islet Inflammation. Cell Metab. 2012, 15, 518–533. [Google Scholar] [CrossRef]
- Escobar-Morreale, H.F. Polycystic ovary syndrome: Definition, aetiology, diagnosis and treatment. Nat. Rev. Endocrinol. 2018, 14, 270–284. [Google Scholar] [CrossRef]
- Chang, J.; Azziz, R.; Legro, R.; Dewailly, D.; Franks, S.; Tarlatzis, R.; Fauser, B.; Balen, A.; Bouchard, P.; Dalgien, E.; et al. Revised 2003 consensus on diagnostic criteria and long-term health risks related to polycystic ovary syndrome. Fertil. Steril. 2004, 81, 19–25. [Google Scholar] [CrossRef]
- Peigné, M.; Dewailly, D. Long term complications of polycystic ovary syndrome (PCOS). Ann. d’Endocrinol. 2014, 75, 194–199. [Google Scholar] [CrossRef]
- Sanchez-Garrido, M.A.; Tena-Sempere, M. Metabolic dysfunction in polycystic ovary syndrome: Pathogenic role of androgen excess and potential therapeutic strategies. Mol. Metab. 2020, 35, 100937. [Google Scholar] [CrossRef]
- Goodarzi, M.O.; Dumesic, D.A.; Chazenbalk, G.; Azziz, R. Polycystic ovary syndrome: Etiology, pathogenesis and diagnosis. Nat. Rev. Endocrinol. 2011, 7, 219–231. [Google Scholar] [CrossRef]
- Antoniou-Tsigkos, A.; Macut, D.; Mastorakos, G. Physiopathology, Diagnosis, and Treatment of Secondary Female Hypogonadism BT-Hypothalamic-Pituitary Diseases; Casanueva, F.F., Ghigo, E., Eds.; Springer International Publishing: Cham, Switzerland; Berlin/Heidelberg, Germany, 2018; pp. 247–287. ISBN 978-3-319-44444-4. [Google Scholar]
- Valsamakis, G.; Lois, K.; Kumar, S.; Mastorakos, G. Metabolic and other effects of pioglitazone as an add-on therapy to metformin in the treatment of polycystic ovary syndrome (PCOS). Hormones 2013, 12, 363–378. [Google Scholar] [CrossRef]
- Fioramonti, X.; Contie, S.; Song, Z.; Routh, V.H.; Lorsignol, A.; Pénicaud, L. Characterization of Glucosensing Neuron Subpopulations in the Arcuate Nucleus: Integration in Neuropeptide Y and Pro-Opio Melanocortin Networks? Diabetes 2007, 56, 1219–1227. [Google Scholar] [CrossRef]
- Zhang, X.; Zhang, G.; Zhang, H.; Karin, M.; Bai, H.; Cai, D. Hypothalamic IKKβ/NF-κB and ER Stress Link Overnutrition to Energy Imbalance and Obesity. Cell 2008, 135, 61–73. [Google Scholar] [CrossRef] [PubMed]
- Gao, Y.; Bielohuby, M.; Fleming, T.; Grabner, G.F.; Foppen, E.; Bernhard, W.; Guzmán-Ruiz, M.; Layritz, C.; Legutko, B.; Zinser, E.; et al. Dietary sugars, not lipids, drive hypothalamic inflammation. Mol. Metab. 2017, 6, 897–908. [Google Scholar] [CrossRef]
- Fuente-Martín, E.; García-Cáceres, C.; Díaz, F.; Argente-Arizón, P.; Granado, M.; Barrios, V.; Argente, J.; Chowen, J.A. Hypothalamic Inflammation Without Astrogliosis in Response to High Sucrose Intake Is Modulated by Neonatal Nutrition in Male Rats. Endocrinology 2013, 154, 2318–2330. [Google Scholar] [CrossRef] [PubMed]
- Cavaliere, G.; Viggiano, E.; Trinchese, G.; De Filippo, C.; Messina, A.; Monda, V.; Valenzano, A.; Cincione, R.I.; Zammit, C.; Cimmino, F.; et al. Long Feeding High-Fat Diet Induces Hypothalamic Oxidative Stress and Inflammation, and Prolonged Hypothalamic AMPK Activation in Rat Animal Model. Front. Physiol. 2018, 9. [Google Scholar] [CrossRef] [PubMed]
- Posey, K.A.; Clegg, D.J.; Printz, R.L.; Byun, J.; Morton, G.J.; Vivekanandan-Giri, A.; Pennathur, S.; Baskin, D.G.; Heinecke, J.W.; Woods, S.C.; et al. Hypothalamic proinflammatory lipid accumulation, inflammation, and insulin resistance in rats fed a high-fat diet. Am. J. Physiol. Metab. 2009, 296, E1003–E1012. [Google Scholar] [CrossRef]
- Cintra, D.E.; Ropelle, E.R.; Moraes, J.C.; Pauli, J.R.; Morari, J.; De Souza, C.T.; Grimaldi, R.; Stahl, M.; Carvalheira, J.B.; Saad, M.J.; et al. Unsaturated Fatty Acids Revert Diet-Induced Hypothalamic Inflammation in Obesity. PLoS ONE 2012, 7, e30571. [Google Scholar] [CrossRef]
- Morselli, E.; Fuente-Martín, E.; Finan, B.; Kim, M.; Frank, A.; García-Cáceres, C.; Navas, C.R.; Gordillo, R.; Neinast, M.; Kalainayakan, S.P.; et al. Hypothalamic PGC-1α protects against high-fat diet exposure by regulating ERα. Cell Rep. 2014, 9, 633–645. [Google Scholar] [CrossRef] [PubMed]
- Hotamisligil, G.S. Endoplasmic Reticulum Stress and the Inflammatory Basis of Metabolic Disease. Cell 2010, 140, 900–917. [Google Scholar] [CrossRef] [PubMed]
- Vijay-Kumar, M.; Aitken, J.D.; Carvalho, F.A.; Cullender, T.C.; Mwangi, S.; Srinivasan, S.; Sitaraman, S.V.; Knight, R.; Ley, R.E.; Gewirtz, A.T. Metabolic Syndrome and Altered Gut Microbiota in Mice Lacking Toll-Like Receptor 5. Science 2010, 328, 228–231. [Google Scholar] [CrossRef]
- Lu, W.; Xu, Y.; Shao, X.; Gao, F.; Li, Y.; Hu, J.; Zuo, Z.; Shao, X.; Zhou, L.; Zhao, Y.; et al. Uric Acid Produces an Inflammatory Response through Activation of NF-κB in the Hypothalamus: Implications for the Pathogenesis of Metabolic Disorders. Sci. Rep. 2015, 5, 12144. [Google Scholar] [CrossRef] [PubMed]
- Duque, E.D.A.; Munhoz, C.D. The Pro-inflammatory Effects of Glucocorticoids in the Brain. Front. Endocrinol. 2016, 7, 78. [Google Scholar] [CrossRef]
- Chrousos, G.P. Stress, chronic inflammation, and emotional and physical well-being: Concurrent effects and chronic sequelae. J. Allergy Clin. Immunol. 2000, 106, S275–S291. [Google Scholar] [CrossRef]
- McEwen, B.S. Physiology and Neurobiology of Stress and Adaptation: Central Role of the Brain. Physiol. Rev. 2007, 87, 873–904. [Google Scholar] [CrossRef]
- Merkulov, V.M.; Merkulova, T.I.; Bondar, N.P. Mechanisms of brain glucocorticoid resistance in stress-induced psychopathologies. Biochemistry 2017, 82, 351–365. [Google Scholar] [CrossRef] [PubMed]
- Mastorakos, G.; Ilias, I. Interleukin-6: A cytokine and/or a major modulator of the response to somatic stress. Ann. N. Y. Acad. Sci. 2006, 373–381. [Google Scholar] [CrossRef] [PubMed]
- Wohleb, E.S.; Fenn, A.M.; Pacenta, A.M.; Powell, N.D.; Sheridan, J.F.; Godbout, J.P. Peripheral innate immune challenge exaggerated microglia activation, increased the number of inflammatory CNS macrophages, and prolonged social withdrawal in socially defeated mice. Psychoneuroendocrinology 2012, 37, 1491–1505. [Google Scholar] [CrossRef]
- Frank, M.G.; Hershman, S.A.; Weber, M.D.; Watkins, L.R.; Maier, S.F. Chronic exposure to exogenous glucocorticoids primes microglia to pro-inflammatory stimuli and induces NLRP3 mRNA in the hippocampus. Psychoneuroendocrinology 2014, 40, 191–200. [Google Scholar] [CrossRef] [PubMed]
- Busillo, J.M.; Azzam, K.M.; Cidlowski, J.A. Glucocorticoids Sensitize the Innate Immune System through Regulation of the NLRP3 Inflammasome. J. Biol. Chem. 2011, 286, 38703–38713. [Google Scholar] [CrossRef]
- Kopp, B.L.; Wick, D.M.; Herman, J.P. Differential effects of homotypic vs. heterotypic chronic stress regimens on microglial activation in the prefrontal cortex. Physiol. Behav. 2013, 122, 246–252. [Google Scholar] [CrossRef] [PubMed]
- Mastorakos, G.; Chrousos, G.P.; Weber, J.S. Recombinant interleukin-6 activates the hypothalamic-pituitary-adrenal axis in humans. J. Clin. Endocrinol. Metab. 1993, 77, 1690–1694. [Google Scholar] [CrossRef] [PubMed]
- Goshen, I.; Yirmiya, R. Interleukin-1 (IL-1): A central regulator of stress responses. Front. Neuroendocr. 2009, 30, 30–45. [Google Scholar] [CrossRef]
- Yildiz, B.O.; Bozdag, G.; Yapici, Z.; Esinler, I.; Yarali, H. Prevalence, phenotype and cardiometabolic risk of polycystic ovary syndrome under different diagnostic criteria. Hum. Reprod. 2012, 27, 3067–3073. [Google Scholar] [CrossRef]
- Chaudhari, A.P.; Mazumdar, K.; Mehta, P.D. Anxiety, Depression, and Quality of Life in Women with Polycystic Ovarian Syndrome. Indian J. Psychol. Med. 2018, 40, 239–246. [Google Scholar] [CrossRef]
- Lee, I.; Cooney, L.G.; Saini, S.; Sammel, M.D.; Allison, K.C.; Dokras, A. Increased odds of disordered eating in polycystic ovary syndrome: A systematic review and meta-analysis. Eat. Weight. Disord. 2018, 24, 787–797. [Google Scholar] [CrossRef]
- Laven, J.S.E.; Imani, B.; Eijkemans, M.J.C.; Fauser, B.C.J.M. New Approach to Polycystic Ovary Syndrome and Other Forms of Anovulatory Infertility. Obstet. Gynecol. Surv. 2002, 57, 755–767. [Google Scholar] [CrossRef] [PubMed]
- Kyrkou, G.; Trakakis, E.; Attilakos, A.; Panagopoulos, P.; Chrelias, C.; Papadimitriou, A.; Vaggopoulos, V.; Alexiou, E.; Mastorakos, G.; Lykeridou, A.; et al. Metabolic syndrome in Greek women with polycystic ovary syndrome: Prevalence, characteristics and associations with body mass index. A prospective controlled study. Arch. Gynecol. Obstet. 2015, 293, 915–923. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.J.; Choi, Y.M. Dyslipidemia in women with polycystic ovary syndrome. Obstet. Gynecol. Sci. 2013, 56, 137–142. [Google Scholar] [CrossRef] [PubMed]
- Palomba, S.; Falbo, A.; Chiossi, G.; Muscogiuri, G.; Fornaciari, E.; Orio, F.; Tolino, A.; Colao, A.; La Sala, G.B.; Zullo, F. Lipid profile in nonobese pregnant women with polycystic ovary syndrome: A prospective controlled clinical study. Steroids 2014, 88, 36–43. [Google Scholar] [CrossRef]
- Blank, S.; McCartney, C.; Marshall, J. The origins and sequelae of abnormal neuroendocrine function in polycystic ovary syndrome. Hum. Reprod. Updat. 2006, 12, 351–361. [Google Scholar] [CrossRef] [PubMed]
- Yao, B.; Liu, H.-Y.; Gu, Y.-C.; Shi, S.-S.; Tao, X.-Q.; Li, X.-J.; Ge, Y.-F.; Cui, Y.-X.; Yang, G.-B. Gonadotropin-releasing hormone positively regulates steroidogenesis via extracellular signal-regulated kinase in rat Leydig cells. Asian J. Androl. 2011, 13, 438–445. [Google Scholar] [CrossRef]
- Han, S.-K.; Gottsch, M.L.; Lee, K.J.; Popa, S.M.; Smith, J.T.; Jakawich, S.K.; Clifton, D.K.; Steiner, R.A.; Herbison, A.E. Activation of Gonadotropin-Releasing Hormone Neurons by Kisspeptin as a Neuroendocrine Switch for the Onset of Puberty. J. Neurosci. 2005, 25, 11349–11356. [Google Scholar] [CrossRef]
- Marshall, J.C.; Dunaif, A. Should all women with PCOS be treated for insulin resistance? Fertil. Steril. 2012, 97, 18–22. [Google Scholar] [CrossRef]
- Tosi, F.; Negri, C.; Perrone, F.; Dorizzi, R.; Castello, R.; Bonora, E.; Moghetti, P. Hyperinsulinemia Amplifies GnRH Agonist Stimulated Ovarian Steroid Secretion in Women with Polycystic Ovary Syndrome. J. Clin. Endocrinol. Metab. 2012, 97, 1712–1719. [Google Scholar] [CrossRef]
- Baillargeon, J.P.; Nestler, J.E. Commentary: Polycystic ovary syndrome: A syndrome of ovarian hypersensitivity to insulin? J. Clin. Endocrinol. Metab. 2006, 91, 22–24. [Google Scholar] [CrossRef]
- Schneider, J.E. Energy balance and reproduction. Physiol. Behav. 2004, 81, 289–317. [Google Scholar] [CrossRef] [PubMed]
- Chou, S.H.; Mantzoros, C.S. 20 YEARS OF LEPTIN: Role of leptin in human reproductive disorders. J. Endocrinol. 2014, 223, T49–T62. [Google Scholar] [CrossRef] [PubMed]
- Gordon, C.M.; Ackerman, K.E.; Berga, S.L.; Kaplan, J.R.; Mastorakos, G.; Misra, M.; Murad, M.H.; Santoro, N.F.; Warren, M.P. Functional Hypothalamic Amenorrhea: An Endocrine Society Clinical Practice Guideline. J. Clin. Endocrinol. Metab. 2017, 102, 1413–1439. [Google Scholar] [CrossRef]
- Quennell, J.H.; Howell, C.S.; Roa, J.; Augustine, R.A.; Grattan, D.R.; Anderson, G.M. Leptin Deficiency and Diet-Induced Obesity Reduce Hypothalamic Kisspeptin Expression in Mice. Endocrinology 2011, 152, 1541–1550. [Google Scholar] [CrossRef]
- Brown, R.; Imran, S.; Ur, E.; Wilkinson, M. KiSS-1 mRNA in adipose tissue is regulated by sex hormones and food intake. Mol. Cell. Endocrinol. 2008, 281, 64–72. [Google Scholar] [CrossRef] [PubMed]
- Araujo, E.P.; Moraes, J.C.; Cintra, D.E.; Velloso, L.A. Mechanisms in endocrinology: Hypothalamic inflammation and nutrition. Eur. J. Endocrinol. 2016, 175, R97–R105. [Google Scholar] [CrossRef]
- Valdearcos, M.; Xu, A.W.; Koliwad, S.K. Hypothalamic Inflammation in the Control of Metabolic Function. Annu. Rev. Physiol. 2015, 77, 131–160. [Google Scholar] [CrossRef] [PubMed]
- Gropp, E.; Shanabrough, M.; Borok, E.; Xu, A.W.; Janoschek, R.; Buch, T.; Plum, L.; Balthasar, N.; Hampel, B.; Waisman, A.; et al. Agouti-related peptide–expressing neurons are mandatory for feeding. Nat. Neurosci. 2005, 8, 1289–1291. [Google Scholar] [CrossRef]
- Balthasar, N.; Dalgaard, L.T.; Lee, C.E.; Yu, J.; Funahashi, H.; Williams, T.; Ferreira, M.; Tang, V.; McGovern, R.A.; Kenny, C.D.; et al. Divergence of Melanocortin Pathways in the Control of Food Intake and Energy Expenditure. Cell 2005, 123, 493–505. [Google Scholar] [CrossRef] [PubMed]
- Mastorakos, P.; McGAVERN, D.B. The anatomy and immunology of vasculature in the central nervous system. Sci. Immunol. 2019, 4, eaav0492. [Google Scholar] [CrossRef]
- Papargyri, P.; Zapanti, E.; Salakos, N.; Papargyris, L.; Bargiota, A.; Mastorakos, G. Links between HPA axis and adipokines: Clinical implications in paradigms of stress-related disorders. Expert Rev. Endocrinol. Metab. 2018, 13, 317–332. [Google Scholar] [CrossRef] [PubMed]
- Kleinridders, A.; Könner, A.C.; Bruning, J.C. CNS-targets in control of energy and glucose homeostasis. Curr. Opin. Pharmacol. 2009, 9, 794–804. [Google Scholar] [CrossRef] [PubMed]
- Könner, A.C.; Klöckener, T.; Brüning, J.C. Control of energy homeostasis by insulin and leptin: Targeting the arcuate nucleus and beyond. Physiol. Behav. 2009, 97, 632–638. [Google Scholar] [CrossRef] [PubMed]
- Williams, K.W.; Margatho, L.O.; Lee, C.E.; Choi, M.; Lee, S.; Scott, M.M.; Elias, C.F.; Elmquist, J.K. Segregation of Acute Leptin and Insulin Effects in Distinct Populations of Arcuate Proopiomelanocortin Neurons. J. Neurosci. 2010, 30, 2472–2479. [Google Scholar] [CrossRef] [PubMed]
- Ollmann, M.M.; Wilson, B.D.; Yang, Y.-K.; Kerns, J.A.; Chen, Y.; Gantz, I.; Barsh, G.S. Antagonism of Central Melanocortin Receptors in Vitro and in Vivo by Agouti-Related Protein. Science 1997, 278, 135–138. [Google Scholar] [CrossRef] [PubMed]
- André, C.; Guzman-Quevedo, O.; Rey, C.; Rémus-Borel, J.; Clark, S.; Castellanos-Jankiewicz, A.; Ladeveze, E.; Leste-Lasserre, T.; Nadjar, A.; Abrous, D.N.; et al. Inhibiting Microglia Expansion Prevents Diet-Induced Hypothalamic and Peripheral Inflammation. Diabetes 2016, 66, 908–919. [Google Scholar] [CrossRef]
- Dalvi, P.S.; A Chalmers, J.; Luo, V.; Han, D.-Y.; Wellhauser, L.; Liu, Y.; Tran, D.Q.; Castel, J.; Luquet, S.; Wheeler, M.B.; et al. High fat induces acute and chronic inflammation in the hypothalamus: Effect of high-fat diet, palmitate and TNF-α on appetite-regulating NPY neurons. Int. J. Obes. 2017, 41, 149–158. [Google Scholar] [CrossRef]
- Tsaousidou, E.; Paeger, L.; Belgardt, B.F.; Pal, M.; Wunderlich, C.M.; Brönneke, H.; Collienne, U.; Hampel, B.; Wunderlich, F.T.; Schmidt-Supprian, M.; et al. Distinct Roles for JNK and IKK Activation in Agouti-Related Peptide Neurons in the Development of Obesity and Insulin Resistance. Cell Rep. 2014, 9, 1495–1506. [Google Scholar] [CrossRef]
- Ernst, M.B.; Wunderlich, C.M.; Hess, S.; Paehler, M.; Mesaros, A.; Koralov, S.B.; Kleinridders, A.; Husch, A.; Münzberg, H.; Hampel, B.; et al. Enhanced Stat3 Activation in POMC Neurons Provokes Negative Feedback Inhibition of Leptin and InsulinSignaling in Obesity. J. Neurosci. 2009, 29, 11582–11593. [Google Scholar] [CrossRef]
- Shadel, G.S.; Horvath, T.L. Mitochondrial ROS Signaling in Organismal Homeostasis. Cell 2015, 163, 560–569. [Google Scholar] [CrossRef]
- Yang, L.; Qi, Y.; Yang, Y. Astrocytes Control Food Intake by Inhibiting AGRP Neuron Activity via Adenosine A1 Receptors. Cell Rep. 2015, 11, 798–807. [Google Scholar] [CrossRef]
- Yan, J.; Zhang, H.; Yin, Y.; Li, J.; Tang, Y.; Purkayastha, S.; Li, L.; Cai, D. Obesity- and aging-induced excess of central transforming growth factor-β potentiates diabetic development via an RNA stress response. Nat. Med. 2014, 20, 1001–1008. [Google Scholar] [CrossRef] [PubMed]
- De Araujo, E.P.; De Souza, C.T.; Velloso, L. Atypical transforming growth factor–β signaling in the hypothalamus is linked to diabetes. Nat. Med. 2014, 20, 985–987. [Google Scholar] [CrossRef]
- Kaushik, S.; Rodriguez-Navarro, J.A.; Arias, E.; Kiffin, R.; Sahu, S.; Schwartz, G.J.; Cuervo, A.M.; Singh, R. Autophagy in Hypothalamic AgRP Neurons Regulates Food Intake and Energy Balance. Cell Metab. 2011, 14, 173–183. [Google Scholar] [CrossRef] [PubMed]
- Kaushik, S.; Arias, E.; Kwon, H.; Lopez, N.M.; Athonvarangkul, D.; Sahu, S.; Schwartz, G.J.; E Pessin, J.; Singh, R. Loss of autophagy in hypothalamic POMC neurons impairs lipolysis. EMBO Rep. 2012, 13, 258–265. [Google Scholar] [CrossRef] [PubMed]
- Schneeberger, M.; Altirriba, J.; García, A.; Esteban, Y.; Castaño, C.; García-Lavandeira, M.; Alvarez, C.V.; Gomis, R.; Claret, M. Deletion of miRNA processing enzyme Dicer in POMC-expressing cells leads to pituitary dysfunction, neurodegeneration and development of obesity. Mol. Metab. 2013, 2, 74–85. [Google Scholar] [CrossRef] [PubMed]
- Meng, Q.; Cai, D. Defective Hypothalamic Autophagy Directs the Central Pathogenesis of Obesity via the IκB Kinase β (IKKβ)/NF-κB Pathway. J. Biol. Chem. 2011, 286, 32324–32332. [Google Scholar] [CrossRef]
- Portovedo, M.; Ignacio-Souza, L.M.; Bombassaro, B.; Coope, A.; Reginato, A.; Razolli, D.S.; Torsoni, M.A.; Torsoni, A.S.; Leal, R.F.; Velloso, L.A.; et al. Saturated Fatty Acids Modulate Autophagy’s Proteins in the Hypothalamus. PLoS ONE 2015, 10, e0119850. [Google Scholar] [CrossRef]
- Mendes, N.F.; Kim, Y.-B.; Velloso, L.A.; De Araujo, E.P. Hypothalamic Microglial Activation in Obesity: A Mini-Review. Front. Neurosci. 2018, 12, 846. [Google Scholar] [CrossRef]
- Han, C.; Rice, M.W.; Cai, D. Neuroinflammatory and autonomic mechanisms in diabetes and hypertension. Am. J. Physiol. Metab. 2016, 311, E32–E41. [Google Scholar] [CrossRef]
- Ulrich-Lai, Y.M.; Herman, J.P. Neural regulation of endocrine and autonomic stress responses. Nat. Rev. Neurosci. 2009, 10, 397–409. [Google Scholar] [CrossRef] [PubMed]
- Scherer, T.; Lindtner, C.; Zielinski, E.; O’Hare, J.; Filatova, N.; Buettner, C. Short Term Voluntary Overfeeding Disrupts Brain Insulin Control of Adipose Tissue Lipolysis. J. Biol. Chem. 2012, 287, 33061–33069. [Google Scholar] [CrossRef] [PubMed]
- Van Santbrink, E.J.P.; Eijkemans, M.J.; Laven, J.S.E.; Fauser, B.C.J.M. Patient-tailored conventional ovulation induction algorithms in anovulatory infertility. Trends Endocrinol. Metab. 2005, 16, 381–389. [Google Scholar] [CrossRef]
- Rivest, S.; Lee, S.; Attardi, B.; Rivier, C. The chronic intracerebroventricular infusion of interleukin-1 beta alters the activity of the hypothalamic-pituitary-gonadal axis of cycling rats. I. Effect on LHRH and gonadotropin biosynthesis and secretion. Endocrinology 1993, 133, 2424–2430. [Google Scholar] [CrossRef] [PubMed]
- Hohos, N.M.; Skaznik-Wikiel, M.E. High-Fat Diet and Female Fertility. Endocrinology 2017, 158, 2407–2419. [Google Scholar] [CrossRef]
- Roberts, J.S.; Perets, R.A.; Sarfert, K.S.; Bowman, J.J.; Ozark, P.A.; Whitworth, G.B.; Blythe, S.N.; Toporikova, N. High-fat high-sugar diet induces polycystic ovary syndrome in a rodent model†. Biol. Reprod. 2017, 96, 551–562. [Google Scholar] [CrossRef]
- Diamanti-Kandarakis, E.; Katsikis, I.; Piperi, C.; Kandaraki, E.; Piouka, A.; Papavassiliou, A.G.; Panidis, D. Increased serum advanced glycation end-products is a distinct finding in lean women with polycystic ovary syndrome (PCOS). Clin. Endocrinol. 2008, 69, 634–641. [Google Scholar] [CrossRef]
- DiamantiKandarakis, E.; Piperi, C.; Patsouris, E.; Korkolopoulou, P.; Panidis, D.; Pawelczyk, L.; Papavassiliou, A.G.; Duleba, A.J. Immunohistochemical localization of advanced glycation end-products (AGEs) and their receptor (RAGE) in polycystic and normal ovaries. Histochem. Cell Biol. 2007, 127, 581–589. [Google Scholar] [CrossRef]
- Tatone, C.; Eichenlaub-Ritter, U.; Amicarelli, F. Dicarbonyl stress and glyoxalases in ovarian function. Biochem. Soc. Trans. 2014, 42, 433–438. [Google Scholar] [CrossRef]
- Valsamakis, G.; Chrousos, G.; Mastorakos, G. Stress, female reproduction and pregnancy. Psychoneuroendocrinology 2019, 100, 48–57. [Google Scholar] [CrossRef]
- Burfeind, K.G.; Michaelis, K.A.; Marks, D.L. The central role of hypothalamic inflammation in the acute illness response and cachexia. Semin. Cell Dev. Biol. 2016, 54, 42–52. [Google Scholar] [CrossRef]
- Tatomir, A.; Micu, C.; Crivii, C.B. THE IMPACT OF STRESS AND GLUCOCORTICOIDS ON MEMORY. Clujul Med. 2014, 87, 3–6. [Google Scholar] [CrossRef]
- Cernackova, A.; Durackova, Z.; Trebaticka, J.; Mravec, B. Neuroinflammation and depressive disorder: The role of the hypothalamus. J. Clin. Neurosci. 2020, 75, 5–10. [Google Scholar] [CrossRef]
- Tafet, G.E.; Nemeroff, C.B. The Links Between Stress and Depression: Psychoneuroendocrinological, Genetic, and Environmental Interactions. J. Neuropsychiatry Clin. Neurosci. 2016, 28, 77–88. [Google Scholar] [CrossRef] [PubMed]
- Lainez, N.M.; Jonak, C.R.; Nair, M.G.; Ethell, I.M.; Wilson, E.H.; Carson, M.J.; Coss, D. Diet-Induced Obesity Elicits Macrophage Infiltration and Reduction in Spine Density in the Hypothalami of Male but Not Female Mice. Front. Immunol. 2018, 9, 1992. [Google Scholar] [CrossRef]
- Imai, Y.; Ibata, I.; Ito, D.; Ohsawa, K.; Kohsaka, S. A Novel Geneiba1in the Major Histocompatibility Complex Class III Region Encoding an EF Hand Protein Expressed in a Monocytic Lineage. Biochem. Biophys. Res. Commun. 1996, 224, 855–862. [Google Scholar] [CrossRef] [PubMed]
- Moenter, S.M. GnRH Neurons on LSD: A Year of Rejecting Hypotheses That May Have Made Karl Popper Proud. Endocrinology 2017, 159, 199–205. [Google Scholar] [CrossRef] [PubMed]
- Ciofi, P.; Garret, M.; Lapirot, O.; Lafon, P.; Loyens, A.; Prévot, V.; Levine, J.E. Brain-Endocrine Interactions: A Microvascular Route in the Mediobasal Hypothalamus. Endocrinology 2009, 150, 5509–5519. [Google Scholar] [CrossRef] [PubMed]
- McKinley, M.J.; Denton, D.A.; Ryan, P.J.; Yao, S.T.; Stefanidis, A.; Oldfield, B.J. From sensory circumventricular organs to cerebral cortex: Neural pathways controlling thirst and hunger. J. Neuroendocr. 2019, 31, e12689. [Google Scholar] [CrossRef]
- Hall, J.E.; Taylor, A.E.; Hayes, F.J.; Crowley, W.F. Insights into hypothalamic-pituitary dysfunction in polycystic ovary syndrome. J. Endocrinol. Investig. 1998, 21, 602–611. [Google Scholar] [CrossRef] [PubMed]
- Wu, L.L.-Y.; Dunning, K.R.; Yang, X.; Russell, D.L.; Lane, M.; Norman, R.J.; Robker, R.L. High-Fat Diet Causes Lipotoxicity Responses in Cumulus–Oocyte Complexes and Decreased Fertilization Rates. Endocrinology 2010, 151, 5438–5445. [Google Scholar] [CrossRef] [PubMed]
- Dragano, N.R.V.; Solon, C.; Ramalho, A.F.; De Moura, R.F.; Razolli, D.S.; Christiansen, E.; Azevedo, C.; Ulven, T.; Velloso, L.A. Polyunsaturated fatty acid receptors, GPR40 and GPR120, are expressed in the hypothalamus and control energy homeostasis and inflammation. J. Neuroinflamm. 2017, 14, 1–16. [Google Scholar] [CrossRef]
- Sadagurski, M.; Cady, G.; Miller, R.A. Anti-aging drugs reduce hypothalamic inflammation in a sex-specific manner. Aging Cell 2017, 16, 652–660. [Google Scholar] [CrossRef]
- Samodien, E.; Johnson, R.; Pheiffer, C.; Mabasa, L.; Erasmus, M.; Louw, J.; Chellan, N. Diet-induced hypothalamic dysfunction and metabolic disease, and the therapeutic potential of polyphenols. Mol. Metab. 2019, 27, 1–10. [Google Scholar] [CrossRef]
- Yi, C.-X.; Al-Massadi, O.; Donelan, E.; Lehti, M.; Weber, J.; Ress, C.; Trivedi, C.; Müller, T.D.; Woods, S.C.; Hofmann, S.M. Exercise protects against high-fat diet-induced hypothalamic inflammation. Physiol. Behav. 2012, 106, 485–490. [Google Scholar] [CrossRef]
- Holscher, C. Central effects of GLP-1: New opportunities for treatments of neurodegenerative diseases. J. Endocrinol. 2013, 221, T31–T41. [Google Scholar] [CrossRef]
- Lamos, E.M.; Malek, R.; Davis, S.N. GLP-1 receptor agonists in the treatment of polycystic ovary syndrome. Expert Rev. Clin. Pharmacol. 2017, 10, 401–408. [Google Scholar] [CrossRef] [PubMed]


Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Barlampa, D.; Bompoula, M.S.; Bargiota, A.; Kalantaridou, S.; Mastorakos, G.; Valsamakis, G. Hypothalamic Inflammation as a Potential Pathophysiologic Basis for the Heterogeneity of Clinical, Hormonal, and Metabolic Presentation in PCOS. Nutrients 2021, 13, 520. https://doi.org/10.3390/nu13020520
Barlampa D, Bompoula MS, Bargiota A, Kalantaridou S, Mastorakos G, Valsamakis G. Hypothalamic Inflammation as a Potential Pathophysiologic Basis for the Heterogeneity of Clinical, Hormonal, and Metabolic Presentation in PCOS. Nutrients. 2021; 13(2):520. https://doi.org/10.3390/nu13020520
Chicago/Turabian StyleBarlampa, Danai, Maria Sotiria Bompoula, Alexandra Bargiota, Sophia Kalantaridou, George Mastorakos, and Georgios Valsamakis. 2021. "Hypothalamic Inflammation as a Potential Pathophysiologic Basis for the Heterogeneity of Clinical, Hormonal, and Metabolic Presentation in PCOS" Nutrients 13, no. 2: 520. https://doi.org/10.3390/nu13020520
APA StyleBarlampa, D., Bompoula, M. S., Bargiota, A., Kalantaridou, S., Mastorakos, G., & Valsamakis, G. (2021). Hypothalamic Inflammation as a Potential Pathophysiologic Basis for the Heterogeneity of Clinical, Hormonal, and Metabolic Presentation in PCOS. Nutrients, 13(2), 520. https://doi.org/10.3390/nu13020520