
The Role of Gut Bacterial Metabolites in Brain Development, Aging and Disease


Abstract
:1. Introduction
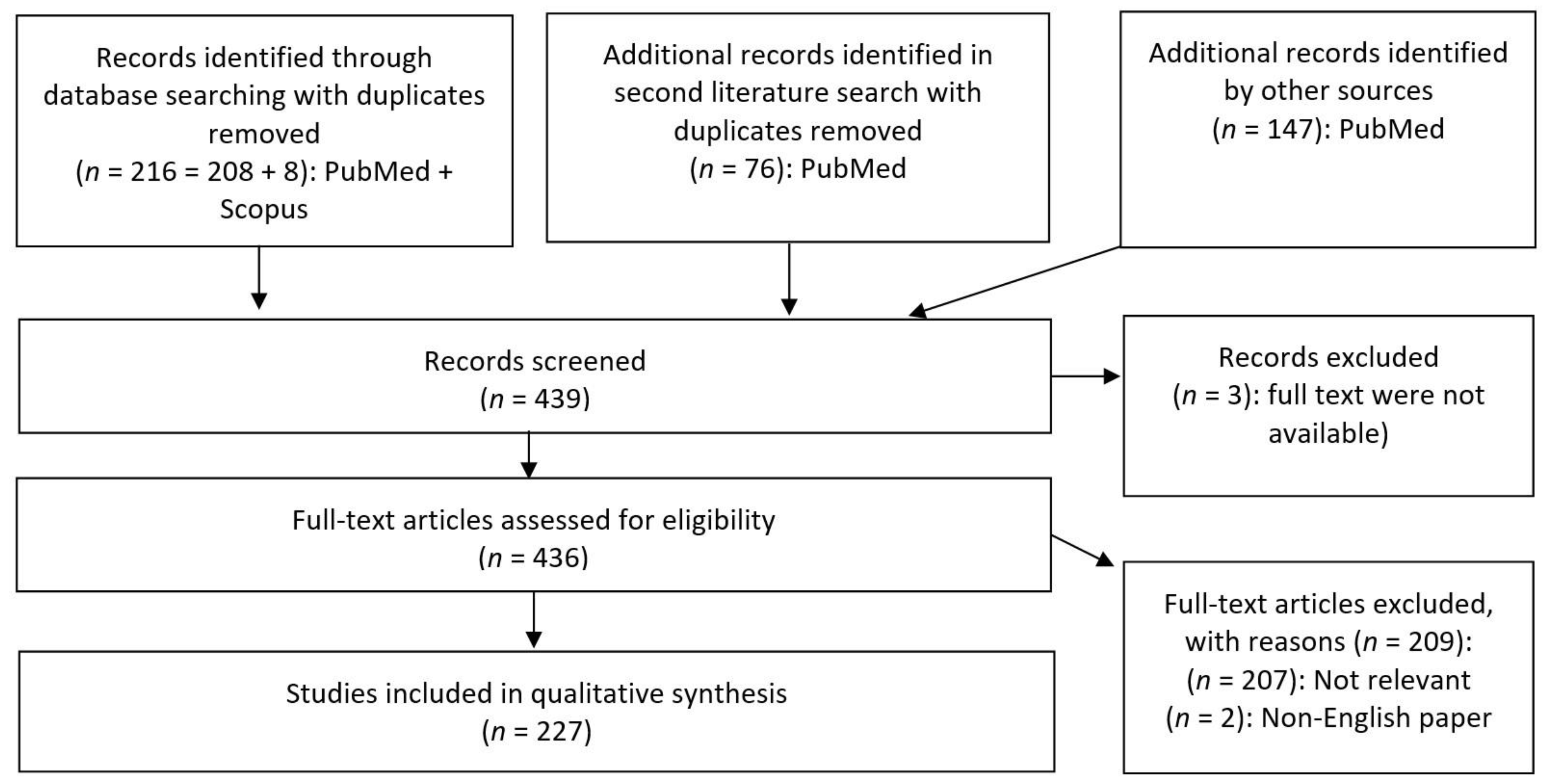
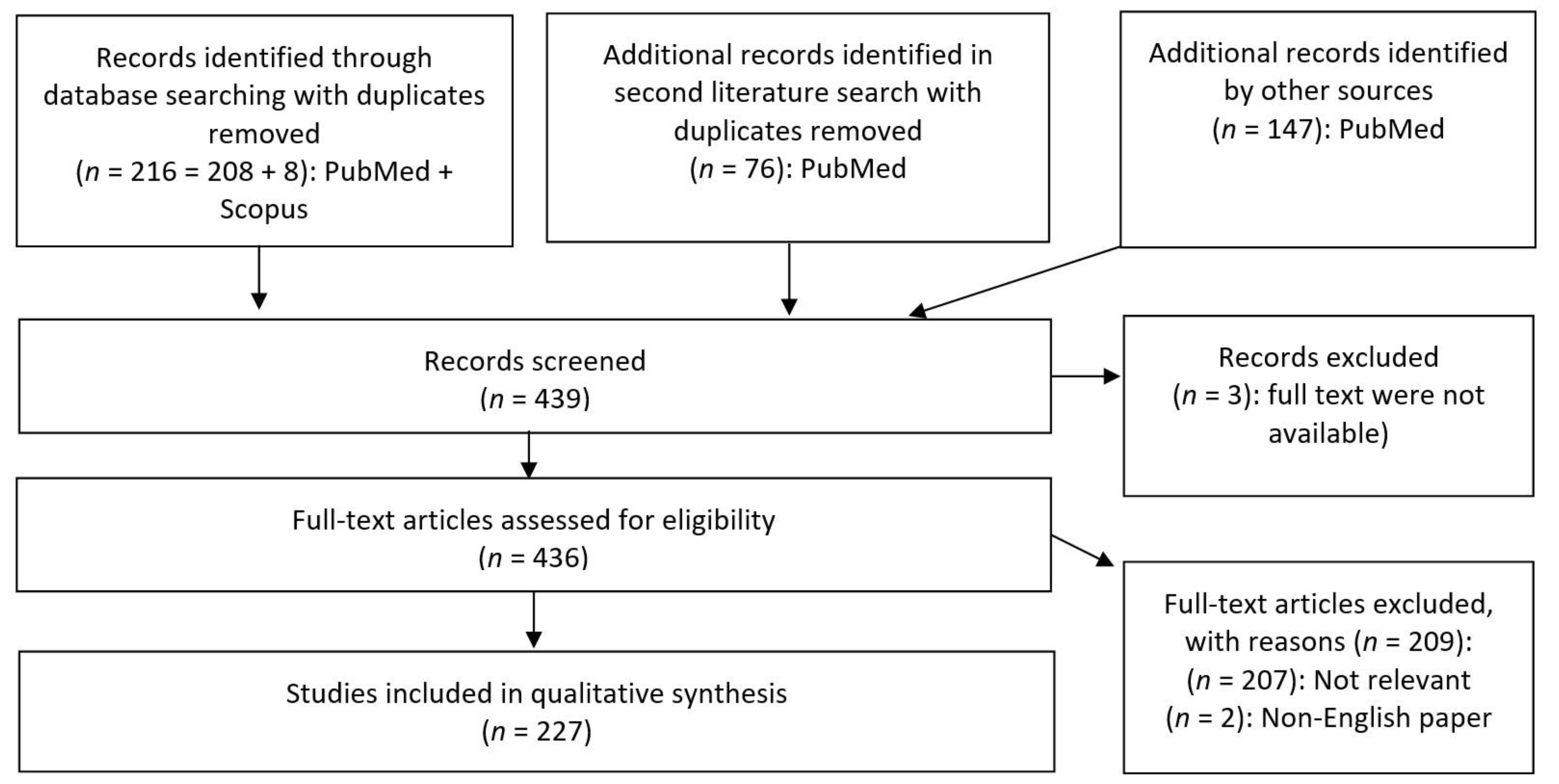
2. Materials and Methods
- Published in a peer-reviewed article;
- Paper available in full-text PDF;
- Paper available in English;
- Paper discussing metabolites from bacteria found in gastrointestinal tracts of animals.
3. Short-Chain Fatty Acids
- Histone deacetylase inhibition (HDACI) through BA, PA and AA, resulting in upregulated gene transcriptions in the context of epigenetic modulation [23,24]. As extensively reviewed by Stilling et al. [24] with a focus on BA, studies on this subject are mainly conducted in animal models and in supraphysiological concentrations, thus the validity of any conclusions drawn from the current evidence is promising, yet limited for human application as of now.
- Agonistic effects on G-protein-coupled receptors (GPCRs), namely free fatty acid receptors FFAR2 (GPR43), FFAR3 (GPR41) and the niacin receptor 1 (NIACR1, also known as hydroxycarboxylic acid receptor 2 (HCAR2) or GPR109A) [15,25]. Whether these effects are relevant in humans is to be determined, since current findings on these GPCRs are mostly based on rodent or cell models. FFAR3 for example, was found in the CNS and sympathetic ganglia of rats, and in the peripheral nervous system of mice [15]. Moreover, results linking these GPCRs with microglia cell morphology and growth hormone secretion in pituitary cells [25,26] call for further research with a focus on SCFAs as potential bacterial mediators of brain function.
- Modifications of cellular metabolism and activity in immune cells [27,28]. Similar to points 1 and 2, findings on these SCFA-mediated mechanisms are currently derived from animal and cell-based models. Nevertheless, studies have demonstrated striking results on BA promoting cell metabolism and differentiation in memory T cells [27,28], which underlines the importance of this mechanism.
3.1. SCFA and Autism Spectrum Disorder
3.2. SCFAs and Affective Disorders
3.3. SCFAs and Autoimmune Diseases of the Brain: Multiple Sclerosis (MS)
3.4. SCFAs and Neurodegenerative Diseases of the Brain
3.4.1. General Findings on Neurodegenerative Processes
3.4.2. SCFAs and Alzheimer’s Disease
3.4.3. SCFAs and Parkinson’s Disease
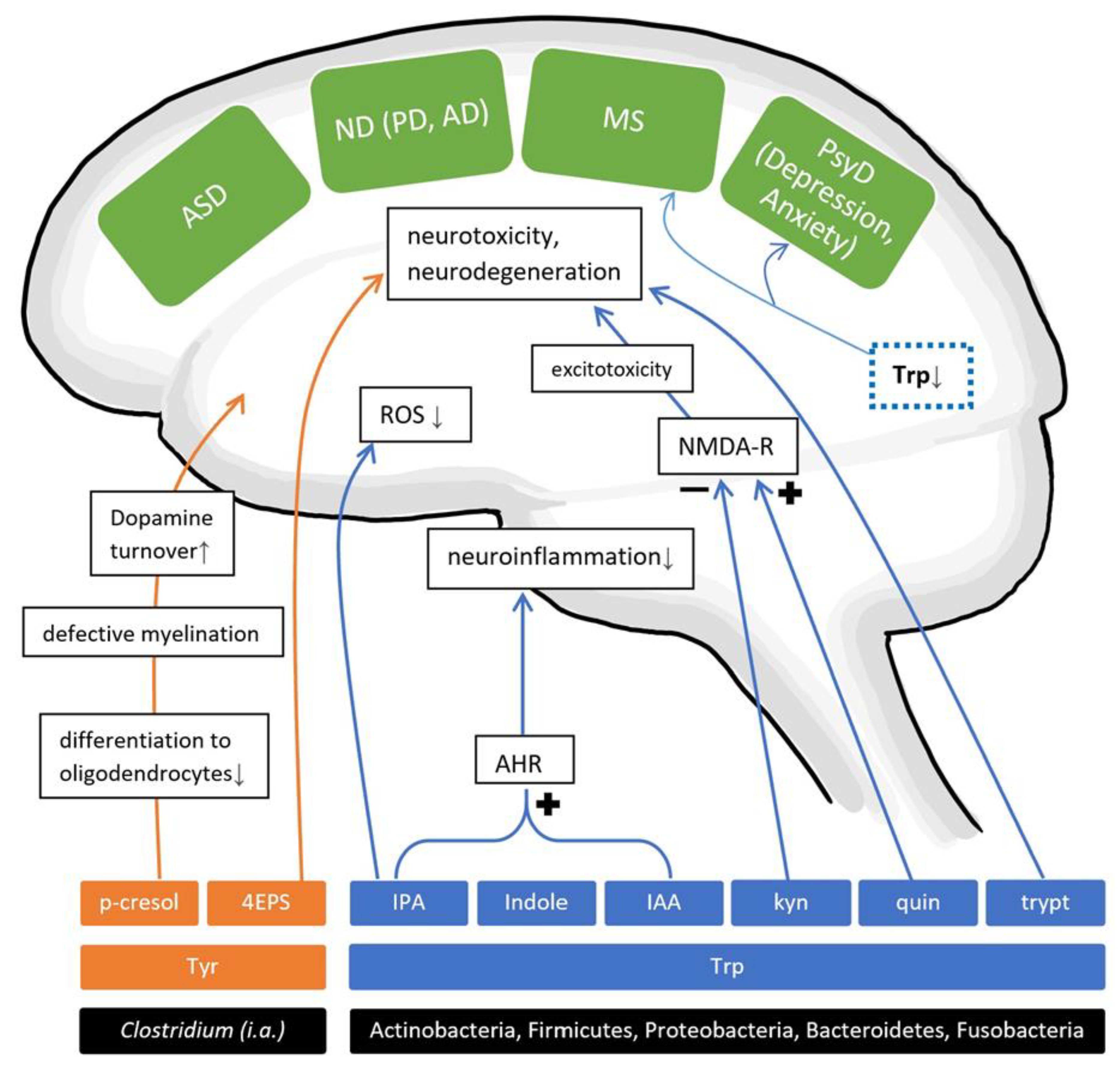
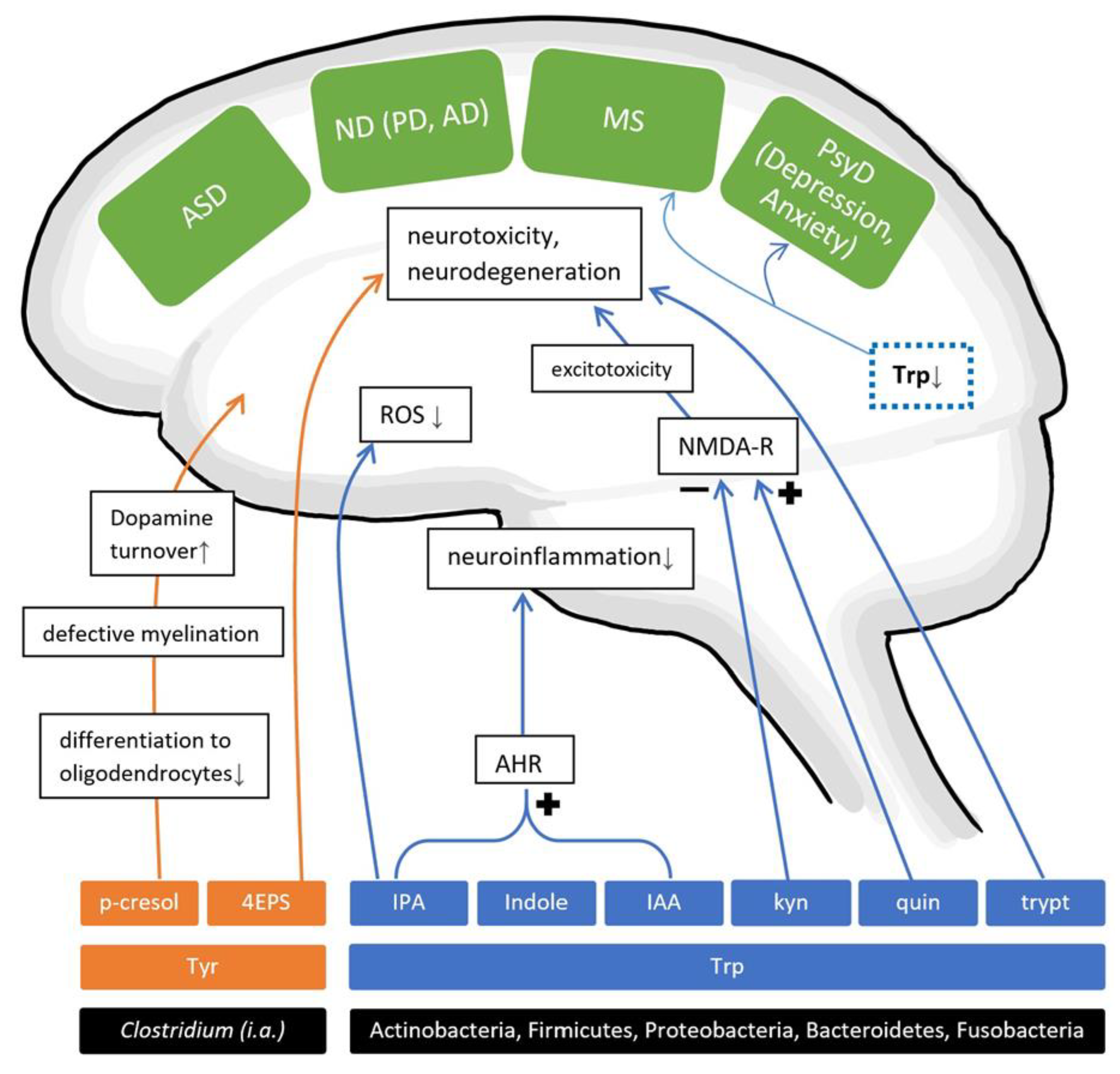
4. Non-SCFA Bacterial Metabolites
4.1. Amino Acid Metabolites
4.1.1. AAMs and Neurodevelopmental Disorders
4.1.2. AAMs and Psychiatric Disorders
4.1.3. AAMs and Neurodegenerative Diseases
Alzheimer’s Disease
Parkinson’s Disease
4.1.4. AAMs and Autoimmune Diseases of the Brain
4.2. Other Metabolites
4.2.1. Trimethylamine N Oxide (TMAO)
Carnitine Analogues
4.2.2. Polyphenolic Metabolites
Phenolic Compounds
4.2.3. Bacterial Amyloid Proteins
5. Discussion and Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| CNS | central nervous system |
| SCFA | short-chain fatty acids |
| AAM | amino acid metabolites |
| ASD | autism spectrum disorder |
| MS | multiple sclerosis |
| PD | Parkinson’s disease |
| AD | Alzheimer’s disease |
| GIT | gastrointestinal tract |
| GBA | gut–brain axis |
| BBB | blood–brain barrier |
| BA | butyric acid |
| PA | propionic acid |
| AA | acetic acid |
| VA | valeric acid |
| TMAO | trimethylamine N-oxide |
| IS | indoxyl sulphate |
| 4EPS | 4-ethylphenylsulfate |
| IAA | indole acetic acid |
| IPA | indole propionic acid |
| IBS | irritable bowel syndrome |
| GF | germ free |
| WT | wild type |
| HDACI | histone deacetylase inhibition/inhibitor |
| NT | neurotypical |
| FMT | faecal microbiome transplants |
| HC | healthy controls |
| DBH | dopamine beta-hydroxylase |
| MDD | major depressive disorder |
| HAB | high anxiety-like behaviour |
| NaB | sodium butyrate |
| αSyn | alpha-synuclein protein |
| AAA | aromatic amino acids |
| Tyr | tyrosine |
| Phe | phenylalanine |
| Trp | tryptophan |
| 5-HT | serotonin |
| ATD | acute tryptophan depletion |
| DA | dopamine |
| AHR | aryl hydrogen receptor |
References
- Martin, R.; Makino, H.; Cetinyurek Yavuz, A.; Ben-Amor, K.; Roelofs, M.; Ishikawa, E.; Kubota, H.; Swinkels, S.; Sakai, T.; Oishi, K.; et al. Early-Life Events, Including Mode of Delivery and Type of Feeding, Siblings and Gender, Shape the Developing Gut Microbiota. PLoS ONE 2016, 11, e0158498. [Google Scholar] [CrossRef] [Green Version]
- Jasarevic, E.; Rodgers, A.B.; Bale, T.L. A novel role for maternal stress and microbial transmission in early life programming and neurodevelopment. Neurobiol. Stress 2015, 1, 81–88. [Google Scholar] [CrossRef] [Green Version]
- Rybnikova, E. Brain, antibiotics, and microbiota—How do they interplay? An Editorial for ‘Antibiotics-induced modulation of large intestinal microbiota altered aromatic amino acid profile and expression of neurotransmitters in the hypothalamus of piglets’ on page 219. J. Neurochem. 2018, 146, 208–210. [Google Scholar] [CrossRef] [Green Version]
- Mehrian-Shai, R.; Reichardt, J.K.V.; Harris, C.C.; Toren, A. The Gut-Brain Axis, Paving the Way to Brain Cancer. Trends Cancer 2019, 5, 200–207. [Google Scholar] [CrossRef] [PubMed]
- Moher, D.; Liberati, A.; Tetzlaff, J.; Altman, D.G. Preferred reporting items for systematic reviews and meta-analyses: The PRISMA statement. PLoS Med. 2009, 6, e1000097. [Google Scholar] [CrossRef] [Green Version]
- Miller, T.L.; Wolin, M.J. Pathways of acetate, propionate, and butyrate formation by the human fecal microbial flora. Appl. Environ. Microbiol. 1996, 62, 1589–1592. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rooks, M.G.; Garrett, W.S. Gut microbiota, metabolites and host immunity. Nat. Rev. Immunol. 2016, 16, 341–352. [Google Scholar] [CrossRef]
- Westfall, S.; Lomis, N.; Kahouli, I.; Dia, S.Y.; Singh, S.P.; Prakash, S. Microbiome, probiotics and neurodegenerative diseases: Deciphering the gut brain axis. Cell. Mol. Life Sci. 2017, 74, 3769–3787. [Google Scholar] [CrossRef]
- Labus, J.S.; Hollister, E.B.; Jacobs, J.; Kirbach, K.; Oezguen, N.; Gupta, A.; Acosta, J.; Luna, R.A.; Aagaard, K.; Versalovic, J.; et al. Differences in gut microbial composition correlate with regional brain volumes in irritable bowel syndrome. Microbiome 2017, 5, 49. [Google Scholar] [CrossRef]
- Mayer, E.A.; Savidge, T.; Shulman, R.J. Brain-gut microbiome interactions and functional bowel disorders. Gastroenterology 2014, 146, 1500–1512. [Google Scholar] [CrossRef] [Green Version]
- Theodorou, V.; Ait Belgnaoui, A.; Agostini, S.; Eutamene, H. Effect of commensals and probiotics on visceral sensitivity and pain in irritable bowel syndrome. Gut Microbes 2014, 5, 430–436. [Google Scholar] [CrossRef] [PubMed]
- Lin, L.; Zhang, J. Role of intestinal microbiota and metabolites on gut homeostasis and human diseases. BMC Immunol. 2017, 18, 2. [Google Scholar] [CrossRef] [Green Version]
- Haghikia, A.; Jorg, S.; Duscha, A.; Berg, J.; Manzel, A.; Waschbisch, A.; Hammer, A.; Lee, D.H.; May, C.; Wilck, N.; et al. Dietary Fatty Acids Directly Impact Central Nervous System Autoimmunity via the Small Intestine. Immunity 2015, 43, 817–829. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hijova, E. Gut bacterial metabolites of indigestible polysaccharides in intestinal fermentation as mediators of public health. Bratisl. Lek. Listy 2019, 120, 807–812. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dalile, B.; Van Oudenhove, L.; Vervliet, B.; Verbeke, K. The role of short-chain fatty acids in microbiota-gut-brain communication. Nat. Rev. Gastroenterol. Hepatol. 2019, 16, 461–478. [Google Scholar] [CrossRef]
- Lewis, K.; Lutgendorff, F.; Phan, V.; Söderholm, J.D.; Sherman, P.M.; McKay, D.M. Enhanced translocation of bacteria across metabolically stressed epithelia is reduced by butyrate. Inflamm. Bowel Dis. 2010, 16, 1138–1148. [Google Scholar] [CrossRef]
- Peng, L.; Li, Z.R.; Green, R.S.; Holzman, I.R.; Lin, J. Butyrate enhances the intestinal barrier by facilitating tight junction assembly via activation of AMP-activated protein kinase in Caco-2 cell monolayers. J. Nutr. 2009, 139, 1619–1625. [Google Scholar] [CrossRef]
- Obrenovich, M.E.M. Leaky Gut, Leaky Brain? Microorganisms 2018, 6, 107. [Google Scholar] [CrossRef] [Green Version]
- Sampson, T.R.; Mazmanian, S.K. Control of brain development, function, and behavior by the microbiome. Cell Host Microbe 2015, 17, 565–576. [Google Scholar] [CrossRef] [Green Version]
- Braniste, V.; Al-Asmakh, M.; Kowal, C.; Anuar, F.; Abbaspour, A.; Tóth, M.; Korecka, A.; Bakocevic, N.; Ng, L.G.; Kundu, P.; et al. The gut microbiota influences blood-brain barrier permeability in mice. Sci. Transl. Med. 2014, 6, 263ra158. [Google Scholar] [CrossRef] [Green Version]
- Hoyles, L.; Snelling, T.; Umlai, U.K.; Nicholson, J.K.; Carding, S.R.; Glen, R.C.; McArthur, S. Microbiome-host systems interactions: Protective effects of propionate upon the blood-brain barrier. Microbiome 2018, 6, 55. [Google Scholar] [CrossRef] [Green Version]
- Parker, A.; Fonseca, S.; Carding, S.R. Gut microbes and metabolites as modulators of blood-brain barrier integrity and brain health. Gut Microbes 2020, 11, 135–157. [Google Scholar] [CrossRef] [Green Version]
- Nankova, B.B.; Agarwal, R.; MacFabe, D.F.; La Gamma, E.F. Enteric bacterial metabolites propionic and butyric acid modulate gene expression, including CREB-dependent catecholaminergic neurotransmission, in PC12 cells—Possible relevance to autism spectrum disorders. PLoS ONE 2014, 9, e103740. [Google Scholar] [CrossRef] [PubMed]
- Stilling, R.M.; van de Wouw, M.; Clarke, G.; Stanton, C.; Dinan, T.G.; Cryan, J.F. The neuropharmacology of butyrate: The bread and butter of the microbiota-gut-brain axis? Neurochem. Int. 2016, 99, 110–132. [Google Scholar] [CrossRef] [PubMed]
- Fernandes, M.F.; de Oliveira, S.; Portovedo, M.; Rodrigues, P.B.; Vinolo, M.A.R. Effect of Short Chain Fatty Acids on Age-Related Disorders. Adv. Exp. Med. Biol. 2020, 1260, 85–105. [Google Scholar] [CrossRef] [PubMed]
- Silva, Y.P.; Bernardi, A.; Frozza, R.L. The Role of Short-Chain Fatty Acids from Gut Microbiota in Gut-Brain Communication. Front. Endocrinol. 2020, 11, 25. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bachem, A.; Makhlouf, C.; Binger, K.J.; de Souza, D.P.; Tull, D.; Hochheiser, K.; Whitney, P.G.; Fernandez-Ruiz, D.; Dähling, S.; Kastenmüller, W.; et al. Microbiota-Derived Short-Chain Fatty Acids Promote the Memory Potential of Antigen-Activated CD8+ T Cells. Immunity 2019, 51, 285–297.e5. [Google Scholar] [CrossRef]
- Erny, D.; Hrabě de Angelis, A.L.; Jaitin, D.; Wieghofer, P.; Staszewski, O.; David, E.; Keren-Shaul, H.; Mahlakoiv, T.; Jakobshagen, K.; Buch, T.; et al. Host microbiota constantly control maturation and function of microglia in the CNS. Nat. Neurosci. 2015, 18, 965–977. [Google Scholar] [CrossRef] [PubMed]
- Park, J.; Wang, Q.; Wu, Q.; Mao-Draayer, Y.; Kim, C.H. Bidirectional regulatory potentials of short-chain fatty acids and their G-protein-coupled receptors in autoimmune neuroinflammation. Sci. Rep. 2019, 9, 8837. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hug, H.; Mohajeri, M.H.; La Fata, G. Toll-Like Receptors: Regulators of the Immune Response in the Human Gut. Nutrients 2018, 10, 203. [Google Scholar] [CrossRef] [Green Version]
- Oleskin, A.V.; Shenderov, B.A. Neuromodulatory effects and targets of the SCFAs and gasotransmitters produced by the human symbiotic microbiota. Microb. Ecol. Health Dis. 2016, 27, 30971. [Google Scholar] [CrossRef] [PubMed]
- Varela, R.B.; Valvassori, S.S.; Lopes-Borges, J.; Mariot, E.; Dal-Pont, G.C.; Amboni, R.T.; Bianchini, G.; Quevedo, J. Sodium butyrate and mood stabilizers block ouabain-induced hyperlocomotion and increase BDNF, NGF and GDNF levels in brain of Wistar rats. J. Psychiatr. Res. 2015, 61, 114–121. [Google Scholar] [CrossRef] [PubMed]
- Barichello, T.; Generoso, J.S.; Simões, L.R.; Faller, C.J.; Ceretta, R.A.; Petronilho, F.; Lopes-Borges, J.; Valvassori, S.S.; Quevedo, J. Sodium Butyrate Prevents Memory Impairment by Re-establishing BDNF and GDNF Expression in Experimental Pneumococcal Meningitis. Mol. Neurobiol. 2015, 52, 734–740. [Google Scholar] [CrossRef] [PubMed]
- Costa-Mattioli, M.; Sonenberg, N.; Richter, J.D. Translational regulatory mechanisms in synaptic plasticity and memory storage. Prog. Mol. Biol. Transl. Sci. 2009, 90, 293–311. [Google Scholar] [CrossRef] [PubMed]
- Buffington, S.A.; Huang, W.; Costa-Mattioli, M. Translational control in synaptic plasticity and cognitive dysfunction. Annu. Rev. Neurosci. 2014, 37, 17–38. [Google Scholar] [CrossRef] [Green Version]
- Srikantha, P.; Mohajeri, M.H. The Possible Role of the Microbiota-Gut-Brain-Axis in Autism Spectrum Disorder. Int. J. Mol. Sci. 2019, 20, 2115. [Google Scholar] [CrossRef] [Green Version]
- Nitschke, A.; Deonandan, R.; Konkle, A.T. The link between autism spectrum disorder and gut microbiota: A scoping review. Autism 2020, 24, 1328–1344. [Google Scholar] [CrossRef] [PubMed]
- Averina, O.V.; Kovtun, A.S.; Polyakova, S.I.; Savilova, A.M.; Rebrikov, D.V.; Danilenko, V.N. The bacterial neurometabolic signature of the gut microbiota of young children with autism spectrum disorders. J. Med. Microbiol. 2020, 69, 558–571. [Google Scholar] [CrossRef] [PubMed]
- De Angelis, M.; Piccolo, M.; Vannini, L.; Siragusa, S.; De Giacomo, A.; Serrazzanetti, D.I.; Cristofori, F.; Guerzoni, M.E.; Gobbetti, M.; Francavilla, R. Fecal microbiota and metabolome of children with autism and pervasive developmental disorder not otherwise specified. PLoS ONE 2013, 8, e76993. [Google Scholar] [CrossRef] [Green Version]
- Carlson, A.L.; Xia, K.; Azcarate-Peril, M.A.; Goldman, B.D.; Ahn, M.; Styner, M.A.; Thompson, A.L.; Geng, X.; Gilmore, J.H.; Knickmeyer, R.C. Infant Gut Microbiome Associated with Cognitive Development. Biol. Psychiatry 2018, 83, 148–159. [Google Scholar] [CrossRef]
- Kang, D.W.; Ilhan, Z.E.; Isern, N.G.; Hoyt, D.W.; Howsmon, D.P.; Shaffer, M.; Lozupone, C.A.; Hahn, J.; Adams, J.B.; Krajmalnik-Brown, R. Differences in fecal microbial metabolites and microbiota of children with autism spectrum disorders. Anaerobe 2018, 49, 121–131. [Google Scholar] [CrossRef]
- Wang, L.; Christophersen, C.T.; Sorich, M.J.; Gerber, J.P.; Angley, M.T.; Conlon, M.A. Elevated fecal short chain fatty acid and ammonia concentrations in children with autism spectrum disorder. Dig. Dis. Sci. 2012, 57, 2096–2102. [Google Scholar] [CrossRef] [PubMed]
- Adams, J.B.; Johansen, L.J.; Powell, L.D.; Quig, D.; Rubin, R.A. Gastrointestinal flora and gastrointestinal status in children with autism—Comparisons to typical children and correlation with autism severity. BMC Gastroenterol. 2011, 11, 22. [Google Scholar] [CrossRef] [Green Version]
- Liu, S.; Li, E.; Sun, Z.; Fu, D.; Duan, G.; Jiang, M.; Yu, Y.; Mei, L.; Yang, P.; Tang, Y.; et al. Altered gut microbiota and short chain fatty acids in Chinese children with autism spectrum disorder. Sci. Rep. 2019, 9, 287. [Google Scholar] [CrossRef]
- Sharon, G.; Cruz, N.J.; Kang, D.W.; Gandal, M.J.; Wang, B.; Kim, Y.M.; Zink, E.M.; Casey, C.P.; Taylor, B.C.; Lane, C.J.; et al. Human Gut Microbiota from Autism Spectrum Disorder Promote Behavioral Symptoms in Mice. Cell 2019, 177, 1600–1618.e17. [Google Scholar] [CrossRef] [Green Version]
- El-Ansary, A.; Shaker, G.H.; El-Gezeery, A.R.; Al-Ayadhi, L. The neurotoxic effect of clindamycin-induced gut bacterial imbalance and orally administered propionic acid on DNA damage assessed by the comet assay: Protective potency of carnosine and carnitine. Gut Pathog. 2013, 5, 9. [Google Scholar] [CrossRef] [Green Version]
- Al-Mosalem, O.A.; El-Ansary, A.; Attas, O.; Al-Ayadhi, L. Metabolic biomarkers related to energy metabolism in Saudi autistic children. Clin. Biochem. 2009, 42, 949–957. [Google Scholar] [CrossRef]
- MacFabe, D.F.; Cain, D.P.; Rodriguez-Capote, K.; Franklin, A.E.; Hoffman, J.E.; Boon, F.; Taylor, A.R.; Kavaliers, M.; Ossenkopp, K.P. Neurobiological effects of intraventricular propionic acid in rats: Possible role of short chain fatty acids on the pathogenesis and characteristics of autism spectrum disorders. Behav. Brain Res. 2007, 176, 149–169. [Google Scholar] [CrossRef]
- Alfawaz, H.; Al-Onazi, M.; Bukhari, S.I.; Binobead, M.; Othman, N.; Algahtani, N.; Bhat, R.S.; Moubayed, N.M.S.; Alzeer, H.S.; El-Ansary, A. The Independent and Combined Effects of Omega-3 and Vitamin B12 in Ameliorating Propionic Acid Induced Biochemical Features in Juvenile Rats as Rodent Model of Autism. J. Mol. Neurosci. 2018, 66, 403–413. [Google Scholar] [CrossRef] [PubMed]
- Choi, J.; Lee, S.; Won, J.; Jin, Y.; Hong, Y.; Hur, T.Y.; Kim, J.H.; Lee, S.R. Pathophysiological and neurobehavioral characteristics of a propionic acid-mediated autism-like rat model. PLoS ONE 2018, 13, e0192925. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kratsman, N.; Getselter, D.; Elliott, E. Sodium butyrate attenuates social behavior deficits and modifies the transcription of inhibitory/excitatory genes in the frontal cortex of an autism model. Neuropharmacology 2016, 102, 136–145. [Google Scholar] [CrossRef]
- Harrington, R.A.; Lee, L.C.; Crum, R.M.; Zimmerman, A.W.; Hertz-Picciotto, I. Serotonin hypothesis of autism: Implications for selective serotonin reuptake inhibitor use during pregnancy. Autism Res. 2013, 6, 149–168. [Google Scholar] [CrossRef]
- Ching, M.S.; Shen, Y.; Tan, W.H.; Jeste, S.S.; Morrow, E.M.; Chen, X.; Mukaddes, N.M.; Yoo, S.Y.; Hanson, E.; Hundley, R.; et al. Deletions of NRXN1 (neurexin-1) predispose to a wide spectrum of developmental disorders. Am. J. Med. Genet. B Neuropsychiatr. Genet. 2010, 153, 937–947. [Google Scholar] [CrossRef] [PubMed]
- Kim, H.G.; Kishikawa, S.; Higgins, A.W.; Seong, I.S.; Donovan, D.J.; Shen, Y.; Lally, E.; Weiss, L.A.; Najm, J.; Kutsche, K.; et al. Disruption of neurexin 1 associated with autism spectrum disorder. Am. J. Hum. Genet. 2008, 82, 199–207. [Google Scholar] [CrossRef] [Green Version]
- Szatmari, P.; Paterson, A.D.; Zwaigenbaum, L.; Roberts, W.; Brian, J.; Liu, X.Q.; Vincent, J.B.; Skaug, J.L.; Thompson, A.P.; Senman, L.; et al. Mapping autism risk loci using genetic linkage and chromosomal rearrangements. Nat. Genet. 2007, 39, 319–328. [Google Scholar] [CrossRef] [Green Version]
- Boccuto, L.; Lauri, M.; Sarasua, S.M.; Skinner, C.D.; Buccella, D.; Dwivedi, A.; Orteschi, D.; Collins, J.S.; Zollino, M.; Visconti, P.; et al. Prevalence of SHANK3 variants in patients with different subtypes of autism spectrum disorders. Eur. J. Hum. Genet. 2013, 21, 310–316. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mohajeri, M.H.; La Fata, G.; Steinert, R.E.; Weber, P. Relationship between the gut microbiome and brain function. Nutr. Rev. 2018, 76, 481–496. [Google Scholar] [CrossRef] [PubMed]
- Liu, F.; Horton-Sparks, K.; Hull, V.; Li, R.W.; Martínez-Cerdeño, V. The valproic acid rat model of autism presents with gut bacterial dysbiosis similar to that in human autism. Mol. Autism 2018, 9, 61. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Christensen, J.; Grønborg, T.K.; Sørensen, M.J.; Schendel, D.; Parner, E.T.; Pedersen, L.H.; Vestergaard, M. Prenatal valproate exposure and risk of autism spectrum disorders and childhood autism. Jama 2013, 309, 1696–1703. [Google Scholar] [CrossRef] [Green Version]
- Ingram, J.L.; Peckham, S.M.; Tisdale, B.; Rodier, P.M. Prenatal exposure of rats to valproic acid reproduces the cerebellar anomalies associated with autism. Neurotoxicol. Teratol. 2000, 22, 319–324. [Google Scholar] [CrossRef]
- Cohen, O.S.; Varlinskaya, E.I.; Wilson, C.A.; Glatt, S.J.; Mooney, S.M. Acute prenatal exposure to a moderate dose of valproic acid increases social behavior and alters gene expression in rats. Int. J. Dev. Neurosci. 2013, 31, 740–750. [Google Scholar] [CrossRef] [Green Version]
- D’Souza, A.; Onem, E.; Patel, P.; La Gamma, E.F.; Nankova, B.B. Valproic acid regulates catecholaminergic pathways by concentration-dependent threshold effects on TH mRNA synthesis and degradation. Brain Res. 2009, 1247, 1–10. [Google Scholar] [CrossRef]
- Fukuchi, M.; Nii, T.; Ishimaru, N.; Minamino, A.; Hara, D.; Takasaki, I.; Tabuchi, A.; Tsuda, M. Valproic acid induces up- or down-regulation of gene expression responsible for the neuronal excitation and inhibition in rat cortical neurons through its epigenetic actions. Neurosci. Res. 2009, 65, 35–43. [Google Scholar] [CrossRef] [PubMed]
- Stilling, R.M.; Ryan, F.J.; Hoban, A.E.; Shanahan, F.; Clarke, G.; Claesson, M.J.; Dinan, T.G.; Cryan, J.F. Microbes & neurodevelopment—Absence of microbiota during early life increases activity-related transcriptional pathways in the amygdala. Brain Behav. Immun. 2015, 50, 209–220. [Google Scholar] [CrossRef] [PubMed]
- AbuHasan, Q.; Reddy, V.; Siddiqui, W. Neuroanatomy, Amygdala. In StatPearls; StatPearls Publishing LLC.: Treasure Island, FL, USA, 2020. [Google Scholar]
- Lu, J.; Lu, L.; Yu, Y.; Cluette-Brown, J.; Martin, C.R.; Claud, E.C. Effects of Intestinal Microbiota on Brain Development in Humanized Gnotobiotic Mice. Sci. Rep. 2018, 8, 5443. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Skonieczna-Żydecka, K.; Grochans, E.; Maciejewska, D.; Szkup, M.; Schneider-Matyka, D.; Jurczak, A.; Łoniewski, I.; Kaczmarczyk, M.; Marlicz, W.; Czerwińska-Rogowska, M.; et al. Faecal Short Chain Fatty Acids Profile is Changed in Polish Depressive Women. Nutrients 2018, 10, 1939. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Deng, F.L.; Pan, J.X.; Zheng, P.; Xia, J.J.; Yin, B.M.; Liang, W.W.; Li, Y.F.; Wu, J.; Xu, F.; Wu, Q.Y.; et al. Metabonomics reveals peripheral and central short-chain fatty acid and amino acid dysfunction in a naturally occurring depressive model of macaques. Neuropsychiatr. Dis. Treat. 2019, 15, 1077–1088. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kelly, J.R.; Borre, Y.; O’Brien, C.; Patterson, E.; El Aidy, S.; Deane, J.; Kennedy, P.J.; Beers, S.; Scott, K.; Moloney, G.; et al. Transferring the blues: Depression-associated gut microbiota induces neurobehavioural changes in the rat. J. Psychiatr. Res. 2016, 82, 109–118. [Google Scholar] [CrossRef] [PubMed]
- Caspani, G.; Kennedy, S.; Foster, J.A.; Swann, J. Gut microbial metabolites in depression: Understanding the biochemical mechanisms. Microb. Cell 2019, 6, 454–481. [Google Scholar] [CrossRef] [PubMed]
- Fond, G.; Loundou, A.; Hamdani, N.; Boukouaci, W.; Dargel, A.; Oliveira, J.; Roger, M.; Tamouza, R.; Leboyer, M.; Boyer, L. Anxiety and depression comorbidities in irritable bowel syndrome (IBS): A systematic review and meta-analysis. Eur. Arch. Psychiatry Clin. Neurosci. 2014, 264, 651–660. [Google Scholar] [CrossRef]
- Schmidtner, A.K.; Slattery, D.A.; Gläsner, J.; Hiergeist, A.; Gryksa, K.; Malik, V.A.; Hellmann-Regen, J.; Heuser, I.; Baghai, T.C.; Gessner, A.; et al. Minocycline alters behavior, microglia and the gut microbiome in a trait-anxiety-dependent manner. Transl. Psychiatry 2019, 9, 223. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pryde, S.E.; Duncan, S.H.; Hold, G.L.; Stewart, C.S.; Flint, H.J. The microbiology of butyrate formation in the human colon. FEMS Microbiol. Lett. 2002, 217, 133–139. [Google Scholar] [CrossRef]
- Haase, S.; Haghikia, A.; Wilck, N.; Muller, D.N.; Linker, R.A. Impacts of microbiome metabolites on immune regulation and autoimmunity. Immunology 2018, 154, 230–238. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, C.H.; Park, J.; Kim, M. Gut microbiota-derived short-chain Fatty acids, T cells, and inflammation. Immune Netw. 2014, 14, 277–288. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Miller, A.H.; Raison, C.L. The role of inflammation in depression: From evolutionary imperative to modern treatment target. Nat. Rev. Immunol. 2016, 16, 22–34. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Van de Wouw, M.; Boehme, M.; Lyte, J.M.; Wiley, N.; Strain, C.; O’Sullivan, O.; Clarke, G.; Stanton, C.; Dinan, T.G.; Cryan, J.F. Short-chain fatty acids: Microbial metabolites that alleviate stress-induced brain-gut axis alterations. J. Physiol. 2018, 596, 4923–4944. [Google Scholar] [CrossRef]
- Jamshidian, A.; Shaygannejad, V.; Pourazar, A.; Zarkesh-Esfahani, S.H.; Gharagozloo, M. Biased Treg/Th17 balance away from regulatory toward inflammatory phenotype in relapsed multiple sclerosis and its correlation with severity of symptoms. J. Neuroimmunol. 2013, 262, 106–112. [Google Scholar] [CrossRef]
- Jangi, S.; Gandhi, R.; Cox, L.M.; Li, N.; von Glehn, F.; Yan, R.; Patel, B.; Mazzola, M.A.; Liu, S.; Glanz, B.L.; et al. Alterations of the human gut microbiome in multiple sclerosis. Nat. Commun. 2016, 7, 12015. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.; Chia, N.; Kalari, K.R.; Yao, J.Z.; Novotna, M.; Paz Soldan, M.M.; Luckey, D.H.; Marietta, E.V.; Jeraldo, P.R.; Chen, X.; et al. Multiple sclerosis patients have a distinct gut microbiota compared to healthy controls. Sci. Rep. 2016, 6, 28484. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zeng, Q.; Junli, G.; Liu, X.; Chen, C.; Sun, X.; Li, H.; Zhou, Y.; Cui, C.; Wang, Y.; Yang, Y.; et al. Gut dysbiosis and lack of short chain fatty acids in a Chinese cohort of patients with multiple sclerosis. Neurochem. Int. 2019, 129, 104468. [Google Scholar] [CrossRef]
- Xiao, S.; Jiang, S.; Qian, D.; Duan, J. Modulation of microbially derived short-chain fatty acids on intestinal homeostasis, metabolism, and neuropsychiatric disorder. Appl. Microbiol. Biotechnol. 2020, 104, 589–601. [Google Scholar] [CrossRef] [PubMed]
- Melbye, P.; Olsson, A.; Hansen, T.H.; Sondergaard, H.B.; Bang Oturai, A. Short-chain fatty acids and gut microbiota in multiple sclerosis. Acta Neurol. Scand. 2019, 139, 208–219. [Google Scholar] [CrossRef] [PubMed]
- Chen, T.; Noto, D.; Hoshino, Y.; Mizuno, M.; Miyake, S. Butyrate suppresses demyelination and enhances remyelination. J. Neuroinflamm. 2019, 16, 165. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dopkins, N.; Nagarkatti, P.S.; Nagarkatti, M. The role of gut microbiome and associated metabolome in the regulation of neuroinflammation in multiple sclerosis and its implications in attenuating chronic inflammation in other inflammatory and autoimmune disorders. Immunology 2018, 154, 178–185. [Google Scholar] [CrossRef] [PubMed]
- Erkkinen, M.G.; Kim, M.O.; Geschwind, M.D. Clinical Neurology and Epidemiology of the Major Neurodegenerative Diseases. Cold Spring Harb. Perspect. Biol. 2018, 10. [Google Scholar] [CrossRef] [Green Version]
- Ahmed, S.; Busetti, A.; Fotiadou, P.; Vincy Jose, N.; Reid, S.; Georgieva, M.; Brown, S.; Dunbar, H.; Beurket-Ascencio, G.; Delday, M.I.; et al. In vitro Characterization of Gut Microbiota-Derived Bacterial Strains with Neuroprotective Properties. Front. Cell. Neurosci. 2019, 13, 402. [Google Scholar] [CrossRef] [Green Version]
- Li, J.M.; Yu, R.; Zhang, L.P.; Wen, S.Y.; Wang, S.J.; Zhang, X.Y.; Xu, Q.; Kong, L.D. Dietary fructose-induced gut dysbiosis promotes mouse hippocampal neuroinflammation: A benefit of short-chain fatty acids. Microbiome 2019, 7. [Google Scholar] [CrossRef]
- Vogt, N.M.; Kerby, R.L.; Dill-McFarland, K.A.; Harding, S.J.; Merluzzi, A.P.; Johnson, S.C.; Carlsson, C.M.; Asthana, S.; Zetterberg, H.; Blennow, K.; et al. Gut microbiome alterations in Alzheimer’s disease. Sci. Rep. 2017, 7, 13537. [Google Scholar] [CrossRef] [PubMed]
- Zhuang, Z.Q.; Shen, L.L.; Li, W.W.; Fu, X.; Zeng, F.; Gui, L.; Lü, Y.; Cai, M.; Zhu, C.; Tan, Y.L.; et al. Gut Microbiota is Altered in Patients with Alzheimer’s Disease. J. Alzheimers Dis. 2018, 63, 1337–1346. [Google Scholar] [CrossRef] [Green Version]
- Bostanciklioglu, M. The role of gut microbiota in pathogenesis of Alzheimer’s disease. J. Appl. Microbiol. 2019, 127, 954–967. [Google Scholar] [CrossRef]
- Paley, E.L.; Merkulova-Rainon, T.; Faynboym, A.; Shestopalov, V.I.; Aksenoff, I. Geographical Distribution and Diversity of Gut Microbial NADH:Ubiquinone Oxidoreductase Sequence Associated with Alzheimer’s Disease. J. Alzheimers Dis. 2018, 61, 1531–1540. [Google Scholar] [CrossRef]
- Saji, N.; Niida, S.; Murotani, K.; Hisada, T.; Tsuduki, T.; Sugimoto, T.; Kimura, A.; Toba, K.; Sakurai, T. Analysis of the relationship between the gut microbiome and dementia: A cross-sectional study conducted in Japan. Sci. Rep. 2019, 9, 1008. [Google Scholar] [CrossRef]
- Nie, P.; Li, Z.; Wang, Y.; Zhang, Y.; Zhao, M.; Luo, J.; Du, S.; Deng, Z.; Chen, J.; Chen, S.; et al. Gut microbiome interventions in human health and diseases. Med. Res. Rev. 2019, 39, 2286–2313. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.; Wang, Y.; Xiayu, X.; Shi, C.; Chen, W.; Song, N.; Fu, X.; Zhou, R.; Xu, Y.F.; Huang, L.; et al. Altered Gut Microbiota in a Mouse Model of Alzheimer’s Disease. J. Alzheimers Dis. 2017, 60, 1241–1257. [Google Scholar] [CrossRef]
- Zheng, J.; Zheng, S.J.; Cai, W.J.; Yu, L.; Yuan, B.F.; Feng, Y.Q. Stable isotope labeling combined with liquid chromatography-tandem mass spectrometry for comprehensive analysis of short-chain fatty acids. Anal. Chim. Acta 2019, 1070, 51–59. [Google Scholar] [CrossRef]
- Fujii, Y.; Nguyen, T.T.T.; Fujimura, Y.; Kameya, N.; Nakamura, S.; Arakawa, K.; Morita, H. Fecal metabolite of a gnotobiotic mouse transplanted with gut microbiota from a patient with Alzheimer’s disease. Biosci. Biotechnol. Biochem. 2019, 83, 2144–2152. [Google Scholar] [CrossRef] [PubMed]
- Mohajeri, M.H.; Leuba, G. Prevention of age-associated dementia. Brain Res. Bull. 2009, 80, 315–325. [Google Scholar] [CrossRef]
- Ho, L.; Ono, K.; Tsuji, M.; Mazzola, P.; Singh, R.; Pasinetti, G.M. Protective roles of intestinal microbiota derived short chain fatty acids in Alzheimer’s disease-type beta-amyloid neuropathological mechanisms. Expert Rev. Neurother. 2018, 18, 83–90. [Google Scholar] [CrossRef] [Green Version]
- Sun, J.; Xu, J.; Ling, Y.; Wang, F.; Gong, T.; Yang, C.; Ye, S.; Ye, K.; Wei, D.; Song, Z.; et al. Fecal microbiota transplantation alleviated Alzheimer’s disease-like pathogenesis in APP/PS1 transgenic mice. Transl. Psychiatry 2019, 9, 189. [Google Scholar] [CrossRef] [Green Version]
- Peleg, S.; Sananbenesi, F.; Zovoilis, A.; Burkhardt, S.; Bahari-Javan, S.; Agis-Balboa, R.C.; Cota, P.; Wittnam, J.L.; Gogol-Doering, A.; Opitz, L.; et al. Altered histone acetylation is associated with age-dependent memory impairment in mice. Science 2010, 328, 753–756. [Google Scholar] [CrossRef] [Green Version]
- Govindarajan, N.; Agis-Balboa, R.C.; Walter, J.; Sananbenesi, F.; Fischer, A. Sodium butyrate improves memory function in an Alzheimer’s disease mouse model when administered at an advanced stage of disease progression. J. Alzheimers Dis. 2011, 26, 187–197. [Google Scholar] [CrossRef]
- Bourassa, M.W.; Alim, I.; Bultman, S.J.; Ratan, R.R. Butyrate, neuroepigenetics and the gut microbiome: Can a high fiber diet improve brain health? Neurosci. Lett. 2016, 625, 56–63. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nguyen, T.D.; Prykhodko, O.; Fak Hallenius, F.; Nyman, M. Monovalerin and trivalerin increase brain acetic acid, decrease liver succinic acid, and alter gut microbiota in rats fed high-fat diets. Eur. J. Nutr. 2019, 58, 1545–1560. [Google Scholar] [CrossRef]
- Soliman, M.L.; Combs, C.K.; Rosenberger, T.A. Modulation of inflammatory cytokines and mitogen-activated protein kinases by acetate in primary astrocytes. J. Neuroimmune Pharm. 2013, 8, 287–300. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Marino, B.L.B.; de Souza, L.R.; Sousa, K.P.A.; Ferreira, J.V.; Padilha, E.C.; da Silva, C.H.T.P.; Taft, C.A.; Hage-Melim, L.I.S. Parkinson’s Disease: A Review from Pathophysiology to Treatment. Mini-Rev. Med. Chem. 2020, 20, 754–767. [Google Scholar] [CrossRef]
- Brettschneider, J.; Del Tredici, K.; Lee, V.M.; Trojanowski, J.Q. Spreading of pathology in neurodegenerative diseases: A focus on human studies. Nat. Rev. Neurosci. 2015, 16, 109–120. [Google Scholar] [CrossRef] [PubMed]
- Braak, H.; Del Tredici, K.; Rüb, U.; de Vos, R.A.; Jansen Steur, E.N.; Braak, E. Staging of brain pathology related to sporadic Parkinson’s disease. Neurobiol. Aging 2003, 24, 197–211. [Google Scholar] [CrossRef]
- Van Kessel, S.P.; El Aidy, S. Bacterial Metabolites Mirror Altered Gut Microbiota Composition in Patients with Parkinson’s Disease. J. Parkinsons Dis. 2019, 9, S359–S370. [Google Scholar] [CrossRef] [Green Version]
- Gerhardt, S.; Mohajeri, M.H. Changes of Colonic Bacterial Composition in Parkinson’s Disease and Other Neurodegenerative Diseases. Nutrients 2018, 10, 708. [Google Scholar] [CrossRef] [Green Version]
- Hasegawa, S.; Goto, S.; Tsuji, H.; Okuno, T.; Asahara, T.; Nomoto, K.; Shibata, A.; Fujisawa, Y.; Minato, T.; Okamoto, A.; et al. Intestinal Dysbiosis and Lowered Serum Lipopolysaccharide-Binding Protein in Parkinson’s Disease. PLoS ONE 2015, 10, e0142164. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Scheperjans, F.; Aho, V.; Pereira, P.A.B.; Koskinen, K.; Paulin, L.; Pekkonen, E.; Haapaniemi, E.; Kaakkola, S.; Eerola-Rautio, J.; Pohja, M.; et al. Gut microbiota are related to Parkinson’s disease and clinical phenotype. Mov. Disord. 2015, 30, 350–358. [Google Scholar] [CrossRef] [PubMed]
- Boertien, J.M.; Pereira, P.A.B.; Aho, V.T.E.; Scheperjans, F. Increasing Comparability and Utility of Gut Microbiome Studies in Parkinson’s Disease: A Systematic Review. J. Parkinsons Dis. 2019, 9, S297–S312. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sanguinetti, E.; Collado, M.C.; Marrachelli, V.G.; Monleon, D.; Selma-Royo, M.; Pardo-Tendero, M.M.; Burchielli, S.; Iozzo, P. Microbiome-metabolome signatures in mice genetically prone to develop dementia, fed a normal or fatty diet. Sci. Rep. 2018, 8, 4907. [Google Scholar] [CrossRef] [Green Version]
- Kim, S.; Kwon, S.H.; Kam, T.I.; Panicker, N.; Karuppagounder, S.S.; Lee, S.; Lee, J.H.; Kim, W.R.; Kook, M.; Foss, C.A.; et al. Transneuronal Propagation of Pathologic α-Synuclein from the Gut to the Brain Models Parkinson’s Disease. Neuron 2019, 103, 627–641.e7. [Google Scholar] [CrossRef]
- Houser, M.C.; Tansey, M.G. The gut-brain axis: Is intestinal inflammation a silent driver of Parkinson’s disease pathogenesis? Npj Parkinsons Dis. 2017, 3, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Unger, M.M.; Spiegel, J.; Dillmann, K.U.; Grundmann, D.; Philippeit, H.; Bürmann, J.; Faßbender, K.; Schwiertz, A.; Schäfer, K.H. Short chain fatty acids and gut microbiota differ between patients with Parkinson’s disease and age-matched controls. Parkinsonism Relat. Disord. 2016, 32, 66–72. [Google Scholar] [CrossRef]
- Sampson, T.R.; Debelius, J.W.; Thron, T.; Janssen, S.; Shastri, G.G.; Ilhan, Z.E.; Challis, C.; Schretter, C.E.; Rocha, S.; Gradinaru, V.; et al. Gut Microbiota Regulate Motor Deficits and Neuroinflammation in a Model of Parkinson’s Disease. Cell 2016, 167, 1469–1480.e12. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sun, M.F.; Zhu, Y.L.; Zhou, Z.L.; Jia, X.B.; Xu, Y.D.; Yang, Q.; Cui, C.; Shen, Y.Q. Neuroprotective effects of fecal microbiota transplantation on MPTP-induced Parkinson’s disease mice: Gut microbiota, glial reaction and TLR4/TNF-α signaling pathway. Brain Behav. Immun. 2018, 70, 48–60. [Google Scholar] [CrossRef] [PubMed]
- Wakade, C.; Chong, R. A novel treatment target for Parkinson’s disease. J. Neurol. Sci. 2014, 347, 34–38. [Google Scholar] [CrossRef]
- Donohoe, D.R.; Garge, N.; Zhang, X.; Sun, W.; O’Connell, T.M.; Bunger, M.K.; Bultman, S.J. The microbiome and butyrate regulate energy metabolism and autophagy in the mammalian colon. Cell Metab. 2011, 13, 517–526. [Google Scholar] [CrossRef] [Green Version]
- Stefanko, D.P.; Barrett, R.M.; Ly, A.R.; Reolon, G.K.; Wood, M.A. Modulation of long-term memory for object recognition via HDAC inhibition. Proc. Natl. Acad. Sci. USA 2009, 106, 9447–9452. [Google Scholar] [CrossRef] [Green Version]
- Vecsey, C.G.; Hawk, J.D.; Lattal, K.M.; Stein, J.M.; Fabian, S.A.; Attner, M.A.; Cabrera, S.M.; McDonough, C.B.; Brindle, P.K.; Abel, T.; et al. Histone deacetylase inhibitors enhance memory and synaptic plasticity via CREB:CBP-dependent transcriptional activation. J. Neurosci. 2007, 27, 6128–6140. [Google Scholar] [CrossRef]
- Sherwin, E.; Sandhu, K.V.; Dinan, T.G.; Cryan, J.F. May the Force Be With You: The Light and Dark Sides of the Microbiota-Gut-Brain Axis in Neuropsychiatry. CNS Drugs 2016, 30, 1019–1041. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Paiva, I.; Pinho, R.; Pavlou, M.A.; Hennion, M.; Wales, P.; Schütz, A.L.; Rajput, A.; Szego, M.É.; Kerimoglu, C.; Gerhardt, E.; et al. Sodium butyrate rescues dopaminergic cells from alpha-synuclein-induced transcriptional deregulation and DNA damage. Hum. Mol. Genet. 2017, 26, 2231–2246. [Google Scholar] [CrossRef]
- Kidd, S.K.; Schneider, J.S. Protection of dopaminergic cells from MPP+-mediated toxicity by histone deacetylase inhibition. Brain Res. 2010, 1354, 172–178. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- St. Laurent, R.; O’Brien, L.M.; Ahmad, S.T. Sodium butyrate improves locomotor impairment and early mortality in a rotenone-induced Drosophila model of Parkinson’s disease. Neuroscience 2013, 246, 382–390. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Namkung, H.; Kim, S.H.; Sawa, A. The Insula: An Underestimated Brain Area in Clinical Neuroscience, Psychiatry, and Neurology: (Trends in Neuroscience 40, 200–207, 2017). Trends Neurosci. 2018, 41, 551–554. [Google Scholar] [CrossRef] [Green Version]
- Van Kessel, S.P.; Frye, A.K.; El-Gendy, A.O.; Castejon, M.; Keshavarzian, A.; van Dijk, G.; El Aidy, S. Gut bacterial tyrosine decarboxylases restrict levels of levodopa in the treatment of Parkinson’s disease. Nat. Commun. 2019, 10, 310. [Google Scholar] [CrossRef] [Green Version]
- Marietta, E.; Horwath, I.; Taneja, V. Microbiome, Immunomodulation, and the Neuronal System. Neurotherapeutics 2018, 15, 23–30. [Google Scholar] [CrossRef] [Green Version]
- Rodriguez-Castano, G.P.; Dorris, M.R.; Liu, X.; Bolling, B.W.; Acosta-Gonzalez, A.; Rey, F.E. Bacteroides thetaiotaomicron Starch Utilization Promotes Quercetin Degradation and Butyrate Production by Eubacterium ramulus. Front. Microbiol. 2019, 10, 1145. [Google Scholar] [CrossRef]
- Cirstea, M.S.; Yu, A.C.; Golz, E.; Sundvick, K.; Kliger, D.; Radisavljevic, N.; Foulger, L.H.; Mackenzie, M.; Huan, T.; Finlay, B.B.; et al. Microbiota Composition and Metabolism Are Associated with Gut Function in Parkinson’s Disease. Mov. Disord. 2020. [Google Scholar] [CrossRef]
- Gill, S.R.; Pop, M.; Deboy, R.T.; Eckburg, P.B.; Turnbaugh, P.J.; Samuel, B.S.; Gordon, J.I.; Relman, D.A.; Fraser-Liggett, C.M.; Nelson, K.E. Metagenomic analysis of the human distal gut microbiome. Science 2006, 312, 1355–1359. [Google Scholar] [CrossRef] [Green Version]
- Parthasarathy, A.; Cross, P.J.; Dobson, R.C.J.; Adams, L.E.; Savka, M.A.; Hudson, A.O. A Three-Ring Circus: Metabolism of the Three Proteogenic Aromatic Amino Acids and Their Role in the Health of Plants and Animals. Front. Mol. Biosci. 2018, 5, 29. [Google Scholar] [CrossRef]
- Kaur, H.; Bose, C.; Mande, S.S. Tryptophan Metabolism by Gut Microbiome and Gut-Brain-Axis: An in silico Analysis. Front. Neurosci. 2019, 13, 1365. [Google Scholar] [CrossRef] [PubMed]
- Wikoff, W.R.; Anfora, A.T.; Liu, J.; Schultz, P.G.; Lesley, S.A.; Peters, E.C.; Siuzdak, G. Metabolomics analysis reveals large effects of gut microflora on mammalian blood metabolites. Proc. Natl. Acad. Sci. USA 2009, 106, 3698–3703. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Saito, Y.; Sato, T.; Nomoto, K.; Tsuji, H. Identification of phenol- and p-cresol-producing intestinal bacteria by using media supplemented with tyrosine and its metabolites. FEMS Microbiol. Ecol. 2018, 94. [Google Scholar] [CrossRef]
- Gabriele, S.; Sacco, R.; Cerullo, S.; Neri, C.; Urbani, A.; Tripi, G.; Malvy, J.; Barthelemy, C.; Bonnet-Brihault, F.; Persico, A.M. Urinary p-cresol is elevated in young French children with autism spectrum disorder: A replication study. Biomarkers 2014, 19, 463–470. [Google Scholar] [CrossRef] [PubMed]
- Altieri, L.; Neri, C.; Sacco, R.; Curatolo, P.; Benvenuto, A.; Muratori, F.; Santocchi, E.; Bravaccio, C.; Lenti, C.; Saccani, M.; et al. Urinary p-cresol is elevated in small children with severe autism spectrum disorder. Biomarkers 2011, 16, 252–260. [Google Scholar] [CrossRef]
- Pascucci, T.; Colamartino, M.; Fiori, E.; Sacco, R.; Coviello, A.; Ventura, R.; Puglisi-Allegra, S.; Turriziani, L.; Persico, A.M. P-cresol Alters Brain Dopamine Metabolism and Exacerbates Autism-Like Behaviors in the BTBR Mouse. Brain Sci. 2020, 10, 233. [Google Scholar] [CrossRef]
- Gacias, M.; Gaspari, S.; Santos, P.M.; Tamburini, S.; Andrade, M.; Zhang, F.; Shen, N.; Tolstikov, V.; Kiebish, M.A.; Dupree, J.L.; et al. Microbiota-driven transcriptional changes in prefrontal cortex override genetic differences in social behavior. Elife 2016, 5. [Google Scholar] [CrossRef]
- Hsiao, E.Y.; McBride, S.W.; Hsien, S.; Sharon, G.; Hyde, E.R.; McCue, T.; Codelli, J.A.; Chow, J.; Reisman, S.E.; Petrosino, J.F.; et al. Microbiota modulate behavioral and physiological abnormalities associated with neurodevelopmental disorders. Cell 2013, 155, 1451–1463. [Google Scholar] [CrossRef] [Green Version]
- Gevi, F.; Zolla, L.; Gabriele, S.; Persico, A.M. Urinary metabolomics of young Italian autistic children supports abnormal tryptophan and purine metabolism. Mol. Autism 2016, 7, 47. [Google Scholar] [CrossRef] [Green Version]
- Urrutia, A.; García-Angulo, V.A.; Fuentes, A.; Caneo, M.; Legüe, M.; Urquiza, S.; Delgado, S.E.; Ugalde, J.; Burdisso, P.; Calixto, A. Bacterially produced metabolites protect C. elegans neurons from degeneration. PLoS Biol. 2020, 18, e3000638. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hollister, E.B.; Riehle, K.; Luna, R.A.; Weidler, E.M.; Rubio-Gonzales, M.; Mistretta, T.A.; Raza, S.; Doddapaneni, H.V.; Metcalf, G.A.; Muzny, D.M.; et al. Structure and function of the healthy pre-adolescent pediatric gut microbiome. Microbiome 2015, 3, 36. [Google Scholar] [CrossRef] [Green Version]
- Delgado, P.L.; Charney, D.S.; Price, L.H.; Aghajanian, G.K.; Landis, H.; Heninger, G.R. Serotonin function and the mechanism of antidepressant action. Reversal of antidepressant-induced remission by rapid depletion of plasma tryptophan. Arch. Gen. Psychiatry 1990, 47, 411–418. [Google Scholar] [CrossRef] [PubMed]
- Young, S.N. Acute tryptophan depletion in humans: A review of theoretical, practical and ethical aspects. J. Psychiatry Neurosci. 2013, 38, 294–305. [Google Scholar] [CrossRef] [Green Version]
- Clarke, G.; Grenham, S.; Scully, P.; Fitzgerald, P.; Moloney, R.D.; Shanahan, F.; Dinan, T.G.; Cryan, J.F. The microbiome-gut-brain axis during early life regulates the hippocampal serotonergic system in a sex-dependent manner. Mol. Psychiatry 2013, 18, 666–673. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lukić, I.; Getselter, D.; Koren, O.; Elliott, E. Role of Tryptophan in Microbiota-Induced Depressive-Like Behavior: Evidence from Tryptophan Depletion Study. Front. Behav. Neurosci. 2019, 13, 123. [Google Scholar] [CrossRef] [Green Version]
- Yu, M.; Jia, H.; Zhou, C.; Yang, Y.; Zhao, Y.; Yang, M.; Zou, Z. Variations in gut microbiota and fecal metabolic phenotype associated with depression by 16S rRNA gene sequencing and LC/MS-based metabolomics. J. Pharm. Biomed. Anal. 2017, 138, 231–239. [Google Scholar] [CrossRef]
- Ruddick, J.P.; Evans, A.K.; Nutt, D.J.; Lightman, S.L.; Rook, G.A.; Lowry, C.A. Tryptophan metabolism in the central nervous system: Medical implications. Expert Rev. Mol. Med. 2006, 8, 1–27. [Google Scholar] [CrossRef] [PubMed]
- O’Mahony, S.M.; Clarke, G.; Borre, Y.E.; Dinan, T.G.; Cryan, J.F. Serotonin, tryptophan metabolism and the brain-gut-microbiome axis. Behav. Brain Res. 2015, 277, 32–48. [Google Scholar] [CrossRef] [PubMed]
- Lapin, I.P.; Oxenkrug, G.F. Intensification of the central serotoninergic processes as a possible determinant of the thymoleptic effect. Lancet 1969, 1, 132–136. [Google Scholar] [CrossRef]
- Abramowitz, M.K.; Meyer, T.W.; Hostetter, T.H. Chapter 18—The Pathophysiology of Uremia. In Chronic Kidney Disease, Dialysis, and Transplantation, 3rd ed.; Himmelfarb, J., Sayegh, M.H., Eds.; W.B. Saunders: Philadelphia, PA, USA, 2010; pp. 251–264. [Google Scholar]
- Jaglin, M.; Rhimi, M.; Philippe, C.; Pons, N.; Bruneau, A.; Goustard, B.; Dauge, V.; Maguin, E.; Naudon, L.; Rabot, S. Indole, a Signaling Molecule Produced by the Gut Microbiota, Negatively Impacts Emotional Behaviors in Rats. Front. Neurosci. 2018, 12, 216. [Google Scholar] [CrossRef] [PubMed]
- Buckley, M.M.; O’Brien, R.; Brosnan, E.; Ross, R.P.; Stanton, C.; Buckley, J.M.; O’Malley, D. Glucagon-Like Peptide-1 Secreting L-Cells Coupled to Sensory Nerves Translate Microbial Signals to the Host Rat Nervous System. Front. Cell. Neurosci. 2020, 14, 95. [Google Scholar] [CrossRef] [PubMed]
- Han, W.; Tellez, L.A.; Perkins, M.H.; Perez, I.O.; Qu, T.; Ferreira, J.; Ferreira, T.L.; Quinn, D.; Liu, Z.W.; Gao, X.B.; et al. A Neural Circuit for Gut-Induced Reward. Cell 2018, 175, 665–678.e23. [Google Scholar] [CrossRef] [Green Version]
- Cuomo, A.; Maina, G.; Rosso, G.; Beccarini Crescenzi, B.; Bolognesi, S.; Di Muro, A.; Giordano, N.; Goracci, A.; Neal, S.M.; Nitti, M.; et al. The Microbiome: A New Target for Research and Treatment of Schizophrenia and its Resistant Presentations? A Systematic Literature Search and Review. Front. Pharm. 2018, 9, 1040. [Google Scholar] [CrossRef] [PubMed]
- Paley, E.L.; Denisova, G.; Sokolova, O.; Posternak, N.; Wang, X.; Brownell, A.L. Tryptamine induces tryptophanyl-tRNA synthetase-mediated neurodegeneration with neurofibrillary tangles in human cell and mouse models. Neuromol. Med. 2007, 9, 55–82. [Google Scholar] [CrossRef]
- Paley, E.L. Tryptamine-induced tryptophanyl-tRNAtrp deficiency in neurodifferentiation and neurodegeneration interplay: Progenitor activation with neurite growth terminated in Alzheimer’s disease neuronal vesicularization and fragmentation. J. Alzheimers Dis. 2011, 26, 263–298. [Google Scholar] [CrossRef] [Green Version]
- Paley, E.L.; Perry, G.; Sokolova, O. Tryptamine induces axonopathy and mitochondriopathy mimicking neurodegenerative diseases via tryptophanyl-tRNA deficiency. Curr. Alzheimer Res. 2013, 10, 987–1004. [Google Scholar] [CrossRef] [PubMed]
- Yeh, Y.C.; Huang, M.F.; Liang, S.S.; Hwang, S.J.; Tsai, J.C.; Liu, T.L.; Wu, P.H.; Yang, Y.H.; Kuo, K.C.; Kuo, M.C.; et al. Indoxyl sulfate, not p-cresyl sulfate, is associated with cognitive impairment in early-stage chronic kidney disease. Neurotoxicology 2016, 53, 148–152. [Google Scholar] [CrossRef] [PubMed]
- Sankowski, B.; Księżarczyk, K.; Raćkowska, E.; Szlufik, S.; Koziorowski, D.; Giebułtowicz, J. Higher cerebrospinal fluid to plasma ratio of p-cresol sulfate and indoxyl sulfate in patients with Parkinson’s disease. Clin. Chim. Acta 2020, 501, 165–173. [Google Scholar] [CrossRef] [PubMed]
- Dou, L.; Jourde-Chiche, N.; Faure, V.; Cerini, C.; Berland, Y.; Dignat-George, F.; Brunet, P. The uremic solute indoxyl sulfate induces oxidative stress in endothelial cells. J. Thromb. Haemost. 2007, 5, 1302–1308. [Google Scholar] [CrossRef] [PubMed]
- Adesso, S.; Magnus, T.; Cuzzocrea, S.; Campolo, M.; Rissiek, B.; Paciello, O.; Autore, G.; Pinto, A.; Marzocco, S. Indoxyl Sulfate Affects Glial Function Increasing Oxidative Stress and Neuroinflammation in Chronic Kidney Disease: Interaction between Astrocytes and Microglia. Front. Pharm. 2017, 8, 370. [Google Scholar] [CrossRef]
- Singh, A.; Kukreti, R.; Saso, L.; Kukreti, S. Oxidative Stress: A Key Modulator in Neurodegenerative Diseases. Molecules 2019, 24, 1583. [Google Scholar] [CrossRef] [Green Version]
- Gray, M.T.; Woulfe, J.M. Striatal blood-brain barrier permeability in Parkinson’s disease. J. Cereb. Blood Flow Metab. 2015, 35, 747–750. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chu, C.; Murdock, M.H.; Jing, D.; Won, T.H.; Chung, H.; Kressel, A.M.; Tsaava, T.; Addorisio, M.E.; Putzel, G.G.; Zhou, L.; et al. The microbiota regulate neuronal function and fear extinction learning. Nature 2019, 574, 543–548. [Google Scholar] [CrossRef] [PubMed]
- Zieve, L.; Lyftogt, C.; Raphael, D. Ammonia toxicity: Comparative protective effect of various arginine and ornithine derivatives, aspartate, benzoate, and carbamyl glutamate. Metab. Brain Dis. 1986, 1, 25–35. [Google Scholar] [CrossRef]
- Luan, H.; Liu, L.F.; Tang, Z.; Zhang, M.; Chua, K.K.; Song, J.X.; Mok, V.C.; Li, M.; Cai, Z. Comprehensive urinary metabolomic profiling and identification of potential noninvasive marker for idiopathic Parkinson’s disease. Sci. Rep. 2015, 5, 13888. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Luan, H.; Liu, L.F.; Meng, N.; Tang, Z.; Chua, K.K.; Chen, L.L.; Song, J.X.; Mok, V.C.; Xie, L.X.; Li, M.; et al. LC-MS-based urinary metabolite signatures in idiopathic Parkinson’s disease. J. Proteome Res. 2015, 14, 467–478. [Google Scholar] [CrossRef]
- Hatano, T.; Saiki, S.; Okuzumi, A.; Mohney, R.P.; Hattori, N. Identification of novel biomarkers for Parkinson’s disease by metabolomic technologies. J. Neurol. Neurosurg. Psychiatry 2016, 87, 295–301. [Google Scholar] [CrossRef]
- Okuzumi, A.; Hatano, T.; Ueno, S.I.; Ogawa, T.; Saiki, S.; Mori, A.; Koinuma, T.; Oji, Y.; Ishikawa, K.I.; Fujimaki, M.; et al. Metabolomics-based identification of metabolic alterations in PARK2. Ann. Clin. Transl. Neurol. 2019, 6, 525–536. [Google Scholar] [CrossRef] [Green Version]
- Gao, J.; Xu, K.; Liu, H.; Liu, G.; Bai, M.; Peng, C.; Li, T.; Yin, Y. Impact of the Gut Microbiota on Intestinal Immunity Mediated by Tryptophan Metabolism. Front. Cell. Infect. Microbiol. 2018, 8, 13. [Google Scholar] [CrossRef] [Green Version]
- Kim, D.-C.; Quang, T.H.; Yoon, C.-S.; Ngan, N.T.T.; Lim, S.-I.; Lee, S.-Y.; Kim, Y.-C.; Oh, H. Anti-neuroinflammatory activities of indole alkaloids from kanjang (Korean fermented soy source) in lipopolysaccharide-induced BV2 microglial cells. Food Chem. 2016, 213, 69–75. [Google Scholar] [CrossRef] [PubMed]
- Willkommen, D.; Lucio, M.; Moritz, F.; Forcisi, S.; Kanawati, B.; Smirnov, K.S.; Schroeter, M.; Sigaroudi, A.; Schmitt-Kopplin, P.; Michalke, B. Metabolomic investigations in cerebrospinal fluid of Parkinson’s disease. PLoS ONE 2018, 13, e0208752. [Google Scholar] [CrossRef] [Green Version]
- Southan, C.; DeWolf, W.E., Jr.; Kruse, L.I. Inactivation of dopamine beta-hydroxylase by p-cresol: Evidence for a second, minor site of covalent modification at tyrosine 357. Biochim. Biophys. Acta 1990, 1037, 256–258. [Google Scholar] [CrossRef]
- Crossgrove, J.; Zheng, W. Manganese toxicity upon overexposure. NMR Biomed. 2004, 17, 544–553. [Google Scholar] [CrossRef] [PubMed]
- Reaney, S.H.; Bench, G.; Smith, D.R. Brain accumulation and toxicity of Mn(II) and Mn(III) exposures. Toxicol. Sci. 2006, 93, 114–124. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chi, L.; Gao, B.; Bian, X.; Tu, P.; Ru, H.; Lu, K. Manganese-induced sex-specific gut microbiome perturbations in C57BL/6 mice. Toxicol. Appl. Pharm. 2017, 331, 142–153. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fowlie, G.; Cohen, N.; Ming, X. The Perturbance of Microbiome and Gut-Brain Axis in Autism Spectrum Disorders. Int. J. Mol. Sci. 2018, 19, 2251. [Google Scholar] [CrossRef] [Green Version]
- Rothhammer, V.; Mascanfroni, I.D.; Bunse, L.; Takenaka, M.C.; Kenison, J.E.; Mayo, L.; Chao, C.C.; Patel, B.; Yan, R.; Blain, M.; et al. Type I interferons and microbial metabolites of tryptophan modulate astrocyte activity and central nervous system inflammation via the aryl hydrocarbon receptor. Nat. Med. 2016, 22, 586–597. [Google Scholar] [CrossRef]
- Cekanaviciute, E.; Yoo, B.B.; Runia, T.F.; Debelius, J.W.; Singh, S.; Nelson, C.A.; Kanner, R.; Bencosme, Y.; Lee, Y.K.; Hauser, S.L.; et al. Gut bacteria from multiple sclerosis patients modulate human T cells and exacerbate symptoms in mouse models. Proc. Natl. Acad. Sci. USA 2017, 114, 10713–10718. [Google Scholar] [CrossRef] [Green Version]
- Mohanta, L.; Das, B.C.; Patri, M. Microbial communities modulating brain functioning and behaviors in zebrafish: A mechanistic approach. Microb. Pathog. 2020, 145, 104251. [Google Scholar] [CrossRef]
- Chong-Nguyen, C.; Duboc, H.; Sokol, H. The gut microbiota, a new cardiovascular risk factor? Presse Med. 2017, 46, 708–713. [Google Scholar] [CrossRef]
- Fu, B.C.; Hullar, M.A.J.; Randolph, T.W.; Franke, A.A.; Monroe, K.R.; Cheng, I.; Wilkens, L.R.; Shepherd, J.A.; Madeleine, M.M.; Le Marchand, L.; et al. Associations of plasma trimethylamine N-oxide, choline, carnitine, and betaine with inflammatory and cardiometabolic risk biomarkers and the fecal microbiome in the Multiethnic Cohort Adiposity Phenotype Study. Am. J. Clin. Nutr. 2020, 111, 1226–1234. [Google Scholar] [CrossRef]
- Velasquez, M.T.; Ramezani, A.; Manal, A.; Raj, D.S. Trimethylamine N-Oxide: The Good, the Bad and the Unknown. Toxins 2016, 8, 326. [Google Scholar] [CrossRef] [Green Version]
- Vogt, N.M.; Romano, K.A.; Darst, B.F.; Engelman, C.D.; Johnson, S.C.; Carlsson, C.M.; Asthana, S.; Blennow, K.; Zetterberg, H.; Bendlin, B.B.; et al. The gut microbiota-derived metabolite trimethylamine N-oxide is elevated in Alzheimer’s disease. Alzheimers Res. Ther. 2018, 10, 124. [Google Scholar] [CrossRef] [Green Version]
- Del Rio, D.; Zimetti, F.; Caffarra, P.; Tassotti, M.; Bernini, F.; Brighenti, F.; Zini, A.; Zanotti, I. The Gut Microbial Metabolite Trimethylamine-N-Oxide Is Present in Human Cerebrospinal Fluid. Nutrients 2017, 9, 1053. [Google Scholar] [CrossRef] [Green Version]
- Li, D.; Ke, Y.; Zhan, R.; Liu, C.; Zhao, M.; Zeng, A.; Shi, X.; Ji, L.; Cheng, S.; Pan, B.; et al. Trimethylamine-N-oxide promotes brain aging and cognitive impairment in mice. Aging Cell 2018, 17, e12768. [Google Scholar] [CrossRef] [PubMed]
- Gao, Q.; Wang, Y.; Wang, X.; Fu, S.; Zhang, X.; Wang, R.T. Decreased levels of circulating trimethylamine N-oxide alleviate cognitive and pathological deterioration in transgenic mice: A potential therapeutic approach for Alzheimer’s disease. Aging 2019, 11, 8642–8663. [Google Scholar] [CrossRef]
- Govindarajulu, M.; Pinky, P.D.; Steinke, I.; Bloemer, J.; Ramesh, S.; Kariharan, T.; Rella, R.T.; Bhattacharya, S.; Dhanasekaran, M.; Suppiramaniam, V.; et al. Gut Metabolite TMAO Induces Synaptic Plasticity Deficits by Promoting Endoplasmic Reticulum Stress. Front. Mol. Neurosci. 2020, 13, 138. [Google Scholar] [CrossRef]
- Brunt, V.E.; LaRocca, T.J.; Bazzoni, A.E.; Sapinsley, Z.J.; Miyamoto-Ditmon, J.; Gioscia-Ryan, R.A.; Neilson, A.P.; Link, C.D.; Seals, D.R. The gut microbiome-derived metabolite trimethylamine N-oxide modulates neuroinflammation and cognitive function with aging. Geroscience 2020. [Google Scholar] [CrossRef] [PubMed]
- Alkasir, R.; Li, J.; Li, X.; Jin, M.; Zhu, B. Human gut microbiota: The links with dementia development. Protein Cell 2017, 8, 90–102. [Google Scholar] [CrossRef] [Green Version]
- Wang, Z.; Klipfell, E.; Bennett, B.J.; Koeth, R.; Levison, B.S.; Dugar, B.; Feldstein, A.E.; Britt, E.B.; Fu, X.; Chung, Y.M.; et al. Gut flora metabolism of phosphatidylcholine promotes cardiovascular disease. Nature 2011, 472, 57–63. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gorelick, P.B.; Scuteri, A.; Black, S.E.; Decarli, C.; Greenberg, S.M.; Iadecola, C.; Launer, L.J.; Laurent, S.; Lopez, O.L.; Nyenhuis, D.; et al. Vascular contributions to cognitive impairment and dementia: A statement for healthcare professionals from the american heart association/american stroke association. Stroke 2011, 42, 2672–2713. [Google Scholar] [CrossRef]
- Wu, P.; Chen, J.; Tao, J.; Wu, S.; Xu, G.; Wang, Z.; Wei, D.; Yin, W. Trimethylamine N-oxide promotes apoE(-/-) mice atherosclerosis by inducing vascular endothelial cell pyroptosis via the SDHB/ROS pathway. J. Cell Physiol. 2020, 235, 6582–6591. [Google Scholar] [CrossRef] [PubMed]
- Koeth, R.A.; Wang, Z.; Levison, B.S.; Buffa, J.A.; Org, E.; Sheehy, B.T.; Britt, E.B.; Fu, X.; Wu, Y.; Li, L.; et al. Intestinal microbiota metabolism of L-carnitine, a nutrient in red meat, promotes atherosclerosis. Nat. Med. 2013, 19, 576–585. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhu, W.; Gregory, J.C.; Org, E.; Buffa, J.A.; Gupta, N.; Wang, Z.; Li, L.; Fu, X.; Wu, Y.; Mehrabian, M.; et al. Gut Microbial Metabolite TMAO Enhances Platelet Hyperreactivity and Thrombosis Risk. Cell 2016, 165, 111–124. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Janeiro, M.H.; Ramírez, M.J.; Milagro, F.I.; Martínez, J.A.; Solas, M. Implication of Trimethylamine N-Oxide (TMAO) in Disease: Potential Biomarker or New Therapeutic Target. Nutrients 2018, 10, 1398. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xu, R.; Wang, Q. Towards understanding brain-gut-microbiome connections in Alzheimer’s disease. BMC Syst. Biol. 2016, 10 (Suppl. S3), 63. [Google Scholar] [CrossRef] [Green Version]
- Hulme, H.; Meikle, L.M.; Strittmatter, N.; van der Hooft, J.J.J.; Swales, J.; Bragg, R.A.; Villar, V.H.; Ormsby, M.J.; Barnes, S.; Brown, S.L.; et al. Microbiome-derived carnitine mimics as previously unknown mediators of gut-brain axis communication. Sci. Adv. 2020, 6, eaax6328. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Knobloch, M.; Pilz, G.A.; Ghesquière, B.; Kovacs, W.J.; Wegleiter, T.; Moore, D.L.; Hruzova, M.; Zamboni, N.; Carmeliet, P.; Jessberger, S. A Fatty Acid Oxidation-Dependent Metabolic Shift Regulates Adult Neural Stem Cell Activity. Cell Rep. 2017, 20, 2144–2155. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xie, Z.; Jones, A.; Deeney, J.T.; Hur, S.K.; Bankaitis, V.A. Inborn Errors of Long-Chain Fatty Acid β-Oxidation Link Neural Stem Cell Self-Renewal to Autism. Cell Rep. 2016, 14, 991–999. [Google Scholar] [CrossRef] [Green Version]
- Argou-Cardozo, I.; Zeidán-Chuliá, F. Clostridium Bacteria and Autism Spectrum Conditions: A Systematic Review and Hypothetical Contribution of Environmental Glyphosate Levels. Med. Sci. 2018, 6, 29. [Google Scholar] [CrossRef] [Green Version]
- Ho, L.; Zhao, D.; Ono, K.; Ruan, K.; Mogno, I.; Tsuji, M.; Carry, E.; Brathwaite, J.; Sims, S.; Frolinger, T.; et al. Heterogeneity in gut microbiota drive polyphenol metabolism that influences α-synuclein misfolding and toxicity. J. Nutr. Biochem. 2019, 64, 170–181. [Google Scholar] [CrossRef] [PubMed]
- Wang, D.; Ho, L.; Faith, J.; Ono, K.; Janle, E.M.; Lachcik, P.J.; Cooper, B.R.; Jannasch, A.H.; D’Arcy, B.R.; Williams, B.A.; et al. Role of intestinal microbiota in the generation of polyphenol-derived phenolic acid mediated attenuation of Alzheimer’s disease β-amyloid oligomerization. Mol. Nutr. Food Res. 2015, 59, 1025–1040. [Google Scholar] [CrossRef]
- Reddy, V.P.; Aryal, P.; Robinson, S.; Rafiu, R.; Obrenovich, M.; Perry, G. Polyphenols in Alzheimer’s Disease and in the Gut-Brain Axis. Microorganisms 2020, 8, 199. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Filosa, S.; Di Meo, F.; Crispi, S. Polyphenols-gut microbiota interplay and brain neuromodulation. Neural Regen. Res. 2018, 13, 2055–2059. [Google Scholar] [CrossRef] [PubMed]
- Feng, X.; Li, Y.; Brobbey Oppong, M.; Qiu, F. Insights into the intestinal bacterial metabolism of flavonoids and the bioactivities of their microbe-derived ring cleavage metabolites. Drug Metab. Rev. 2018, 50, 343–356. [Google Scholar] [CrossRef]
- Gasperotti, M.; Passamonti, S.; Tramer, F.; Masuero, D.; Guella, G.; Mattivi, F.; Vrhovsek, U. Fate of microbial metabolites of dietary polyphenols in rats: Is the brain their target destination? ACS Chem. Neurosci. 2015, 6, 1341–1352. [Google Scholar] [CrossRef] [Green Version]
- Cheng, C.Y.; Su, S.Y.; Tang, N.Y.; Ho, T.Y.; Chiang, S.Y.; Hsieh, C.L. Ferulic acid provides neuroprotection against oxidative stress-related apoptosis after cerebral ischemia/reperfusion injury by inhibiting ICAM-1 mRNA expression in rats. Brain Res. 2008, 1209, 136–150. [Google Scholar] [CrossRef] [PubMed]
- Yabe, T.; Hirahara, H.; Harada, N.; Ito, N.; Nagai, T.; Sanagi, T.; Yamada, H. Ferulic acid induces neural progenitor cell proliferation in vitro and in vivo. Neuroscience 2010, 165, 515–524. [Google Scholar] [CrossRef]
- Fuertes, A.; Perez-Burillo, S.; Apaolaza, I.; Valles, Y.; Francino, M.P.; Rufian-Henares, J.A.; Planes, F.J. Adaptation of the Human Gut Microbiota Metabolic Network During the First Year After Birth. Front. Microbiol. 2019, 10, 848. [Google Scholar] [CrossRef] [PubMed]
- Fullana, M.A.; Albajes-Eizagirre, A.; Soriano-Mas, C.; Vervliet, B.; Cardoner, N.; Benet, O.; Radua, J.; Harrison, B.J. Fear extinction in the human brain: A meta-analysis of fMRI studies in healthy participants. Neurosci. Biobehav. Rev. 2018, 88, 16–25. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Singewald, N.; Holmes, A. Rodent models of impaired fear extinction. Psychopharmacology 2019, 236, 21–32. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Friedland, R.P.; Chapman, M.R. The role of microbial amyloid in neurodegeneration. PLoS Pathog. 2017, 13, e1006654. [Google Scholar] [CrossRef]
- Lundmark, K.; Westermark, G.T.; Olsén, A.; Westermark, P. Protein fibrils in nature can enhance amyloid protein A amyloidosis in mice: Cross-seeding as a disease mechanism. Proc. Natl. Acad. Sci. USA 2005, 102, 6098–6102. [Google Scholar] [CrossRef] [Green Version]
- Svensson, E.; Horváth-Puhó, E.; Thomsen, R.W.; Djurhuus, J.C.; Pedersen, L.; Borghammer, P.; Sørensen, H.T. Vagotomy and subsequent risk of Parkinson’s disease. Ann. Neurol. 2015, 78, 522–529. [Google Scholar] [CrossRef]
- Pan-Montojo, F.; Schwarz, M.; Winkler, C.; Arnhold, M.; O’Sullivan, G.A.; Pal, A.; Said, J.; Marsico, G.; Verbavatz, J.M.; Rodrigo-Angulo, M.; et al. Environmental toxins trigger PD-like progression via increased alpha-synuclein release from enteric neurons in mice. Sci. Rep. 2012, 2, 898. [Google Scholar] [CrossRef] [Green Version]
- Miraglia, F.; Colla, E. Microbiome, Parkinson’s Disease and Molecular Mimicry. Cells 2019, 8, 222. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bienenstock, J.; Kunze, W.A.; Forsythe, P. Disruptive physiology: Olfaction and the microbiome-gut-brain axis. Biol. Rev. Camb. Philos. Soc. 2018, 93, 390–403. [Google Scholar] [CrossRef] [PubMed]
- Sampson, T.R.; Challis, C.; Jain, N.; Moiseyenko, A.; Ladinsky, M.S.; Shastri, G.G.; Thron, T.; Needham, B.D.; Horvath, I.; Debelius, J.W.; et al. A gut bacterial amyloid promotes α-synuclein aggregation and motor impairment in mice. Elife 2020, 9. [Google Scholar] [CrossRef] [PubMed]
- Chen, S.G.; Stribinskis, V.; Rane, M.J.; Demuth, D.R.; Gozal, E.; Roberts, A.M.; Jagadapillai, R.; Liu, R.; Choe, K.; Shivakumar, B.; et al. Exposure to the Functional Bacterial Amyloid Protein Curli Enhances Alpha-Synuclein Aggregation in Aged Fischer 344 Rats and Caenorhabditis elegans. Sci. Rep. 2016, 6, 34477. [Google Scholar] [CrossRef]
- Caputi, V.; Giron, M.C. Microbiome-Gut-Brain Axis and Toll-Like Receptors in Parkinson’s Disease. Int. J. Mol. Sci. 2018, 19, 1689. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, D.; Zhao, D.; Shah, S.Z.A.; Wu, W.; Lai, M.; Zhang, X.; Li, J.; Guan, Z.; Zhao, H.; Li, W.; et al. Implications of gut microbiota dysbiosis and metabolic changes in prion disease. Neurobiol. Dis. 2020, 135, 104704. [Google Scholar] [CrossRef]
- Blacher, E.; Bashiardes, S.; Shapiro, H.; Rothschild, D.; Mor, U.; Dori-Bachash, M.; Kleimeyer, C.; Moresi, C.; Harnik, Y.; Zur, M.; et al. Potential roles of gut microbiome and metabolites in modulating ALS in mice. Nature 2019, 572, 474–480. [Google Scholar] [CrossRef]


{kind=link}
{kind=link}
{kind=link}
| Disease | SCFA | Literature | p-Values | |
|---|---|---|---|---|
| ASD | AA | ↓ | f [44], f [43] | p = 0.011, p = 0.0000003 |
| ↑ | f [42], f [39],* u [36] | p = 0.037, p < 0.005, p < 0.005 | ||
| - | f [41] | p = 0.979 | ||
| BA | ↓ | f [44], f [43] | p = 0.005, p = 0.005 | |
| ↑ | f [42] | p = 0.025 | ||
| - | f [41] | p = 0.974 | ||
| Isobutyric acid | ↑ | f [42] | p = 0.022 | |
| Isovaleric acid | ↑ | f [42] | p = 0.038 | |
| PA | ↓ | f [43] | p = 0.002 | |
| ↑ | f [42], f [39] | p = 0.007, p < 0.005 | ||
| - | f [41], f [44] | p = 0.979, p = 0.243 | ||
| VA | ↓ | f [43] | p = 0.005 | |
| ↑ | f [44], f [42] | p < 0.001, p = 0.007 | ||
| MDD | AA | ↓ | f [67] | p = 0.04 |
| ↑ | f [69] | p = 0.65 | ||
| BA | - | f [67], f [69] | p = 0.68, p = 0.867 | |
| Caproic acid | ↑ | f [67] | p = 0.09 | |
| Isobutyric acid | - | f [67] | p = 0.70 | |
| - | f [69] | p = 0.501 | ||
| Isocaproic acid | ↑ | f [67] | p < 0.01 | |
| Isovaleric acid | - | f [67] | p = 0.4 | |
| PA | ↓ | f [67] | p = 0.07 | |
| - | f [69] | p = 0.918 | ||
| VA | ↓ | f [67] | p = 0.56 | |
| MS | AA | ↓ | f [81], s [29] | p < 0.0001, p = 0.001 |
| BA | ↓ | f [81], s [29] | p < 0.05, p = 0.0001 | |
| Isovalerate, valerate, hexanoate, heptanoate | - | s [29] | p > 0.05 | |
| PA | ↓ | f [81], s [29] | p < 0.0001, p = 0.01 | |
| PD | AA | ↓ | f [117], * p [109] | p < 0.01, p = 0.0201 |
| BA | ↓ | f [117] | p < 0.01 | |
| Isobutyric acid | - | f [117] | p > 0.05 | |
| Isovaleric acid | - | f [117] | p > 0.05 | |
| PA | ↓ | f [117] | p < 0.01 | |
| VA | - | f [117] | p > 0.05 |
| SCFA | Taxa | Study |
|---|---|---|
| tSCFA | Faecalibacterium, Ruminococcus, Bifidobacterium | [39] |
| PA | Bacteroides | [39] |
| VA | Acidobacteria, Actinomycetaceae | [44] |
| BA | Streptococcaceae, Peptostreptococcaceae, Lactobacillaceae, Clostridiaceae, Family_XIII, Leuconostocaceae | [44] |
| PA | Desulfovibrionaceae, Streptococcaceae | [44] |
| AA | Desulfovibrionaceae | [44] |
| AA, PA | Prevotella_9 | [81] |
| BA | Clostridium, Eubacterium, Butyrivibrio | [103] |
| AA | Bacteroidetes, B.hydrogenotrophica | [130] |
| BA | Lachnospiraceae, Faecalibacterium prausnitzii, Eubacterium, Roseburia | [130] |
| PA | Bacteroidetes, Proteobacteria, some Lachnospiraceae | [130] |
| BA | Eubacterium ramulus | [131] |
| PA | Clostridium | [46] |
| AA, PA | Parabacteroides distasonis, Megaspheara massiliensis | [87] |
| BA, VA, HA | Parabacteroides distasonis, Megaspheara massiliensis | [87] |
| PA | Lactobacillus, Propionibacterium | [97] |
| BA | Faecalibacterium prausnitzii, Eubacterium rectale, Roserburia, Eubacterium hallii, Ruminococcus bromii | [14] |
| PA | Akkermansia municiphila | [14] |
| BA | Blautia, Lachnospiraceae: Coprococcus, Roseburia, Faecalibacterium, Lachnospira | [132] |
| BA | Clostridia (class) | [73] |
| AA | Blautia hydrogenotrophica, Clostridium, Streptococcus | [82] |
| PA | Salmonella, Roseburia inulinivorans, Ruminococcus obeum, Bacteroides, Phascolarctobacterium succinatutens, Dialister, Veillonella, Megasphaera elsdenii, Coprococcus catus | [82] |
| BA | Anaerostipes, Coprococcus catus, Eubacterium rectale, Eubacterium hallii, Faecalibacterium prausnitzii, Roseburia, Coprococcus comes, Coprococcus eutactus | [82] |
| Factors | Odds Ratio | p-Value |
|---|---|---|
| Enterotype III | 18.5 b | <0.001 b |
| Enterotype I | 0.1 a | <0.001 a |
| ApoE | 3.9 a, 4.4 b | 0.035 a, 0.026 b |
| SLI | 15.0 a | 0.005 a |
| VSRAD | 3.5 a, 4.2 b | <0.001 a,b |
| AAM | Taxa | Study |
|---|---|---|
| Taurine | Alistipes HGB5, Alistipes finegoldii, Bacteroides xylanisolvens | [45] |
| GABA | Bifidobacterium, Bacteroides, Lactobacillus; Lactobacillus brevis | [41,97] |
| GABA, lactate | E. coli HT115-strain | [144] |
| Serotonin | Candida, Streptococcus, Escherichia, Enterococcus, Pseudomonas | [135,184] |
| Serotonin, dopamine, norepinephrine | Streptococcus, Enterococcus, Escherichia | [135] |
| Serotonin, dopamine | Clostridiales incertae sedis | [150] |
| Norepinephrine | Escherichia, Bacillus, Saccharomyces | [184] |
| Dopamine | Bacillus | [184] |
| Acetylcholine | Lactobacillus | [184] |
| 4EPS, p-cresol (sulphate) | Clostridium | [36,140,142] |
| P-cresol (sulphate) | Clostridiaceae (Clostridium I, IV, IX, XI, XIII, XIVa, XVI), Bacteroidaceae, Coriobacteriaceae | [109,137] |
| P-cresol, phenylacetylglutamine | Oscillospira, Ruminococcus, Mogibacteriaceae, Christensellaceae, Clostridiales, Akkermansia | [132] |
| Dextrorphan O-glucuronide, 3-methyldioxyindole(F4) | Christensenella, Candidatus arthromitus | [150] |
| “TRYP6” (Kynurenine, quinolinate, indole, IAA, IPA, tryptamine) | Actinobacteria, Firmicutes, Proteobacteria, Bacteroidetes, Fusobacteria | [135] |
| Quinolinate, indole, IAA, IPA, tryptamine | Clostridium | [135] |
| Kynurenine, quinolinate, indole, IAA, IPA | Burkholderia | [135] |
| Kynurenine, quinolinate, IAA, tryptamine | Streptomyces, Pseudomonas, Bacillus | [135] |
| IAA | Bacillus, Klebsiella, Ralstonia, Staphylococcus | [135] |
| Indole | Bacteroides, Citrobacter, Clostridium_XIX, Desulfitobacterium, Edwardsiella, Escherichia, Fusobacterium, Providencia, Shigella | [135] |
| Parabacteroides distasonis, Megasphaera massiliensis E. coli | [87] [155] | |
| IPA | Clostridium, Escherichia, Proteus | [135] |
| Kynurenine | Pseudomonas, Bacillus, Burkholderia, Streptomyces | [135] |
| Quinolinate | Klebsiella, Bacillus, Burkholderia | [135] |
| Tryptamine | Holdemania, Tyzzerella, Desulfovibrio, Yersinia, Bacillus, Clostridium, Ruminococcus | [135] |
| Indole, indoxyl-(3)-sulphate, IPA, indole-(3)-aldehyde | Lactobacillus reuteri | [182] |
| Faecal Metabolites | Trend in Depressed Rats |
|---|---|
| Nicotinic acid | ↑ |
| Hypoxanthine | ↑ |
| Dextrorphan O-glucuronide | ↑ |
| 3-Methoxytryptophan | ↑ |
| 5-Methoxytryptophan | ↓ |
| L-Urobilin | ↑ |
| MG(0:0/20:3(5Z,8Z,11Z)/0:0) | ↓ |
| PE(14:1(9Z)/14:1(9Z)) | ↑ |
| PS(18:0/22:6(4Z,7Z,10Z,13Z,16Z,19Z)) | ↑ |
| Cholic acid | ↑ |
| MG(0:0/20:4(8Z,11Z,14Z,17Z)/0:0) | ↓ |
| Stearyl citrate | ↓ |
| Hyocholic acid | ↑ |
| L-Urobilinogen | ↑ |
| Deoxycholic acid | ↑ |
| Chenodeoxycholic acid | ↑ |
| Disease | Amino Acid Metabolites | Sample and Literature | p-Values | |
|---|---|---|---|---|
| ASD | P-cresol | - | f [42] | p = 0.884 |
| ↑ | f [39], f [41], * s/p [36], u [138,139] | p < 0.05, p = 0.04, p < 0.05, p < 0.05, p < 0.05 | ||
| IAA, indolyl lactate | ↑ | u [143] | p < 0.001 | |
| IS | ↑ | p < 0.05 | ||
| Indoles (indole, 3-methylindole) | ↑ | f [39] | p < 0.05 | |
| Serotonin, GABA | ↑ | * s/p [36] | p < 0.05 | |
| GABA | ↓ | f [41] | p = 0.077 | |
| 3-(3-hydroxyphenyl)-3-hydroxypropionic acid, 3-hydroxyphenylacetic acid, 3-hydroxyhippuric acid | ↑ | * u [36] | p < 0.05 | |
| PD | P-cresol | ↑ | * c [109], c [176], s [132], ccapa [163] | p < 0.05, p = 0.0002, p = 0.0028, p < 0.05 |
| IAA | ↓ | s [172,173], * s [109] | p = 0.0083, p = 0.0258, p < 0.05 | |
| ↑ | * cp [109], u [170,171] | p < 0.05, p b, p < 0.001 | ||
| Indole | ↑ | * cp [109] | p < 0.05 | |
| IS | ↑ | c a [163] | p < 0.05 | |
| Catechol sulphate, hippuric acid, 3-hydroxyhippuric acid, catechol sulphate, 3-(3-hydroxyphenyl)propionic acid, indole-3-methyl acetate, 2-furoylglycine, phenylethylamine | ↓ | * s [109] | p < 0.05 | |
| Phenylactate, 3-(4-hydroxyphenyl)lactate | ↑ | * s [109] | p < 0.05 | |
| 3-(4-hydroxyphenyl)acetic acid, tryptamine, phenylacetic acid, aminobenzoic acid, hydroxybenzoic acid | ↑ | u [170,171] | p < 0.05 | |
| Phenylacetylglutamine | ↑ | s [132] | p = 0.004 | |
| Quinic acid | ↑ | * c [109] | p < 0.05 | |
| Trimethylamine, threonate | ↓ | * p [109] | p < 0.05 | |
| Benzoic acid, 3-(4-hydroxyphenyl)acetic acid | ↓ | * cp [109] | p < 0.05 | |
| MS | Aryl hydrocarbon receptor agonists | ↓ | s [182] | p < 0.05 |
| Disease | Metabolite | Change | Literature |
|---|---|---|---|
| AD | TMAO | ↑ | c [188] |
| MCI | TMAO | ↑ | c [188] |
| PD | TMAO | ↑ | cappa [163] |
| ASD | Isopropanol | ↑ | f [41] |
| Other Metabolites | Taxa | Study |
|---|---|---|
| TMA(O) | Prevotella, Mitsuokella, Fusobacterium, Desulfovibrio, Methanobrevibacter smithii; some from Lachnospiraceae and Ruminococcaceae | [186] |
| TMA | Anaerococcus, Clostridium, Escherichia, Proteus, Providencia, Edwardsiella | [22] |
| curli | Enterobacteriaceae (E. coli, Salmonella typhimurium, Citrobacter freundii, Cronobacter sakazakii, Proteus mirabilis) | [109] |
| curli | E. coli | [224] |
| curli | Streptococcus, Staphylococcus, Mycobacteria, Klebsiella, Bacillus | [217] |
| Nicotinamide | Akkermansia muciniphila | [227] |
| 3-HBA, 3,4diHBA, DHCA | Bacteroides ovatus | [206] |
| 3-methyl-4-(trimethylammonio)butanoate, 4-(trimethylammonio)pentanoate | Lachnospiraceae (Clostridiales): C.clostridioforme, C.symbosium | [202] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Tran, S.M.-S.; Mohajeri, M.H. The Role of Gut Bacterial Metabolites in Brain Development, Aging and Disease. Nutrients 2021, 13, 732. https://doi.org/10.3390/nu13030732
Tran SM-S, Mohajeri MH. The Role of Gut Bacterial Metabolites in Brain Development, Aging and Disease. Nutrients. 2021; 13(3):732. https://doi.org/10.3390/nu13030732
Chicago/Turabian StyleTran, Shirley Mei-Sin, and M. Hasan Mohajeri. 2021. "The Role of Gut Bacterial Metabolites in Brain Development, Aging and Disease" Nutrients 13, no. 3: 732. https://doi.org/10.3390/nu13030732
APA StyleTran, S. M.-S., & Mohajeri, M. H. (2021). The Role of Gut Bacterial Metabolites in Brain Development, Aging and Disease. Nutrients, 13(3), 732. https://doi.org/10.3390/nu13030732