
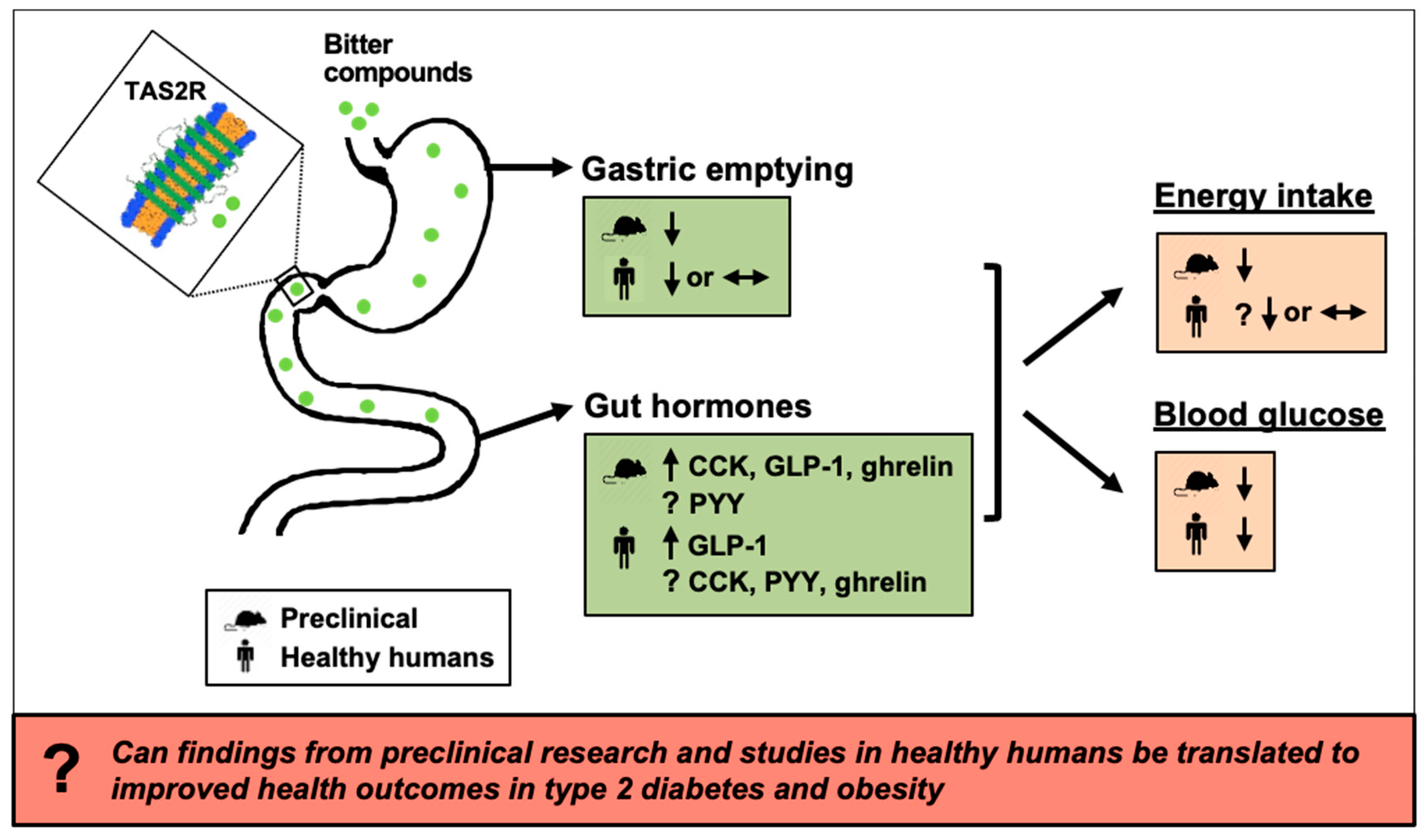
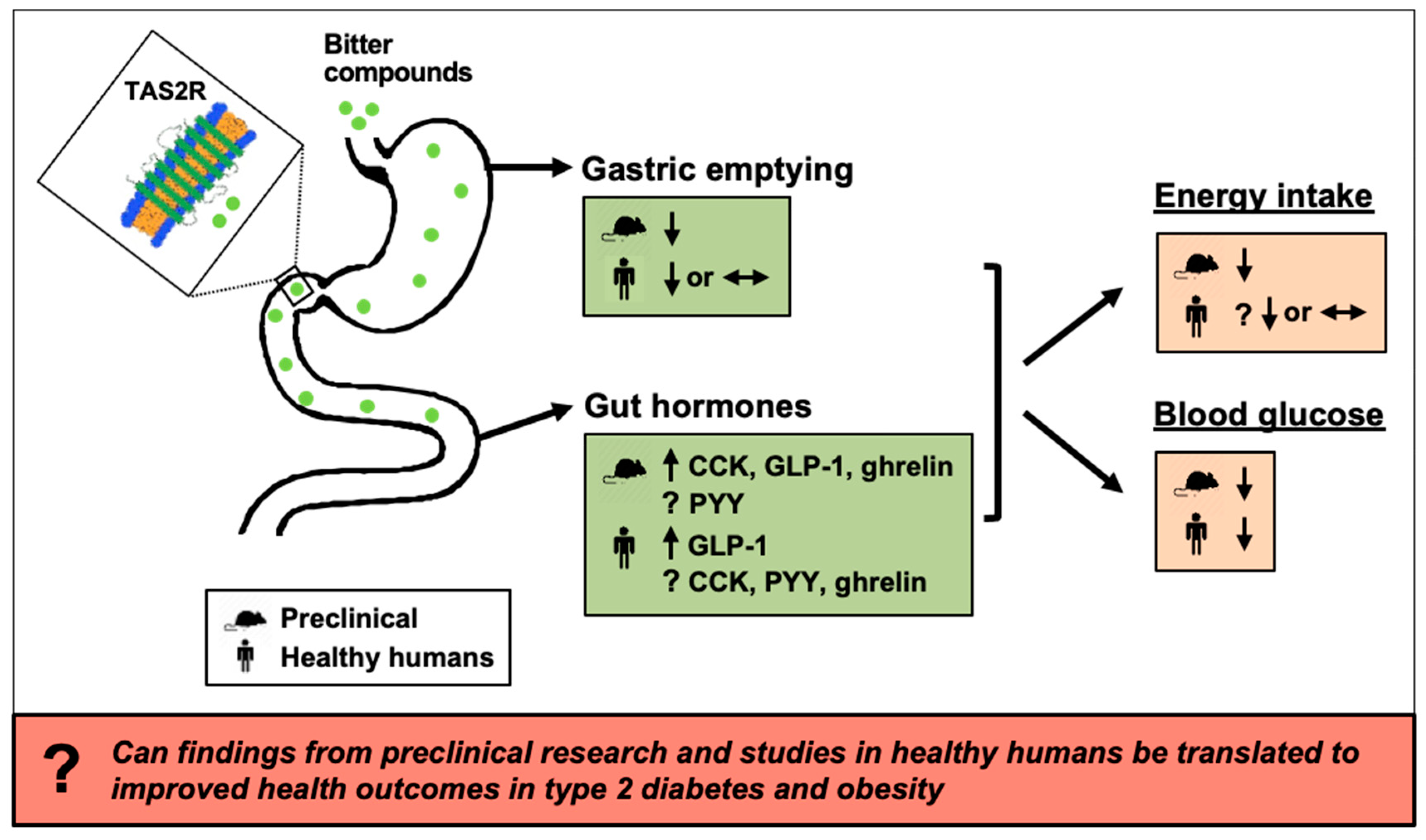
Effects of Bitter Substances on GI Function, Energy Intake and Glycaemia-Do Preclinical Findings Translate to Outcomes in Humans?




Abstract
:1. Introduction
2. Sensing of Bitter Substances in the GI Lumen
2.1. Sources of Bitter Compounds
2.2. Bitter Taste Receptors
3. Effects of Bitter Substances on Gut Hormone Release
3.1. Cholecystokinin
3.1.1. Outcomes of Preclinical Studies

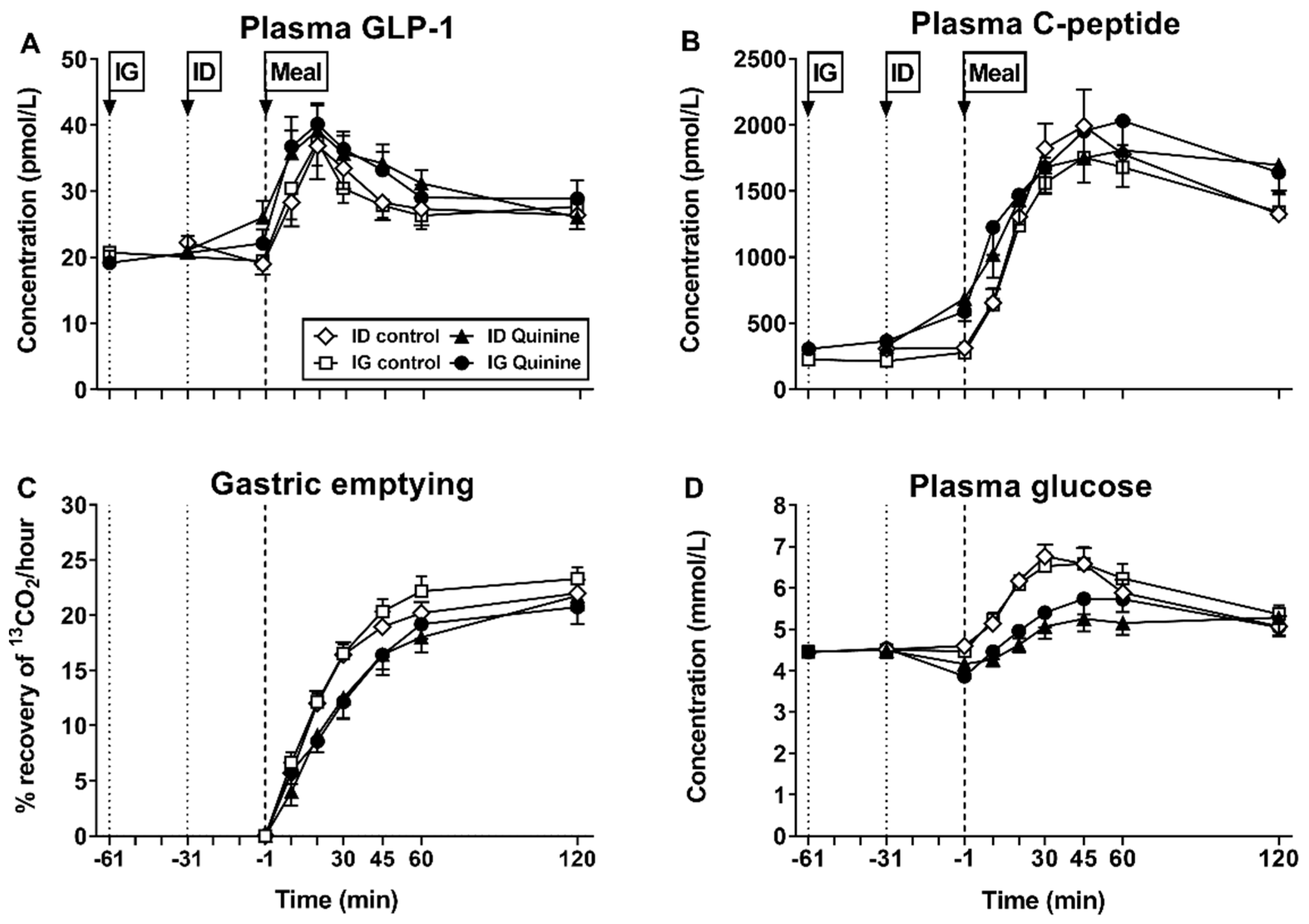{kind=link}
{kind=link}
| Bitter Tastant | Model | Doses Given/Location of Delivery | Approx. Equivalent Dose in a 70-kg Human 1 | Observed Effect | Ref # |
|---|---|---|---|---|---|
| Berberine | STC-1 cells | 1, 10, 100, 200 µM | - | ↑ GLP-1 | [80] |
| NCI-H716 cells | 1, 10, 100, 200 µM | - | ↑ GLP-1 | [81] | |
| Chloroquine | Human fundic cells | 0.3–10 mM | - | ↑ Ghrelin | [52] |
| Denatonium benzoate | STC-1 cells | 1–10 mM | - | ↑ CCK | [2] |
| NCI-H716 cells | 2, 10 mM | - | ↑ GLP-1, PYY | [3] | |
| Human fundic mucosa | 0.5, 1, 5 mM | - | ↑ Ghrelin | [52] | |
| Mice | 1 mg/kg/oral | ≈70 mg | ↑ GLP-1 | [3] | |
| Mice | 60 μmol/kg/IG | ≈1.8 g | ↑ GLP-1 | [1] | |
| Epicatechin gallate | MGN3-1 cells | 10 μM | - | ↓ Ghrelin | [82] |
| 500 μM | - | ↑ Ghrelin | |||
| Erythromycin A | Human fundic mucosa | 0.03, 0.3, 1 mmol/L | - | ↑ Ghrelin | [52] |
| Flufenamic acid | Rat ex-vivo segments: | [79] | |||
| - duodenal | 10 µM | - | ↓ CCK | ||
| - ileal | 10 µM | - | ↑ GLP-1 ↔ PYY | ||
| Gallic acid | MGN3-1 cells | 10 μM | - | ↓ Ghrelin | [82] |
| Gentiana scabra extract | NCI-H716 cells | 100–750 μg/mL | - | ↑ GLP-1 | [83] |
| Hoodia gordonii | HuTu-80 cells | 10 mM | - | ↑ CCK | [13] |
| KDT501 2 | STC-1 cells | 10 μM | - | ↑ GLP-1 | [84] |
| Mice | 150 mg/kg/oral | ≈10 g | ↑ GLP-1 | [84] | |
| Mature hop bitter acids | STC-1 cells | 50, 100, 200 μg/mL | - | ↑ CCK, GLP-1 ↔ PYY | [78] |
| Ofloxacin | NCI-H716 cells | 10, 50, 100 mM | - | ↑ GLP-1 | [85] |
| 1,10-Phenanthroline | NCI-H716 cells | 10–500 µM | - | ↑ GLP-1 | [51] |
| Human fundic mucosa | 0.1, 1 mM | - | ↑ Ghrelin | [52] | |
| Rat ex-vivo segments: | [79] | ||||
| - duodenal | 150 µM | - | ↑ CCK | ||
| - ileal | 150 µM | - | ↑ GLP-1 ↔ PYY | ||
| Phenylthiocarbamide | STC-1 cells | 2, 5, 10 mM | - | ↑ CCK | [2] |
| Caco-2 cells | 10 mM | - | ↑ CCK | [77] | |
| Human fundic cells | 0.3–10 mM | - | ↑ Ghrelin | [52] | |
| Propylthiouracil | Human fundic cells | 0.3–10 mM | - | ↑ Ghrelin | [52] |
| Mice | 200 mg/kg/IG | ≈14 g | ↑ GLP-1 | [50] | |
| Qing-Hua granules | Mice | 3.75, 7.5, 15 g/kg/d/IG | ≈263–1050 g | ↑ GLP-1 | [86] |
| Quinine hydrochloride | NCI-H716 cells | 0.5, 1, 2 mM | - | ↑ GLP-1 | [3] |
| Mice | 160 μmol/kg/IG | ≈4 g | ↔ GLP-1, ghrelin | [1] | |
| Vanillic acid | Rat ileal segments | 151.17 µM | - | ↑ GLP-1 | [79] |
| Wild bitter gourd | STC-1 cells | 100, 500, 1000 µg/mL | - | ↑ GLP-1 | [87] |
| Mice | 5 g/kg/IG | ≈350 g | ↑ GLP-1 | [87] |
3.1.2. Outcomes of Studies in Healthy Humans
| Bitter tastant | Model | Doses Given/Location of Delivery | Observed Effect | Ref # |
|---|---|---|---|---|
| AmarasateTM 1 | Males | 500 mg in acid-resistant or standard capsules/oral | ↑ CCK, GLP-1, PYY | [72] |
| Denatonium benzoate | Females | 1 μmol/kg bolus/IG [≈32 mg] 2 | ↔ Ghrelin | [19] |
| Quinine hydrochloride | Males | 10 µmol/kg bolus/IG [≈270 mg] | ↓ Ghrelin | [18] |
| Males and females | 18 mg in acid-resistant capsule/oral | ↑ CCK | [16] | |
| Males and females | 75 mg/ID over 60 min | ↔ CCK, GLP-1, PYY | [74] | |
| Females | 10 μmol/kg bolus/IG [≈270 mg] | ↓ Ghrelin | [75] | |
| Males | 37.5, 75, 225 mg/ID over 60 min | ↔ CCK | [76] | |
| Males | 275, 600 mg bolus/IG 30 min before meal | ↑ GLP-1 | [17] | |
| Males | 600 mg bolus/IG 60 min before meal, ID 30 min before meal | ↑ GLP-1 | [20] | |
| Secoiridoids 3 | Males and females | 100 mg/oral (microencapsulated) incorporated in custard | ↑ GLP-1 ↔ PYY, ghrelin | [73] |
3.2. Glucagon-Like Peptide-1
3.2.1. Outcomes of Preclinical Studies
3.2.2. Outcomes of Studies in Healthy Humans
3.3. Peptide YY
3.3.1. Outcomes of Preclinical Studies
3.3.2. Outcomes of Studies in Healthy Humans
3.4. Ghrelin
3.4.1. Outcomes of Preclinical Studies
3.4.2. Outcomes of Studies in Healthy Humans
4. Effects of Bitter Substances on Gastric Emptying and Gastrointestinal Motility
4.1. Outcomes of Preclinical Studies
| Bitter Tastants | Model | Doses Given/Location of Delivery | Approx. Equivalent Dose in a 70-kg Human 1 | Observed Effect | Ref # |
|---|---|---|---|---|---|
| Chloroquine | Mouse fundic and antral smooth-muscle strips | 10–100 μM >1 mM | − − | ↑ Phasic antral activity ↔ Tonic fundic contraction ↓ Phasic antral activity ↑ Fundic relaxation | [15] |
| Denatonium benzoate | Mouse fundic and antral smooth-muscle strips | 10–100 μM >1 mM | − − | ↑ Tonic fundic contraction and phasic antral activity ↑ Fundic relaxation ↓ Phasic antral activity | [15] |
| Mice | 60 µmol/kg/IG | ≈1.8 g | ↓ Gastric emptying ↓ Fundic and antral motility | [15] | |
| Mice | 10 mM/IG | ≈0.04 g | ↔ Gastric emptying | [14] | |
| Rats | 10 mM/IG | ≈0.04 g | ↓ Gastric emptying | [102] | |
| Guinea pigs | 0.2 nmol/mL/oral 0.1, 1 nmol/kg/IG 30 µmol/kg/IG | ≈0.003 mg ≈0.003–0.03 mg ≈0.98 g | ↑ Gastric accommodation ↓ Gastric accommodation | [101] | |
| Phenylthiocarbamide | Mouse fundic and antral smooth-muscle strips | 10 μM–10 mM | − | ↑ Fundic relaxation ↓ Antral activity | [15] |
| Mice | 30 µmol/kg/IG | ≈3.2 g | ↓ Gastric emptying ↓ Fundic and antral motility | [15] | |
| Salicin | Mouse fundic and antral smooth-muscle strips | 10 μM–10 mM | − | ↔ Fundic and antral contractility | [15] |
| Swertiamarin | Mice | 250, 500 mg/kg/oral | ≈17.5 and 35 g | ↑ Gastric emptying ↑ Small intestinal motility | [100] |
| Mixture of DB, PTC, PTU, quinine HCl, D-salicin | Mice | DB 10 mM; PTC 10 mM; PTU 5 mM; quinine 1.5 mM; D-salicin 5 mM/IG | DB ≈ 46 mg; PTC ≈ 15 mg; PTU ≈8 mg; quinine ≈ 5 mg; D-salicin ≈ 15 mg | ↓ Gastric emptying | [14] |
4.2. Outcomes of Studies in Healthy Humans
5. Effects of Bitter Substances on Energy Intake
5.1. Outcomes of Preclinical Studies
5.2. Outcomes of Studies in Healthy Humans
| Bitter Tastants | Model | Doses Given/Location of Delivery | Type of Meal or Diet | Observed Effects | Ref # |
|---|---|---|---|---|---|
| AmarasateTM 1 | Males | 500 mg in acid-resistant or standard capsules/oral | Ad libitum lunch and snack | ↓ Energy intake | [72] |
| Denatonium benzoate | Females | 1 µmol/kg bolus/IG (≈30 mg) 2 | Ad libitum meal (2330 kcal, 291 g CHO, 94 g F, 55 g Prot) | Trend for ↓ energy intake | [19] |
| Quinine hydrochloride | Males and Females | 18 mg in acid-resistant capsule/oral | Ad-libitum meal (50% CHO, 31% F, 19% Prot) | ↓ Energy intake | [16] |
| Males and Females | 75 mg/ID over 60 min | Ad libitum meal (160 kcal/100 g; 7.1 g Prot, 11 g CHO, 9.4 g F) | ↔ Energy intake | [74] | |
| Females | 10 μmol/kg bolus/IG (≈250 mg) | Ad libitum palatable chocolate milkshake | ↓ Energy intake | [18] | |
| Males | 37.5, 75, 225 mg/ID over 60 min | Ad libitum meal (2300 kcal, 52% CHO, 27% F, 21% Prot) | ↔ Energy intake | [76] | |
| Males | 275, 600 mg bolus/IG 30 min before meal | Ad libitum meal (2300 kcal, 52% CHO, 27% F, 21% Prot) | ↔ Energy intake | [17] | |
| Secoiridoids 3 | Males and females | 100 mg/oral (micro-encapsulated) incorporated in custard | Ad libitum meal (3 h later) | ↔ Energy intake | [73] |
6. Effects of Bitter Substances on Postprandial Blood Glucose
6.1. Outcomes of Preclinical Studies
| Bitter Tastants | Model | Doses Given/Location of Delivery | Approx. Equivalent Dose in a 70-kg Human | Type of Meal | Observed Effects | Ref # |
|---|---|---|---|---|---|---|
| (A) Preclinical models | ||||||
| Denatonium benzoate | Mice | 1 mg/kg/oral | ≈70 mg | OGTT (5 g glucose/kg BW) | ↓ Blood glucose | [3] |
| Gentiana scabra extract | Mice | 100, 300 mg/kg/oral | ≈7–21 g | OGTT (5 g glucose/kg BW) | ↓ Blood glucose | [83] |
| Isocohumulone 1 | Mice | 10, 100 mg/kg/oral | ≈0.7–7 g | OGTT (1 g glucose/kg BW) | ↓ Plasma glucose | [110] |
| Wild bitter gourd | Mice | High-fat diet containing 5% extract/oral | - | OGTT (2 g glucose/kg BW) | ↓ Blood glucose | [87] |
| (B) Healthy humans | ||||||
| Quinine hydrochloride | Males | 37.5, 75, 225 mg/ID over 60 min | N/A 2 | ↔ Blood glucose (fasting) | [76] | |
| Males | 275, 600 mg/IG 30 min before meal | Mixed-nutrient drink (500 kcal, 74 g CHO) | ↓ Plasma glucose | [17] | ||
| Males | 600 mg/IG 60 min before meal, ID 30 min before meal | Mixed-nutrient drink (500 kcal, 74 g CHO) | ↓ Plasma glucose | [20] | ||
| Secoiridoids 3 | Males and females | 100 mg/oral (micro-encapsulated) incorporated in custard | Custard + biscuits (314 kcal, 45.1 g CHO) | ↔ Blood glucose | [73] | |
6.2. Outcomes of Studies in Healthy Humans
7. Is There Evidence That Bitter Substances Reduce Body Weight and Improve Blood Glucose Control in Obesity and Type 2 Diabetes?
8. Summary and Future Directions
Author Contributions
Funding
Conflicts of Interest
References
- Avau, B.; Bauters, D.; Steensels, S.; Vancleef, L.; Laermans, J.; Lesuisse, J.; Buyse, J.; Lijnen, H.R.; Tack, J.; Depoortere, I. The Gustatory Signaling Pathway and Bitter Taste Receptors Affect the Development of Obesity and Adipocyte Metabolism in Mice. PLoS ONE 2015, 10, e0145538. [Google Scholar] [CrossRef] [Green Version]
- Chen, M.C.; Wu, S.V.; Reeve, J.R.; Rozengurt, E. Bitter Stimuli Induce Ca2+ Signaling and CCK Release in Enteroendocrine STC-1 Cells: Role of L-Type Voltage-Sensitive Ca2+ Channels. Am. J. Physiology-Cell Physiol. 2006, 291, C726–C739. [Google Scholar] [CrossRef] [Green Version]
- Kim, K.-S.; Egan, J.M.; Jang, H.-J. Denatonium Induces Secretion of Glucagon-Like Peptide-1 through Activation of Bitter Taste Receptor Pathways. Diabetologia 2014, 57, 2117–2125. [Google Scholar] [CrossRef] [Green Version]
- Marathe, C.S.; Rayner, C.K.; Jones, K.L.; Horowitz, M. Relationships between Gastric Emptying, Postprandial Glycemia, and Incretin Hormones. Diabetes Care 2013, 36, 1396–1405. [Google Scholar] [CrossRef] [Green Version]
- Steinert, R.E.; Feinle-Bisset, C.; Asarian, L.; Horowitz, M.; Beglinger, C.; Geary, N. Ghrelin, CCK, GLP-1, and PYY(3–36): Secretory Controls and Physiological Roles in Eating and Glycemia in Health, Obesity, and After RYGB. Physiol. Rev. 2017, 97, 411–463. [Google Scholar] [CrossRef] [Green Version]
- Murphy, K.G.; Bloom, S.R. Gut Hormones and the Regulation of Energy Homeostasis. Nat. Cell Biol. 2006, 444, 854–859. [Google Scholar] [CrossRef] [PubMed]
- Seimon, R.V.; Lange, K.; Little, T.J.; Brennan, I.M.; Pilichiewicz, A.N.; Feltrin, K.L.; Smeets, A.J.; Horowitz, M.; Feinle-Bisset, C. Pooled-Data Analysis Identifies Pyloric Pressures and Plasma Cholecystokinin Concentrations as Major Determinants of Acute Energy Intake in Healthy, Lean Men. Am. J. Clin. Nutr. 2010, 92, 61–68. [Google Scholar] [CrossRef] [PubMed]
- Unick, J.L.; Beavers, D.; Bond, D.S.; Clark, J.M.; Jakicic, J.M.; Kitabchi, A.E.; Knowler, W.C.; Wadden, T.A.; Wagenknecht, L.E.; Wing, R.R.; et al. The Long-Term Effectiveness of a Lifestyle Intervention in Severely Obese Individuals. Am. J. Med. 2013, 126, 236–242. [Google Scholar] [CrossRef] [Green Version]
- Bhat, S.P.; Sharma, A. Current Drug Targets in Obesity Pharmacotherapy-A Review. Curr. Drug Targets 2017, 18, 1. [Google Scholar] [CrossRef] [PubMed]
- Drucker, D.J.; Nauck, M.A. The Incretin System: Glucagon-Like Peptide-1 Receptor Agonists and Dipeptidyl Peptidase-4 Inhibitors in Type 2 Diabetes. Lancet 2006, 368, 1696–1705. [Google Scholar] [CrossRef]
- Müller, T.; Finan, B.; Bloom, S.; D’Alessio, D.; Drucker, D.; Flatt, P.; Fritsche, A.; Gribble, F.; Grill, H.; Habener, J.; et al. Glucagon-Like Peptide 1 (GLP-1). Mol. Metab. 2019, 30, 72–130. [Google Scholar] [CrossRef] [PubMed]
- Deacon, C.F.; Mannucci, E.; Ahrén, B. Glycaemic Efficacy of Glucagon-Like Peptide-1 Receptor Agonists and Dipeptidyl Peptidase-4 Inhibitors as Add-on Therapy to Metformin in Subjects with Type 2 Diabetes-a Review and Meta Analysis. Diabetes Obes. Metab. 2012, 14, 762–767. [Google Scholar] [CrossRef] [PubMed]
- Le Nevé, B.; Foltz, M.; Daniel, H.; Gouka, R. The Steroid Glycoside H.g.-12 from Hoodia Gordonii Activates the Human Bitter Receptor TAS2R14 and Induces CCK Release from HuTu-80 cells. Am. J. Physiol. Liver Physiol. 2010, 299, G1368–G1375. [Google Scholar] [CrossRef]
- Janssen, S.; Laermans, J.; Verhulst, P.-J.; Thijs, T.; Tack, J.; Depoortere, I. Bitter Taste Receptors and α-Gustducin Regulate the Secretion of Ghrelin with Functional Effects on Food Intake and Gastric Emptying. Proc. Natl. Acad. Sci. USA 2011, 108, 2094–2099. [Google Scholar] [CrossRef] [Green Version]
- Avau, B.; Rotondo, A.; Thijs, T.; Andrews, C.N.; Janssen, P.; Tack, J.; Depoortere, I. Targeting Extra-Oral Bitter Taste Receptors Modulates Gastrointestinal Motility with Effects on Satiation. Sci. Rep. 2015, 5, 15985. [Google Scholar] [CrossRef] [PubMed]
- Andreozzi, P.; Sarnelli, G.; Pesce, M.; Zito, F.P.; Alessandro, A.D.; Verlezza, V.; Palumbo, I.; Turco, F.; Esposito, K.; Cuomo, R. The Bitter Taste Receptor Agonist Quinine Reduces Calorie Intake and Increases the Postprandial Release of Cholecystokinin in Healthy Subjects. J. Neurogastroenterol. Motil. 2015, 21, 511–519. [Google Scholar] [CrossRef] [Green Version]
- Bitarafan, V.; Fitzgerald, P.C.E.; Little, T.J.; Meyerhof, W.; Jones, K.L.; Wu, T.; Horowitz, M.; Feinle-Bisset, C. Intragastric Administration of the Bitter Tastant Quinine Lowers the Glycemic Response to a Nutrient Drink without Slowing Gastric Emptying in Healthy Men. Am. J. Physiol. Integr. Comp. Physiol. 2020, 318, R263–R273. [Google Scholar] [CrossRef] [PubMed]
- Iven, J.; Biesiekierski, J.R.; Zhao, D.; Deloose, E.; O’Daly, O.G.; Depoortere, I.; Tack, J.; Van Oudenhove, L. Intragastric Quinine Administration decreases Hedonic Eating in Healthy Women through Peptide-Mediated Gut-Brain Signaling Mechanisms. Nutr. Neurosci. 2018, 22, 850–862. [Google Scholar] [CrossRef] [PubMed]
- Deloose, E.; Janssen, P.; Corsetti, M.; Biesiekierski, J.; Masuy, I.; Rotondo, A.; Van Oudenhove, L.; Depoortere, I.; Tack, J. Intragastric Infusion of Denatonium Benzoate Attenuates Interdigestive Gastric Motility and Hunger Scores in Healthy Female Volunteers. Am. J. Clin. Nutr. 2017, 105, 580–588. [Google Scholar] [CrossRef] [Green Version]
- Rose, B.D.; Bitarafan, V.; Rezaie, P.; Fitzgerald, P.C.E.; Horowitz, M.; Feinle-Bisset, C. Comparative Effects of Intragastric and Intraduodenal Administration of Quinine on the Plasma Glucose Response to a Mixed-Nutrient Drink in Healthy Men: Relations with Glucoregulatory Hormones and Gastric Emptying. J. Nutr. 2021. [Google Scholar] [CrossRef]
- Bachmanov, A.A.; Beauchamp, G.K. Taste Receptor Genes. Annu. Rev. Nutr. 2007, 27, 389–414. [Google Scholar] [CrossRef] [Green Version]
- Depoortere, I. Taste Receptors of the Gut: Emerging Roles in Health and Disease. Gut 2014, 63, 179–190. [Google Scholar] [CrossRef] [PubMed]
- Lu, P.; Zhang, C.-H.; Lifshitz, L.M.; Zhuge, R. Extraoral Bitter Taste Receptors in Health and Disease. J. Gen. Physiol. 2017, 149, 181–197. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Adler, E.; Hoon, M.A.; Mueller, K.L.; Chandrashekar, J.; Ryba, N.J.; Zuker, C.S. A Novel Family of Mammalian Taste Receptors. Cell 2000, 100, 693–702. [Google Scholar] [CrossRef] [Green Version]
- Wu, S.V.; Rozengurt, N.; Yang, M.; Young, S.H.; Sinnett-Smith, J.; Rozengurt, E. Expression of Bitter Taste Receptors of the T2R Family in the Gastrointestinal Tract and Enteroendocrine STC-1 cells. Proc. Natl. Acad. Sci. USA 2002, 99, 2392–2397. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Behrens, M.; Meyerhof, W. Oral and Extraoral Bitter Taste Receptors; Meyerhof, W., Beisiegel, U., Joost, H.-G., Eds.; Springer: Berlin, Heidelberg, 2010; pp. 87–99. [Google Scholar]
- Drewnowski, A.; Gomez-Carneros, C. Bitter Taste, Phytonutrients, and the Consumer: A Review. Am. J. Clin. Nutr. 2000, 72, 1424–1435. [Google Scholar] [CrossRef] [PubMed]
- Maehashi, K.; Huang, L. Bitter Peptides and Bitter Taste Receptors. Cell. Mol. Life Sci. 2009, 66, 1661–1671. [Google Scholar] [CrossRef] [PubMed]
- Hofmann, T. Taste-Active Maillard Reaction Products: The “Tasty” World of Nonvolatile Maillard Reaction Products. Ann. N. Y. Acad. Sci. 2005, 1043, 20–29. [Google Scholar] [CrossRef]
- Dubois, G.; DeSimone, J.; Lyall, V. Chemistry of Gustatory Stimuli; Elsevier BV: Amsterdam, The Netherlands, 2020; pp. 24–64. [Google Scholar]
- Belitz, H.; Wieser, H. Bitter Compounds: Occurrence and Structure-Activity Relationships. Food Rev. Int. 1985, 1, 271–354. [Google Scholar] [CrossRef]
- Duffy, V.B.; Davidson, A.C.; Kidd, J.R.; Kidd, K.K.; Speed, W.C.; Pakstis, A.J.; Reed, D.R.; Snyder, D.J.; Bartoshuk, L.M. Bitter Receptor Gene (TAS2R38), 6-n-Propylthiouracil (PROP) Bitterness and Alcohol Intake. Alcohol. Clin. Exp. Res. 2004, 28, 1629–1637. [Google Scholar] [CrossRef] [Green Version]
- Keller, K.L.; Adise, S. Variation in the Ability to Taste Bitter Thiourea Compounds: Implications for Food Acceptance, Dietary Intake, and Obesity Risk in Children. Annu. Rev. Nutr. 2016, 36, 157–182. [Google Scholar] [CrossRef] [PubMed]
- Meyerhof, W.; Batram, C.; Kuhn, C.; Brockhoff, A.; Chudoba, E.; Bufe, B.; Appendino, G.; Behrens, M. The Molecular Receptive Ranges of Human TAS2R Bitter Taste Receptors. Chem. Senses 2010, 35, 157–170. [Google Scholar] [CrossRef]
- Li, F. Taste Perception: From the Tongue to the Testis. Mol. Hum. Reprod. 2013, 19, 349–360. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shi, P.; Zhang, J. Extraordinary Diversity of Chemosensory Receptor Gene Repertoires among Vertebrates. In Chemistry and Biology of Pteridines and Folates; Springer: Köln, Germany, 2009; Volume 47, pp. 57–75. [Google Scholar] [CrossRef] [Green Version]
- Kim, U.; Wooding, S.; Ricci, D.; Jorde, L.B.; Drayna, D. Worldwide Haplotype Diversity and Coding Sequence Variation at Human Bitter Taste Receptor Loci. Hum. Mutat. 2005, 26, 199–204. [Google Scholar] [CrossRef]
- Bufe, B.; Breslin, P.A.; Kuhn, C.; Reed, D.R.; Tharp, C.D.; Slack, J.P.; Kim, U.-K.; Drayna, D.; Meyerhof, W. The Molecular Basis of Individual Differences in Phenylthiocarbamide and Propylthiouracil Bitterness Perception. Curr. Biol. 2005, 15, 322–327. [Google Scholar] [CrossRef] [Green Version]
- Kim, U.-K.; Jorgenson, E.; Coon, H.; Leppert, M.; Risch, N.; Drayna, D. Positional Cloning of the Human Quantitative Trait Locus Underlying Taste Sensitivity to Phenylthiocarbamide. Science 2003, 299, 1221–1225. [Google Scholar] [CrossRef]
- Cvijanovic, N.; Feinle-Bisset, C.; Young, R.L.; Little, T.J. Oral and Intestinal Sweet and Fat Tasting: Impact of Receptor Polymorphisms and Dietary Modulation for Metabolic Disease. Nutr. Rev. 2015, 73, 318–334. [Google Scholar] [CrossRef] [PubMed]
- Hajishafiee, M.; Bitarafan, V.; Feinle-Bisset, C. Gastrointestinal Sensing of Meal-Related Signals in Humans, and Dysregulations in Eating-Related Disorders. Nutrients 2019, 11, 1298. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Latorre, R.; Sternini, C.; de Giorgio, R.; Meerveld, B.G.-V. Enteroendocrine Cells: A Review of their Role in Brain-Gut Communication. Neurogastroenterol. Motil. 2016, 28, 620–630. [Google Scholar] [CrossRef] [Green Version]
- Psichas, A.; Reimann, F.; Gribble, F.M. Gut Chemosensing Mechanisms. J. Clin. Investig. 2015, 125, 908–917. [Google Scholar] [CrossRef] [Green Version]
- Rasoamanana, R.; Darcel, N.; Fromentin, G.; Tomé, D. Nutrient Sensing and Signalling by the Gut. Proc. Nutr. Soc. 2012, 71, 446–455. [Google Scholar] [CrossRef] [Green Version]
- Vella, A.; Camilleri, M. The Gastrointestinal Tract as an Integrator of Mechanical and Hormonal Response to Nutrient Ingestion. Diabetes 2017, 66, 2729–2737. [Google Scholar] [CrossRef] [Green Version]
- Symonds, E.L.; Peiris, M.; Page, A.J.; Chia, B.; Dogra, H.; Masding, A.; Galanakis, V.; Atiba, M.; Bulmer, D.; Young, R.L.; et al. Mechanisms of Activation of Mouse and Human Enteroendocrine Cells by Nutrients. Gut 2015, 64, 618–626. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gribble, F.M.; Reimann, F. Function and Mechanisms of Enteroendocrine Cells and Gut Hormones in Metabolism. Nat. Rev. Endocrinol. 2019, 15, 226–237. [Google Scholar] [CrossRef] [PubMed]
- Sternini, C.; Anselmi, L.; Rozengurt, E. Enteroendocrine Cells: A Site of ‘Taste’ in Gastrointestinal Chemosensing. Curr. Opin. Endocrinol. Diabetes Obes. 2008, 15, 73–78. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Imai, H.; Hakukawa, M.; Hayashi, M.; Iwatsuki, K.; Masuda, K. Expression of Bitter Taste Receptors in the Intestinal Cells of Non-Human Primates. Int. J. Mol. Sci. 2020, 21, 902. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pham, H.; Hui, H.; Morvaridi, S.; Cai, J.; Zhang, S.; Tan, J.; Wu, V.; Levin, N.; Knudsen, B.; Goddard, W.A.; et al. A Bitter Pill for Type 2 Diabetes? The Activation of Bitter Taste Receptor TAS2R38 can Stimulate GLP-1 Release from Enteroendocrine L-Cells. Biochem. Biophys. Res. Commun. 2016, 475, 295–300. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Park, J.; Kim, K.-S.; Kim, K.-H.; Lee, I.-S.; Jeong, H.-S.; Kim, Y.; Jang, H.-J. GLP-1 Secretion is Stimulated by 1,10-Phenanthroline via Colocalized T2R5 Signal Transduction in Human Enteroendocrine L Cell. Biochem. Biophys. Res. Commun. 2015, 468, 306–311. [Google Scholar] [CrossRef]
- Wang, Q.; Liszt, K.I.; Deloose, E.; Canovai, E.; Thijs, T.; Farré, R.; Ceulemans, L.J.; Lannoo, M.; Tack, J.; Depoortere, I. Obesity Alters Adrenergic and Chemosensory Signaling Pathways that Regulate Ghrelin Secretion in the Human Gut. FASEB J. 2019, 33, 4907–4920. [Google Scholar] [CrossRef]
- Holst, J.J.; Gribble, F.; Horowitz, M.; Rayner, C.K. Roles of the Gut in Glucose Homeostasis. Diabetes Care 2016, 39, 884–892. [Google Scholar] [CrossRef] [Green Version]
- Suzuki, K.; Jayasena, C.N.; Bloom, S.R. The Gut Hormones in Appetite Regulation. J. Obes. 2011, 2011, 1–10. [Google Scholar] [CrossRef] [Green Version]
- Deloose, E.; Verbeure, W.; Depoortere, I.; Tack, J. Motilin: From Gastric Motility Stimulation to Hunger Signalling. Nat. Rev. Endocrinol. 2019, 15, 238–250. [Google Scholar] [CrossRef]
- Cummings, D.E.; Overduin, J. Gastrointestinal Regulation of Food Intake. J. Clin. Investig. 2007, 117, 13–23. [Google Scholar] [CrossRef] [PubMed]
- Cummings, D.E. Roles for Ghrelin in the Regulation of Appetite and Body Weight. Arch. Surg. 2003, 138, 389–396. [Google Scholar] [CrossRef] [Green Version]
- Field, B.C.T.; Chaudhri, O.B.; Bloom, S.R. Bowels Control Brain: Gut Hormones and Obesity. Nat. Rev. Endocrinol. 2010, 6, 444–453. [Google Scholar] [CrossRef]
- Abbott, C.R.; Small, C.J.; Kennedy, A.R.; Neary, N.M.; Sajedi, A.; Ghatei, M.A.; Bloom, S.R. Blockade of the Neuropeptide Y Y2 Receptor with the Specific Antagonist BIIE0246 Attenuates the Effect of Endogenous and Exogenous Peptide YY(3–36) on Food Intake. Brain Res. 2005, 1043, 139–144. [Google Scholar] [CrossRef]
- Beglinger, C.; Degen, L.; Matzinger, D.; D’Amato, M.; Drewe, J. Loxiglumide, a CCK-A Receptor Antagonist, Stimulates Calorie Intake and Hunger Feelings in Humans. Am. J. Physiol. Integr. Comp. Physiol. 2001, 280, R1149–R1154. [Google Scholar] [CrossRef]
- Steinert, R.E.; Schirra, J.; Meyer-Gerspach, A.C.; Kienle, P.; Fischer, H.; Schulte, F.; Goeke, B.; Beglinger, C. Effect of Glucagon-Like Peptide-1 Receptor Antagonism on Appetite and Food Intake in Healthy Men. Am. J. Clin. Nutr. 2014, 100, 514–523. [Google Scholar] [CrossRef] [Green Version]
- Degen, L.; Oesch, S.; Casanova, M.; Graf, S.; Ketterer, S.; Drewe, J.; Beglinger, C. Effect of Peptide YY3–36 on Food Intake in Humans. Gastroenterology 2005, 129, 1430–1436. [Google Scholar] [CrossRef]
- Gutzwiller, J.-P.; Göke, B.; Drewe, J.; Hildebrand, P.; Ketterer, S.; Handschin, D.; Winterhalder, R.; Conen, D.; Beglinger, C. Glucagon-Like Peptide-1: A Potent Regulator of Food Intake in Humans. Gut 1999, 44, 81–86. [Google Scholar] [CrossRef]
- MacIntosh, C.G.; Morley, J.E.; Wishart, J.; Morris, H.; Jansen, J.B.M.J.; Horowitz, M.; Chapman, I.M. Effect of Exogenous Cholecystokinin (CCK)-8 on Food Intake and Plasma CCK, Leptin, and Insulin Concentrations in Older and Young Adults: Evidence for Increased CCK Activity as a Cause of the Anorexia of Aging. J. Clin. Endocrinol. Metab. 2001, 86, 5830–5837. [Google Scholar] [CrossRef]
- De Lartigue, G.; Diepenbroek, C. Novel Developments in Vagal Afferent Nutrient Sensing and its Role in Energy Homeostasis. Curr. Opin. Pharmacol. 2016, 31, 38–43. [Google Scholar] [CrossRef] [Green Version]
- Dockray, G.J. Enteroendocrine Cell Signalling via the Vagus Nerve. Curr. Opin. Pharmacol. 2013, 13, 954–958. [Google Scholar] [CrossRef]
- Meloni, A.R.; Deyoung, M.B.; Lowe, C.; Parkes, D.G. GLP-1 Receptor Activated Insulin Secretion from Pancreatic β-Cells: Mechanism and Glucose Dependence. Diabetes Obes. Metab. 2012, 15, 15–27. [Google Scholar] [CrossRef] [Green Version]
- Holst, J.J. The Physiology of Glucagon-like Peptide 1. Physiol. Rev. 2007, 87, 1409–1439. [Google Scholar] [CrossRef]
- Kim, W.; Egan, J.M. The Role of Incretins in Glucose Homeostasis and Diabetes Treatment. Pharmacol. Rev. 2008, 60, 470–512. [Google Scholar] [CrossRef] [Green Version]
- Margolskee, R.F. Molecular Mechanisms of Bitter and Sweet Taste Transduction. J. Biol. Chem. 2002, 277, 1–4. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xie, C.; Wang, X.; Young, R.L.; Horowitz, M.; Rayner, C.K.; Wu, T. Role of Intestinal Bitter Sensing in Enteroendocrine Hormone Secretion and Metabolic Control. Front. Endocrinol. 2018, 9, 576. [Google Scholar] [CrossRef]
- Ingram, J.R.; Walker, E.G.; Pahl, M.C.; Lo, K.R.; Shin, H.S.; Lang, C.; Wohlers, M.W.; Poppitt, S.; Sutton, K.H. Activation of Gastrointestinal Bitter Taste Receptors Suppresses Food Intake and Stimulates Secretion of Gastrointestinal Peptide Hormones in Healthy Men. Obes. Facts 2016, 9 (Suppl. 1), 46. [Google Scholar]
- Mennella, I.; Fogliano, V.; Ferracane, R.; Arlorio, M.; Pattarino, F.; Vitaglione, P. Microencapsulated Bitter Compounds (from Gentiana Lutea) Reduce Daily Energy Intakes in Humans. Br. J. Nutr. 2016, 116, 1841–1850. [Google Scholar] [CrossRef] [Green Version]
- Van Avesaat, M.; Troost, F.J.; Ripken, D.; Peters, J.; Hendriks, H.F.; Masclee, A.A. Intraduodenal Infusion of a Combination of Tastants Decreases Food Intake in Humans. Am. J. Clin. Nutr. 2015, 102, 729–735. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Deloose, E.; Corsetti, M.; Van Oudenhove, L.; Depoortere, I.; Tack, J. Intragastric Infusion of the Bitter Tastant Quinine Suppresses Hormone Release and Antral Motility during the Fasting State in Healthy Female Volunteers. Neurogastroenterol. Motil. 2017, 30, e13171. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bitarafan, V.E.; Fitzgerald, P.C.; Little, T.J.; Meyerhof, W.; Wu, T.; Horowitz, M.; Feinle-Bisset, C. Effects of Intraduodenal Infusion of the Bitter Tastant, Quinine, on Antropyloroduodenal Motility, Plasma Cholecystokinin, and Energy Intake in Healthy Men. J. Neurogastroenterol. Motil. 2019, 25, 413–422. [Google Scholar] [CrossRef] [PubMed]
- Jeon, T.-I.; Seo, Y.-K.; Osborne, T.F. Gut Bitter Taste Receptor Signalling induces ABCB1 through a Mechanism Involving CCK. Biochem. J. 2011, 438, 33–37. [Google Scholar] [CrossRef]
- Yamazaki, T.; Morimoto-Kobayashi, Y.; Koizumi, K.; Takahashi, C.; Nakajima, S.; Kitao, S.; Taniguchi, Y.; Katayama, M.; Ogawa, Y. Secretion of a Gastrointestinal Hormone, Cholecystokinin, by Hop-Derived Bitter Components Activates Sympathetic Nerves in Brown Adipose Tissue. J. Nutr. Biochem. 2019, 64, 80–87. [Google Scholar] [CrossRef]
- Grau-Bové, C.; Miguéns-Gómez, A.; González-Quilen, C.; Fernández-López, J.-A.; Remesar, X.; Torres-Fuentes, C.; Ávila-Román, J.; Rodríguez-Gallego, E.; Beltrán-Debón, R.; Blay, M.T.; et al. Modulation of Food Intake by Differential TAS2R Stimulation in Rat. Nutrients 2020, 12, 3784. [Google Scholar] [CrossRef]
- Yue, X.; Liang, J.; Gu, F.; Du, D.; Chen, F. Berberine Activates Bitter Taste Responses of Enteroendocrine STC-1 Cells. Mol. Cell. Biochem. 2018, 447, 21–32. [Google Scholar] [CrossRef]
- Yu, Y.; Hao, G.; Zhang, Q.; Hua, W.; Wang, M.; Zhou, W.; Zong, S.; Huang, M.; Wen, X. Berberine Induces GLP-1 Secretion through Activation of Bitter Taste Receptor Pathways. Biochem. Pharmacol. 2015, 97, 173–177. [Google Scholar] [CrossRef]
- Serrano, J.; Casanova-Martí, À.; Depoortere, I.; Blay, M.T.; Terra, X.; Pinent, M.; Ardévol, A. Subchronic Treatment with Grape-Seed Phenolics Inhibits Ghrelin Production despite a Short-Term Stimulation of Ghrelin Secretion Produced by Bitter-Sensing Flavanols. Mol. Nutr. Food Res. 2016, 60, 2554–2564. [Google Scholar] [CrossRef]
- Suh, H.-W.; Lee, K.-B.; Kim, K.-S.; Yang, H.J.; Choi, E.-K.; Shin, M.H.; Park, Y.S.; Na, Y.-C.; Ahn, K.S.; Jang, Y.P.; et al. A Bitter Herbal Medicine Gentiana Scabra Root Extract Stimulates Glucagon-Like Peptide-1 Secretion and Regulates Blood Glucose in db/db Mouse. J. Ethnopharmacol. 2015, 172, 219–226. [Google Scholar] [CrossRef]
- Kok, B.P.; Galmozzi, A.; Littlejohn, N.K.; Albert, V.; Godio, C.; Kim, W.; Kim, S.M.; Bland, J.S.; Grayson, N.; Fang, M.; et al. Intestinal Bitter Taste Receptor Activation Alters Hormone Secretion and Imparts Metabolic Benefits. Mol. Metab. 2018, 16, 76–87. [Google Scholar] [CrossRef] [PubMed]
- Dotson, C.D.; Zhang, L.; Xu, H.; Shin, Y.-K.; Vigues, S.; Ott, S.H.; Elson, A.E.T.; Choi, H.J.; Shaw, H.; Egan, J.M.; et al. Bitter Taste Receptors Influence Glucose Homeostasis. PLoS ONE 2008, 3, e3974. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, J.; Xu, J.; Hou, R.; Jin, X.; Wang, J.; Yang, N.; Yang, L.; Liu, L.; Tao, F.; Lu, H. Qing-Hua Granule induces GLP-1 Secretion via Bitter Taste Receptor in db/db Mice. Biomed. Pharmacother. 2017, 89, 10–17. [Google Scholar] [CrossRef] [PubMed]
- Huang, T.-N.; Lu, K.-N.; Pai, Y.-P.; Hsu, C.; Huang, C.-J. Role of GLP-1 in the Hypoglycemic Effects of Wild Bitter Gourd. Evidence-Based Complement. Altern. Med. 2013, 2013, 1–13. [Google Scholar] [CrossRef] [Green Version]
- Habib, A.M.; Richards, P.; Rogers, G.J.; Reimann, F.; Gribble, F.M. Co-Localisation and Secretion of Glucagon-Like Peptide 1 and Peptide YY from Primary Cultured Human L Cells. Diabetologia 2013, 56, 1413–1416. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lossow, K.; Hübner, S.; Roudnitzky, N.; Slack, J.P.; Pollastro, F.; Behrens, M.; Meyerhof, W. Comprehensive Analysis of Mouse Bitter Taste Receptors Reveals Different Molecular Receptive Ranges for Orthologous Receptors in Mice and Humans. J. Biol. Chem. 2016, 291, 15358–15377. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Azpiroz, F.; Malagelada, J.R. Intestinal Control of Gastric Tone. Am. J. Physiol. Liver Physiol. 1985, 249, G501–G509. [Google Scholar] [CrossRef] [PubMed]
- Houghton, L.; Read, N.; Heddle, R.; Horowitz, M.; Collins, P.; Chatterton, B.; Dent, J. Relationship of the Motor Activity of the Antrum, Pylorus, and Duodenum to Gastric Emptying of a Solid-Liquid Mixed Meal. Gastroenterology 1988, 94, 1285–1291. [Google Scholar] [CrossRef]
- Brookes, S.J.; Spencer, N.J.; Costa, M.; Zagorodnyuk, V.P. Extrinsic Primary Afferent Signalling in the Gut. Nat. Rev. Gastroenterol. Hepatol. 2013, 10, 286–296. [Google Scholar] [CrossRef] [Green Version]
- Feinle, C.; Grundy, D.; Read, N.W. Effects of Duodenal Nutrients on Sensory and Motor Responses of the Human Stomach to Distension. Am. J. Physiol. Content 1997, 273, G721–G726. [Google Scholar] [CrossRef]
- Kissileff, H.R.; Carretta, J.C.; Geliebter, A.; Pi-Sunyer, F.X. Cholecystokinin and Stomach Distension Combine to Reduce Food Intake in Humans. Am. J. Physiol. Integr. Comp. Physiol. 2003, 285, R992–R998. [Google Scholar] [CrossRef] [PubMed]
- Sturm, K.; Parker, B.; Wishart, J.; Feinle-Bisset, C.; Jones, K.L.; Chapman, I.; Horowitz, M. Energy Intake and Appetite are Related to Antral Area in Healthy Young and Older Subjects. Am. J. Clin. Nutr. 2004, 80, 656–667. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Horowitz, M.; Edelbroek, M.A.L.; Wishart, J.M.; Straathof, J.W. Relationship between Oral Glucose Tolerance and Gastric Emptying in Normal Healthy Subjects. Diabetologia 1993, 36, 857–862. [Google Scholar] [CrossRef] [PubMed]
- Jones, K.L.; Horowitz, M.I.; Carney, B.; Wishart, J.M.; Guha, S.; Green, L. Gastric Emptying in Early Noninsulin-Dependent Diabetes Mellitus. J. Nucl. Med. 1996, 37, 1643–1648. [Google Scholar]
- Little, T.J.; Pilichiewicz, A.N.; Russo, A.; Phillips, L.; Jones, K.L.; Nauck, M.A.; Wishart, J.; Horowitz, M.; Feinle-Bisset, C. Effects of Intravenous Glucagon-Like Peptide-1 on Gastric Emptying and Intragastric Distribution in Healthy Subjects: Relationships with Postprandial Glycemic and Insulinemic Responses. J. Clin. Endocrinol. Metab. 2006, 91, 1916–1923. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nauck, M.A.; Niedereichholz, U.; Ettler, R.; Holst, J.J.; Ørskov, C.; Ritzel, R.; Schmiegel, W.H. Glucagon-Like Peptide 1 Inhibition of Gastric Emptying Outweighs its Insulinotropic Effects in Healthy Humans. Am. J. Physiol. Metab. 1997, 273, E981–E988. [Google Scholar] [CrossRef] [PubMed]
- Kimura, Y.; Sumiyoshi, M. Effects of Swertia Japonica Extract and its Main Compound Swertiamarin on Gastric Emptying and Gastrointestinal Motility in Mice. Fitoterapia 2011, 82, 827–833. [Google Scholar] [CrossRef]
- Harada, Y.; Koseki, J.; Sekine, H.; Fujitsuka, N.; Kobayashi, H. Role of Bitter Taste Receptors in Regulating Gastric Accommodation in Guinea Pigs. J. Pharmacol. Exp. Ther. 2019, 369, 466–472. [Google Scholar] [CrossRef]
- Glendinning, J.I.; Yiin, Y.-M.; Ackroff, K.; Sclafani, A. Intragastric Infusion of Denatonium Conditions Flavor Aversions and Delays Gastric Emptying in Rodents. Physiol. Behav. 2008, 93, 757–765. [Google Scholar] [CrossRef] [Green Version]
- Little, T.J.; Gupta, N.; Case, R.M.; Thompson, D.G.; McLaughlin, J.T. Sweetness and Bitterness Taste of Meals Per se does not Mediate Gastric Emptying in Humans. Am. J. Physiol. Integr. Comp. Physiol. 2009, 297, R632–R639. [Google Scholar] [CrossRef]
- Wicks, D.; Wright, J.; Rayment, P.; Spiller, R. Impact of Bitter Taste on Gastric Motility. Eur. J. Gastroenterol. Hepatol. 2005, 17, 961–965. [Google Scholar] [CrossRef]
- Sumiyoshi, M.; Kimura, Y. Hop (Humulus Lupulus L.) Extract Inhibits Obesity in Mice Fed a High-Fat Diet over the Long Term. Br. J. Nutr. 2013, 109, 162–172. [Google Scholar] [CrossRef] [Green Version]
- Yajima, H.; Noguchi, T.; Ikeshima, E.; Shiraki, M.; Kanaya, T.; Tsuboyama-Kasaoka, N.; Ezaki, O.; Oikawa, S.; Kondo, K. Prevention of Diet-Induced Obesity by Dietary Isomerized Hop Extract Containing Isohumulones, in Rodents. Int. J. Obes. 2005, 29, 991–997. [Google Scholar] [CrossRef] [Green Version]
- Van Heerden, F.R.; Horak, R.M.; Maharaj, V.J.; Vleggaar, R.; Senabe, J.V.; Gunning, P.J. An Appetite Suppressant from Hoodia Species. Phytochemistry 2007, 68, 2545–2553. [Google Scholar] [CrossRef]
- Leng, S.-H.; Lu, F.-E.; Xu, L.-J. Therapeutic Effects of Berberine in Impaired Glucose Tolerance Rats and its Influence on Insulin Secretion. Acta Pharmacol. Sin. 2004, 25, 496–502. [Google Scholar] [PubMed]
- Kratz, C.M.; Levitsky, D.; Lustick, S.L. Long Term Effects of Quinine on Food Intake and Body Weight in the Rat. Physiol. Behav. 1978, 21, 321–324. [Google Scholar] [CrossRef]
- Yajima, H.; Ikeshima, E.; Shiraki, M.; Kanaya, T.; Fujiwara, D.; Odai, H.; Tsuboyama-Kasaoka, N.; Ezaki, O.; Oikawa, S.; Kondo, K. Isohumulones, Bitter Acids Derived from Hops, Activate Both Peroxisome Proliferator-Activated Receptor α and γ and Reduce Insulin Resistance. J. Biol. Chem. 2004, 279, 33456–33462. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nauck, M.A.; Kleine, N.; Holst, J.J.; Willms, B.; Creutzfeldt, W. Normalization of Fasting Hyperglycaemia by Exogenous Glucagon-Like Peptide 1 (7-36 Amide) in Type 2 (Non-Insulin-Dependent) Diabetic Patients. Diabetologia 1993, 36, 741–744. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Henquin, J.; Horemans, B.; Nenquin, M.; Verniers, J.; Lambert, A. Quinine-Induced Modifications of Insulin Release and Glucose Metabolism by Isolated Pancreatic Islets. FEBS Lett. 1975, 57, 280–284. [Google Scholar] [CrossRef] [Green Version]
- Vignini, A.; Borroni, F.; Sabbatinelli, J.; Pugnaloni, S.; Alia, S.; Taus, M.; Ferrante, L.; Mazzanti, L.; Fabri, M. General Decrease of Taste Sensitivity Is Related to Increase of BMI: A Simple Method to Monitor Eating Behavior. Dis. Markers 2019, 2019, 1–8. [Google Scholar] [CrossRef] [Green Version]
- Bianchi, L.L.; Galmarini, M.; García-Burgos, D.; Zamora, M. Time-Intensity and Reaction-Time Methodology Applied to the Dynamic Perception and Liking of Bitterness in Relation to Body Mass Index. Food Res. Int. 2018, 109, 606–613. [Google Scholar] [CrossRef]
- Garcia-Burgos, D.; Zamora, M. Facial Affective Reactions to Bitter-Tasting Foods and Body Mass Index in Adults. Appetite 2013, 71, 178–186. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Simchen, U.; Koebnick, C.; Hoyer, S.; Issanchou, S.; Zunft, H.-J.F. Odour and Taste Sensitivity is Associated with Body Weight and Extent of Misreporting of Body Weight. Eur. J. Clin. Nutr. 2006, 60, 698–705. [Google Scholar] [CrossRef]
- Overberg, J.; Hummel, T.; Krude, H.; Wiegand, S. Differences in Taste Sensitivity between Obese and Non-obese Children and Adolescents. Arch. Dis. Child. 2012, 97, 1048–1052. [Google Scholar] [CrossRef]
- De Carli, L.; Gambino, R.; Lubrano, C.; Rosato, R.; Bongiovanni, D.; Lanfranco, F.; Broglio, F.; Ghigo, E.; Bo, S. Impaired Taste Sensation in Type 2 Diabetic Patients without Chronic Complications: A Case–Control Study. J. Endocrinol. Investig. 2017, 41, 765–772. [Google Scholar] [CrossRef] [PubMed]
- Matsugasumi, M.; Hashimoto, Y.; Okada, H.; Tanaka, M.; Kimura, T.; Kitagawa, N.; Tanaka, Y.; Fukuda, Y.; Sakai, R.; Yamazaki, M.; et al. The Association between Taste Impairment and Serum Zinc Concentration in Adult Patients with Type 2 Diabetes. Can. J. Diabetes 2018, 42, 520–524. [Google Scholar] [CrossRef] [PubMed]
- Pugnaloni, S.; Alia, S.; Mancini, M.; Santoro, V.; Di Paolo, A.; Rabini, R.A.; Fiorini, R.; Sabbatinelli, J.; Fabri, M.; Mazzanti, L.; et al. A Study on the Relationship between Type 2 Diabetes and Taste Function in Patients with Good Glycemic Control. Nutrients 2020, 12, 1112. [Google Scholar] [CrossRef] [PubMed]
- Gondivkar, S.M.; Indurkar, A.; Degwekar, S.; Bhowate, R. Evaluation of Gustatory Function in Patients with Diabetes Mellitus Type 2. Oral Surg. Oral Med. Oral Pathol. Oral Radiol. Endodontol. 2009, 108, 876–880. [Google Scholar] [CrossRef]
- Wölnerhanssen, B.K.; Moran, A.W.; Burdyga, G.; Meyer-Gerspach, A.C.; Peterli, R.; Manz, M.; Thumshirn, M.; Daly, K.; Beglinger, C.; Shirazi-Beechey, S.P. Deregulation of Transcription Factors Controlling Intestinal Epithelial Cell Differentiation; a Predisposing Factor for Reduced Enteroendocrine Cell Number in Morbidly Obese Individuals. Sci. Rep. 2017, 7, 1–13. [Google Scholar] [CrossRef] [Green Version]
- Latorre, R.; Huynh, J.; Mazzoni, M.; Gupta, A.; Bonora, E.; Clavenzani, P.; Chang, L.; Mayer, E.A.; de Giorgio, R.; Sternini, C. Expression of the Bitter Taste Receptor, T2R38, in Enteroendocrine Cells of the Colonic Mucosa of Overweight/Obese vs. Lean Subjects. PLoS ONE 2016, 11, e0147468. [Google Scholar] [CrossRef]
- Keller, M.; Liu, X.; Wohland, T.; Rohde, K.; Gast, M.-T.; Stumvoll, M.; Kovacs, P.; Tönjes, A.; Böttcher, Y. TAS2R38 and Its Influence on Smoking Behavior and Glucose Homeostasis in the German Sorbs. PLoS ONE 2013, 8, e80512. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Spector, A.C.; Kopka, S.L. Rats Fail to Discriminate Quinine from Denatonium: Implications for the Neural Coding of Bitter-Tasting Compounds. J. Neurosci. 2002, 22, 1937–1941. [Google Scholar] [CrossRef] [PubMed] [Green Version]
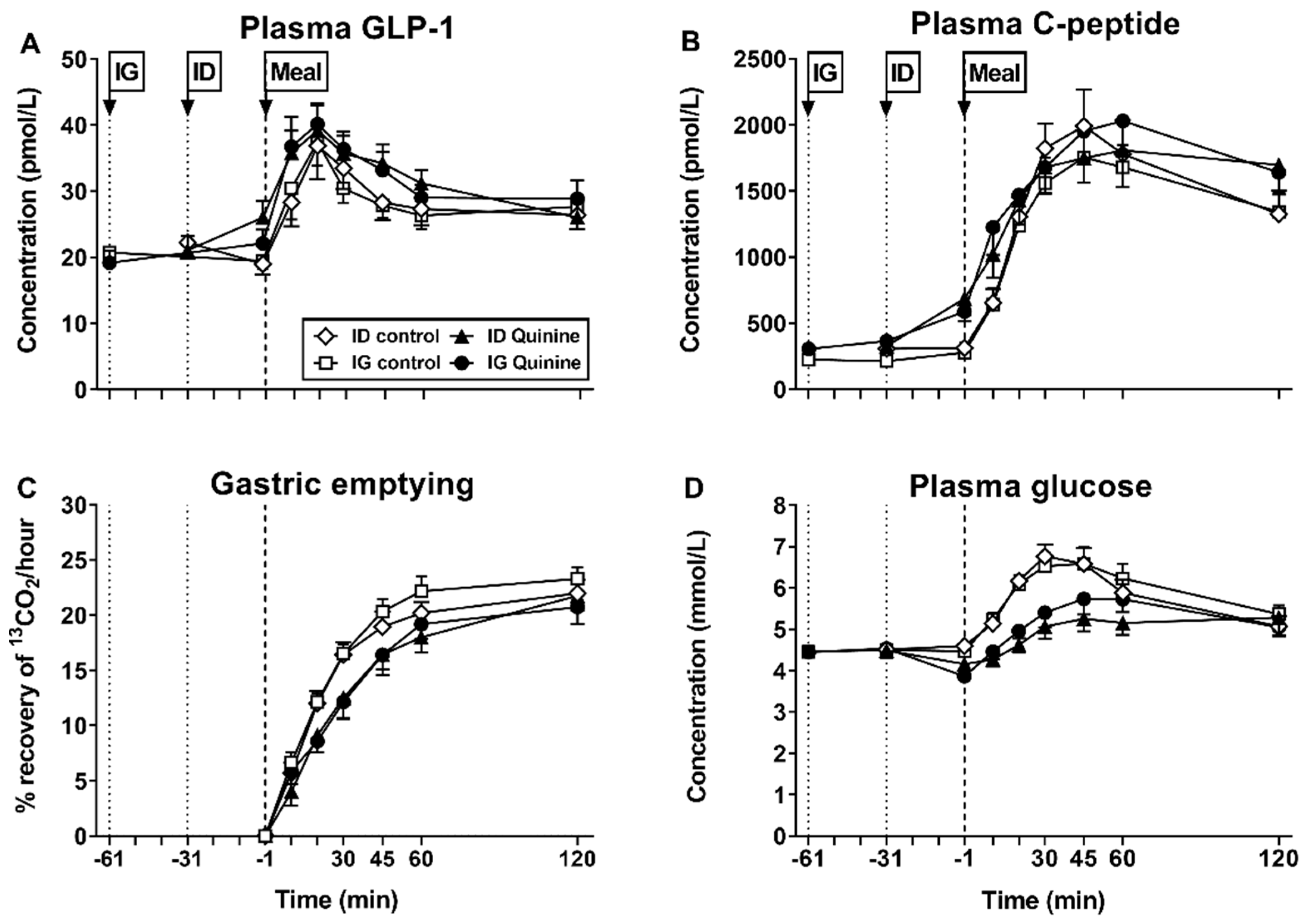
| Bitter Tastants | Model | Doses Given/Location of Delivery | Observed Effect | Ref # |
|---|---|---|---|---|
| Denatonium benzoate | Females | 1 μmol/kg bolus/IG (≈30 mg) 1 | ↔ Gastric emptying | [19] |
| Males and females | 1 µmol/kg bolus/IG | ↓ Fundic relaxation | [15] | |
| Naringin | Males and females | 1 mM bolus (≈580 mg)/IG | ↔ Gastric emptying | [103] |
| Quinine hydrochloride | Males and females | 18 mg in acid-resistant capsule/oral | ↔ Gastric emptying | [16] |
| Females | 10 μmol/kg bolus/IG [≈270 mg] | ↓ ‘Fluctuations’ in antral motility ↔ Duodenal motility | [75] | |
| Males and females | 0.198 mM [≈72 mg]/IG | ↔ Gastric emptying | [103] | |
| Males | 37.5, 75, 225 mg/ID over 60 min | ↔ Antropyloroduodenal motility | [76] | |
| Males | 275, 600 mg bolus/IG 30 min before meal | ↔ Gastric emptying | [17] | |
| Males | 600 mg bolus/IG 60 min before meal, ID 30 min before meal | ↓ Gastric emptying | [20] | |
| Quinine sulphate | Females | 10 mg bolus/oral | ↓ Gastric emptying | [104] |
| Bitter Tastants | Model | Doses Given/Location of Delivery | Approx. Equivalent Dose in a 70-kg Human | Type of Meal or Diet | Observed Effects | Ref # |
|---|---|---|---|---|---|---|
| Berberine | Rats | 93.75, 187.5, 562.5 mg/kg/oral | ≈6.5, 13, 39 g | Ad libitum high-fat chow | ↓ Food intake ↓ Weight gain | [108] |
| Denatonium benzoate | Mice | 60 μmol/kg/IG | ≈1.8 g | Mixed-nutrient liquid meal | ↓ Food intake ↓ Weight gain | [1] |
| Epicatechin | Rats | 300 mg/kg/IG | ≈21 g | Ad libitum standard chow diet | ↓ Food intake | [79] |
| Hoodia gordonii extract | Rats | 6.25–50 mg/kg/oral | ≈0.4–3.5 g | Ad libitum standard diet (55% CHO, 15% Prot, 3% F) | ↓ Food intake ↓ Body weight | [107] |
| Humulus lupulus L. extract | Mice | 2–5% of diet/oral | - | Ad libitum standard (77% CHO, 9.7% F, 13.9% Prot) or high-fat diet (546 kcal/100 g) | ↓ Food intake ↔ Weight gain | [105] |
| Rodents | 0.2–1.2% of diet/oral | - | Ad libitum standard diet (77% CHO, 9.7% F, 13.9% Prot) or high-fat diet (60% F, 14% CHO, 26% Prot) | ↓ Food intake ↓ Weight gain | [106] | |
| 1,10-Phenanthroline | Rats | 200 mg/kg/IG | ≈14 g | Ad libitum standard chow diet | ↓ Food intake | [79] |
| Quinine hydrochloride | Mice | 160 μmol/kg/IG | ≈4 g | Mixed-nutrient liquid meal | ↔ Food intake ↓ Weight gain | [1] |
| Quinine sulphate | Rats | 0.75% of diet/oral | - | Ad libitum powdered chow diet | ↓ Food intake ↓ Body weight | [109] |
| Vanillic acid | Rats | 252 mg/kg/IG | ≈17 g | Ad libitum standard chow diet | ↓ Food intake | [79] |
| Mixture of DB, PTC, PTU, quinine HCl, D-salicin | Mice | DB 10 mM; PTC 10 mM; PTU 5 mM; quinine 1.5 mM; D-salicin 5 mM/IG | DB ≈ 46 mg; PTC ≈ 15 mg; PTU, ≈8 mg; quinine ≈ 5 mg; Salicin ≈ 15 mg | Ad libitum food | ↑ Food intake (first 30 min) ↓ Food intake (next 4 h) | [14] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Rezaie, P.; Bitarafan, V.; Horowitz, M.; Feinle-Bisset, C. Effects of Bitter Substances on GI Function, Energy Intake and Glycaemia-Do Preclinical Findings Translate to Outcomes in Humans? Nutrients 2021, 13, 1317. https://doi.org/10.3390/nu13041317
Rezaie P, Bitarafan V, Horowitz M, Feinle-Bisset C. Effects of Bitter Substances on GI Function, Energy Intake and Glycaemia-Do Preclinical Findings Translate to Outcomes in Humans? Nutrients. 2021; 13(4):1317. https://doi.org/10.3390/nu13041317
Chicago/Turabian StyleRezaie, Peyman, Vida Bitarafan, Michael Horowitz, and Christine Feinle-Bisset. 2021. "Effects of Bitter Substances on GI Function, Energy Intake and Glycaemia-Do Preclinical Findings Translate to Outcomes in Humans?" Nutrients 13, no. 4: 1317. https://doi.org/10.3390/nu13041317
APA StyleRezaie, P., Bitarafan, V., Horowitz, M., & Feinle-Bisset, C. (2021). Effects of Bitter Substances on GI Function, Energy Intake and Glycaemia-Do Preclinical Findings Translate to Outcomes in Humans? Nutrients, 13(4), 1317. https://doi.org/10.3390/nu13041317