
Palmitoylethanolamide and Its Biobehavioral Correlates in Autism Spectrum Disorder: A Systematic Review of Human and Animal Evidence




Abstract
1. Introduction
Objectives
2. Experimental Procedures
2.1. Inclusion and Exclusion Criteria
2.2. Search Strategy and Data Extraction
2.3. Risk of Bias
3. Results
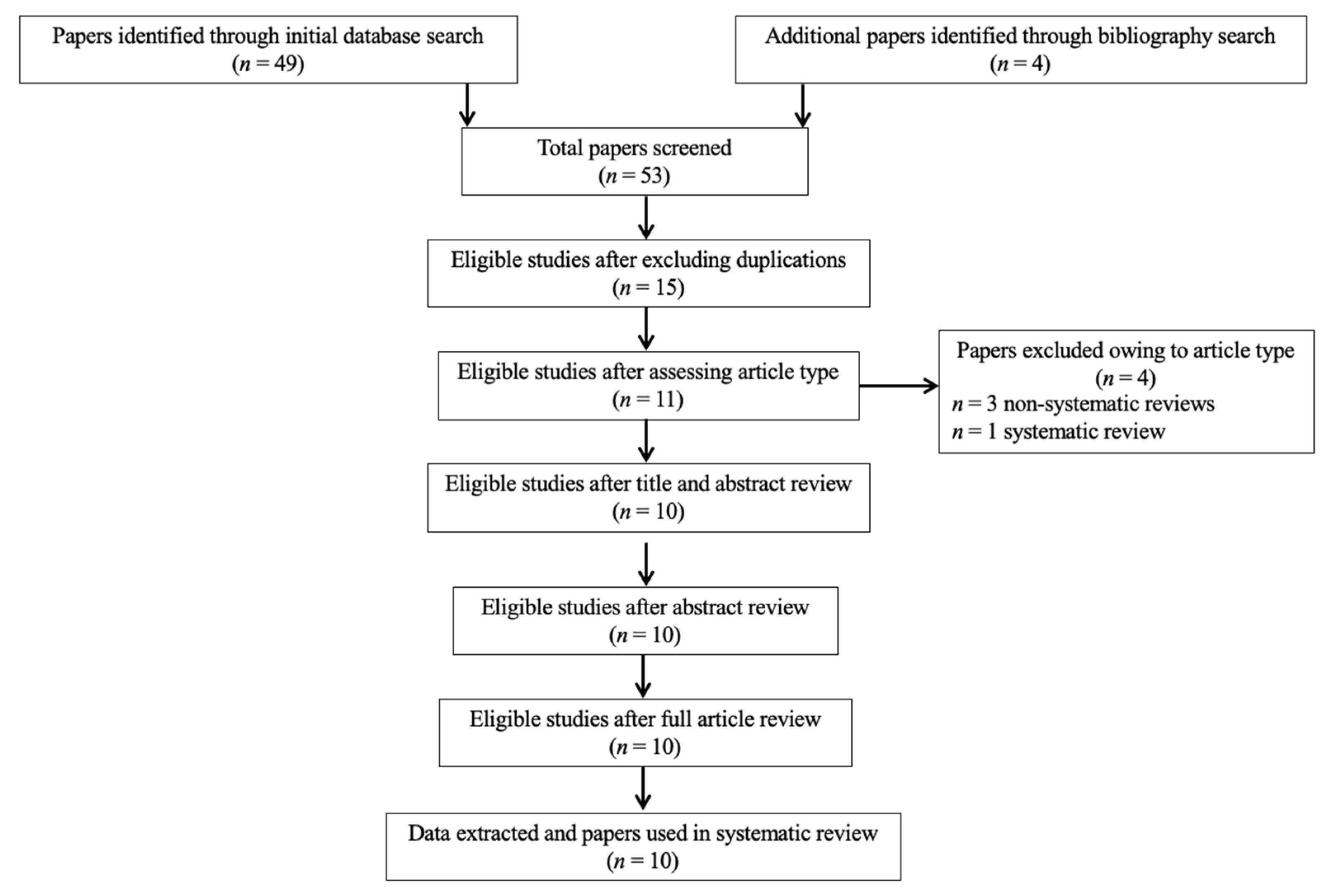
3.1. Study Selection
3.2. In Vivo PEA Treatment Exposure in Children and Adolescents with Autism Spectrum Disorder
3.3. PEA Levels in Children and Adolescents with Autism Spectrum Disorder as Compared to Healthy Controls
3.4. In Vivo PEA Postnatal Exposure in Animal Models of Autism and Perinatal Brain Disorders
3.5. Pea and Related Enzymes and Receptors Quantitative Brain Assessment in Animal Models of Autism, Perinatal Brain Disorders, and Stress Following Exposure to Exogenous Cannabinoids
4. Discussion
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Saghazadeh, A.; Ahangari, N.; Hendi, K.; Saleh, F.; Rezaei, N. Status of essential elements in autism spectrum disorder: Systematic review and meta-analysis. Rev. Neurosci. 2017, 28, 783–809. [Google Scholar] [CrossRef]
- First, M.B.; Williams, J.B.W.; Karg, R.S.; Spitzer, R.L. Structured Clinical Interview for DSM-5 Disorders, Clinician Version (SCID-5-CV); American Psychiatric Association: Arlington, VA, USA, 2015. [Google Scholar]
- Zeng, K.; Kang, J.; Ouyang, G.; Li, J.; Han, J.; Wang, Y.; Sokhadze, E.M.; Casanova, M.F.; Li, X. Disrupted Brain Network in Children with Autism Spectrum Disorder. Sci. Rep. 2017, 7, 1–12. [Google Scholar] [CrossRef]
- Kern, J.K.; Geier, D.A.; King, P.G.; Sykes, L.K.; Mehta, J.A.; Geier, M.R. Shared Brain Connectivity Issues, Symptoms, and Comorbidities in Autism Spectrum Disorder, Attention Deficit/Hyperactivity Disorder, and Tourette Syndrome. Brain Connect. 2015, 5, 321–335. [Google Scholar] [CrossRef]
- Lukito, S.; Norman, L.; Carlisi, C.; Radua, J.; Hart, H.; Simonoff, E.; Rubia, K. Comparative meta-analyses of brain structural and functional abnormalities during cognitive control in attention-deficit/hyperactivity disorder and autism spectrum disorder. Psychol. Med. 2020, 50, 894–919. [Google Scholar] [CrossRef]
- Dewey, D. What Is Comorbidity and Why Does It Matter in Neurodevelopmental Disorders? Curr. Dev. Disord. Rep. 2018, 5, 235–242. [Google Scholar] [CrossRef]
- Masi, A.; Quintana, D.S.; Glozier, N.; Lloyd, A.R.; Hickie, I.B.; Guastella, A.J. Cytokine aberrations in autism spectrum disorder: A systematic review and meta-analysis. Mol. Psychiatry 2014, 20, 440–446. [Google Scholar] [CrossRef] [PubMed]
- Purcell, A.E.; Jeon, O.H.; Zimmerman, A.W.; Blue, M.E.; Pevsner, J. Postmortem brain abnormalities of the glutamate neurotransmitter system in autism. Neurology 2001, 57, 1618–1628. [Google Scholar] [CrossRef] [PubMed]
- Shimmura, C.; Suda, S.; Tsuchiya, K.J.; Hashimoto, K.; Ohno, K.; Matsuzaki, H.; Iwata, K.; Matsumoto, K.; Wakuda, T.; Kameno, Y.; et al. Alteration of Plasma Glutamate and Glutamine Levels in Children with High-Functioning Autism. PLoS ONE 2011, 6, e25340. [Google Scholar] [CrossRef]
- Shinohe, A.; Hashimoto, K.; Nakamura, K.; Tsujii, M.; Iwata, Y.; Tsuchiya, K.J.; Sekine, Y.; Suda, S.; Suzuki, K.; Sugihara, G.-I.; et al. Increased serum levels of glutamate in adult patients with autism. Prog. Neuro Psychopharmacol. Biol. Psychiatry 2006, 30, 1472–1477. [Google Scholar] [CrossRef] [PubMed]
- Blaylock, R.L.; Strunecka, A. Immune-glutamatergic dysfunction as a central mechanism of the autism spectrum disorders. Curr. Med. Chem. 2009, 16, 157–170. [Google Scholar] [CrossRef] [PubMed]
- Walter, L.; Stella, N. Cannabinoids and neuroinflammation. Br. J. Pharmacol. 2004, 141, 775–785. [Google Scholar] [CrossRef]
- Colizzi, M.; McGuire, P.; Pertwee, R.G.; Bhattacharyya, S. Effect of cannabis on glutamate signalling in the brain: A systematic review of human and animal evidence. Neurosci. Biobehav. Rev. 2016, 64, 359–381. [Google Scholar] [CrossRef]
- Colizzi, M.; Ruggeri, M.; Bhattacharyya, S. Unraveling the Intoxicating and Therapeutic Effects of Cannabis Ingredients on Psychosis and Cognition. Front. Psychol. 2020, 11, 833. [Google Scholar] [CrossRef]
- Freitas, H.R.; Isaac, A.R.; Malcher-Lopes, R.; Diaz, B.L.; Trevenzoli, I.H.; Reis, R.A.D.M. Polyunsaturated fatty acids and endocannabinoids in health and disease. Nutr. Neurosci. 2017, 21, 695–714. [Google Scholar] [CrossRef]
- Aran, A.; Harel, M.; Cassuto, H.; Polyansky, L.; Schnapp, A.; Wattad, N.; Shmueli, D.; Golan, D.; Castellanos, F.X. Cannabinoid treatment for autism: A proof-of-concept randomized trial. Mol. Autism 2021, 12, 1–11. [Google Scholar] [CrossRef]
- Tsuboi, K.; Uyama, T.; Okamoto, Y.; Ueda, N. Endocannabinoids and related N-acylethanolamines: Biological activities and metabolism. Inflamm. Regen. 2018, 38, 1–10. [Google Scholar] [CrossRef]
- Rankin, L.; Fowler, C.J. The basal pharmacology of palmitoylethanolamide. Int. J. Mol. Sci. 2020, 21, 7942. [Google Scholar] [CrossRef] [PubMed]
- Solorzano, C.; Zhu, C.; Battista, N.; Astarita, G.; Lodola, A.; Rivara, S.; Mor, M.; Russo, R.; Maccarrone, M.; Antonietti, F.; et al. Selective N-acylethanolamine-hydrolyzing acid amidase inhibition reveals a key role for endogenous palmitoylethanolamide in inflammation. Proc. Natl. Acad. Sci. USA 2009, 106, 20966–20971. [Google Scholar] [CrossRef] [PubMed]
- Verme, J.L.; Fu, J.; Astarita, G.; La Rana, G.; Russo, R.; Calignano, A.; Piomelli, D. The nuclear receptor peroxisome proliferator-activated receptor-α mediates the anti-inflammatory actions of palmitoylethanolamide. Mol. Pharmacol. 2005, 67, 15–19. [Google Scholar] [CrossRef] [PubMed]
- Yu, H.-L.; Deng, X.-Q.; Li, Y.-J.; Quan, Z.-S.; Sun, X.-Y.; Li, Y.-C. Short communication–N-palmitoylethanolamide, an endocannabinoid, exhibits antidepressant effects in the forced swim test and the tail suspension test in mice. Pharmacol. Rep. 2011, 63, 834–839. [Google Scholar] [CrossRef]
- Lambert, D.M.; Vandevoorde, S.; Diependaele, G.; Govaerts, S.J.; Robert, A.R. Anticonvulsant activity of N-palmitoylethanolamide, a putative endocannabinoid, in mice. Epilepsia 2002, 42, 321–327. [Google Scholar] [CrossRef]
- Jaggar, S.I.; Hasnie, F.S.; Sellaturay, S.; Rice, A.S. The anti-hyperalgesic actions of the cannabinoid anandamide and the putative CB2 receptor agonist palmitoylethanolamide in visceral and somatic inflammatory pain. Pain 1998, 76, 189–199. [Google Scholar] [CrossRef]
- West, S.; King, V.; Carey, T.S.; Lohr, K.N.; McKoy, N.; Sutton, S.F.; Lux, L. Systems to rate the strength of scientific evidence. Évid. Rep. Assess. 2002, 47, 1–11. [Google Scholar]
- Antonucci, N.; Cirillo, A.; Siniscalco, D. Beneficial Effects of Palmitoylethanolamide on Expressive Language, Cognition, and Behaviors in Autism: A Report of Two Cases. Case Rep. Psychiatry 2015, 2015, 1–6. [Google Scholar] [CrossRef] [PubMed]
- Bertolino, B.; Crupi, R.; Impellizzeri, D.; Bruschetta, G.; Cordaro, M.; Siracusa, R.; Esposito, E.; Cuzzocrea, S. Beneficial Effects of Co-Ultramicronized Palmitoylethanolamide/Luteolin in a Mouse Model of Autism and in a Case Report of Autism. CNS Neurosci. Ther. 2016, 23, 87–98. [Google Scholar] [CrossRef] [PubMed]
- Khalaj, M.; Saghazadeh, A.; Shirazi, E.; Shalbafan, M.-R.; Alavi, K.; Shooshtari, M.H.; Laksari, F.Y.; Hosseini, M.; Mohammadi, M.-R.; Akhondzadeh, S. Palmitoylethanolamide as adjunctive therapy for autism: Efficacy and safety results from a randomized controlled trial. J. Psychiatr. Res. 2018, 103, 104–111. [Google Scholar] [CrossRef]
- Aran, A.; Eylon, M.; Harel, M.; Polianski, L.; Nemirovski, A.; Tepper, S.; Schnapp, A.; Cassuto, H.; Wattad, N.; Tam, J. Lower circulating endocannabinoid levels in children with autism spectrum disorder. Mol. Autism 2019, 10, 1–11. [Google Scholar] [CrossRef]
- Cristiano, C.; Pirozzi, C.; Coretti, L.; Cavaliere, G.; Lama, A.; Russo, R.; Lembo, F.; Mollica, M.P.; Meli, R.; Calignano, A.; et al. Palmitoylethanolamide counteracts autistic-like behaviours in BTBR T+tf/J mice: Contribution of central and peripheral mechanisms. Brain Behav. Immun. 2018, 74, 166–175. [Google Scholar] [CrossRef]
- Herrera, M.I.; Udovin, L.D.; Toro-Urrego, N.; Kusnier, C.F.; Luaces, J.P.; Capani, F. Palmitoylethanolamide Ameliorates Hippocampal Damage and Behavioral Dysfunction After Perinatal Asphyxia in the Immature Rat Brain. Front. Neurosci. 2018, 12, 145. [Google Scholar] [CrossRef]
- Udovin, L.D.; Kobiec, T.; Herrera, M.I.; Toro-Urrego, N.; Kusnier, C.F.; Kölliker-Frers, R.A.; Ramos-Hryb, A.B.; Luaces, J.P.; Otero-Losada, M.; Capani, F. Partial Reversal of Striatal Damage by Palmitoylethanolamide Administration Following Perinatal Asphyxia. Front. Neurosci. 2020, 13, 1345. [Google Scholar] [CrossRef]
- Kerr, D.; Downey, L.; Conboy, M.; Finn, D.; Roche, M. Alterations in the endocannabinoid system in the rat valproic acid model of autism. Behav. Brain Res. 2013, 249, 124–132. [Google Scholar] [CrossRef]
- Blanco, E.; Galeano, P.; Holubiec, M.I.; Romero, J.I.; Logica, T.; Rivera, P.; Pavón, F.J.; Suarez, J.; Capani, F.; De Fonseca, F.R. Perinatal asphyxia results in altered expression of the hippocampal acylethanolamide/endocannabinoid signaling system associated to memory impairments in postweaned rats. Front. Neuroanat. 2015, 9, 141. [Google Scholar] [CrossRef]
- Tomas-Roig, J.; Piscitelli, F.; Gil, V.; Quintana, E.; Ramió-Torrentà, L.L.; Del Río, J.A.; Moore, T.P.; Agbemenyah, H.; Salinas, G.; Pommerenke, C.; et al. Effects of repeated long-term psychosocial stress and acute cannabinoid exposure on mouse corticostriatal circuitries: Implications for neuropsychiatric disorders. CNS Neurosci. Ther. 2018, 24, 528–538. [Google Scholar] [CrossRef] [PubMed]
- Savino, R.; Carotenuto, M.; Polito, A.N.; Di Noia, S.; Albenzio, M.; Scarinci, A.; Ambrosi, A.; Sessa, F.; Tartaglia, N.; Messina, G. Analyzing the Potential Biological Determinants of Autism Spectrum Disorder: From Neuroinflammation to the Kynurenine Pathway. Brain Sci. 2020, 10, 631. [Google Scholar] [CrossRef] [PubMed]
- Wójtowicz, S.; Strosznajder, J.B.; Jeżyna, M. The Novel Role of PPAR Alpha in the Brain: Promising Target in Therapy of Alzheimer’s Disease and Other Neurodegenerative Disorders. Neurochem. Res. 2020, 45, 972–988. [Google Scholar] [CrossRef] [PubMed]
- Hay, I.; Hynes, K.L.; Burgess, J.R. Mild-to-Moderate Gestational Iodine Deficiency Processing Disorder. Nutrients 2019, 11, 1974. [Google Scholar] [CrossRef] [PubMed]
- Li, D.; Karnath, H.-O.; Xu, X. Candidate Biomarkers in Children with Autism Spectrum Disorder: A Review of MRI Studies. Neurosci. Bull. 2017, 33, 219–237. [Google Scholar] [CrossRef]
- Siafis, S.; Çıray, O.; Schneider-Thoma, J.; Bighelli, I.; Krause, M.; Rodolico, A.; Ceraso, A.; Deste, G.; Huhn, M.; Fraguas, D.; et al. Placebo response in pharmacological and dietary supplement trials of autism spectrum disorder (ASD): Systematic review and meta-regression analysis. Mol. Autism 2020, 11, 1–19. [Google Scholar] [CrossRef]


{kind=link}
| Study (Country) | Aim of Study | PEA Type of Study | Population | N | Outcome Measure (Test Name or Description) | Results |
|---|---|---|---|---|---|---|
| Antonucci et al. (2015) (Italy) | To assess the effect of PEA on language, behavior, and immune chemistry | In vivo treatment exposure in humans | 2 ASD adolescents | 2 | 1. Expressive language (MLU); 2. overall autism severity (CARS-2; ATEC); 3. immune response (blood tests) | 1. MLU: ↑ from 3.0 to 5.4; 2. CARS-2 test: ↓ from 43.5 to 32, ATEC: ↓ from 25 to 12; 3. vitamin D-OH25 level: ↑, CD57+ NK: ↑, total lymphocytes: ↓, total serum IgE: ↓ by ~50% (only in 1 case, in the other case NS), atopic illnesses: ↓, total WC: unchanged |
| Aran et al. (2019) (Israel) | To assess PEA and other ECBs/AEs blood levels and their association with behavior | Quantitative blood assessment in humans | 1. 93 ASD children/adolescents; 2. 93 HCs | 186 | Serum ECBs/AEs levels (LC-MS/MS) | 1. Serum levels of AEA, OEA and PEA: ASD < HC (after Bonferroni correction, still significant), 2-AG: ASD vs. HC, NS; 2. ↓ age or ↓ BMI: ↓serum levels of AEA (after Bonferroni correction, NS; correlations with other ECBs/AEs, NS); 3. ↑ APSI severity score or ↑ use of antipsychotics: ↓ serum levels of AEA (trend effect; correlations with other ECBs/AEs and/or other symptoms, NS) |
| Bertolino et al. (2016) (Italy) | To assess the effect of PEA on behavior | In vivo treatment exposure in humans | 1 ASD child | 1 | 1. Overall autism severity (ATEC); 2. Motor stereotypic behaviors (Quarterly questionnaire) | 1. ATEC: ↓ of both total and subgroup scores (Speech, Sociability, Sensory/Cognitive, Health/Physical/Behavior; improved of about 23%); 2. motor stereotypies: ↓; enuresis: ↓ from 91% to 2.4% after 14 months |
| Khalaj et al. (2018) (Iran) | To assess the effect of PEA add-on to risperidone on language and behavior | In vivo treatment exposure in humans | 1. 31 ASD (Arm A, risperidone + PEA); 2. 31 ASD children (Arm B, risperidone + placebo) | 62 (out of 70 randomized) | Behavior (ABC-C) | 1. ABC-C over time: irritability and hyperactivity, risperidone + PEA < risperidone + placebo (other domains NS); 2. ABC-C at 10 weeks: irritability, hyperactivity, and inappropriate speech (trend), risperidone + PEA < risperidone + placebo (other domains NS); 3. ABC-C at 5 weeks: hyperactivity, stereotypic behavior (trend), and inappropriate speech (trend), risperidone + PEA < risperidone + placebo (other domains NS); 4. ABC-C response rate at 10 weeks, hyperactivity, irritability, and inappropriate speech, risperidone + PEA > risperidone + placebo (other domains NS); 5. ABC-C response rate at 5 weeks, NS; Adverse events, NS |
| Bertolino et al. (2016) (Italy) | To assess the effect of PEA on behavior, neuroinflammation, neuromodulation, and neurogenesis | In vivo postnatal exposure in animals | 1. 60 SHAM+VHI; 2. 60 SHAM+PEA; 3. 60 VPA+VHI; 60 VPA+PEA | 240 | 1. Behavior (SIT; EPM); 2. Neuroinflammation, neuromodulation, and neurogenesis (Immunohistochemistry (Chymase, Tryptase, TNF-α, IL-1β); Western Blot (Bax, Bcl-2, iNOS, IкBα, NF-kB, GFAP); Neurogenesis (BrdU and DCX Immunohistochemistry and Golgi impregnation)) | 1. SIT: stay duration in stranger side, VPA < VPA+PEA; stay duration in central area, VPA > VPA+PEA; SI, VPA < VPA+PEA; EPM: time spent in the open arm, VPA < VPA+PEA; 2. chymase, tryptase, IL-1 and TNF-α expression to mast cells in hippocampus and cerebellum: VPA > VPA+PEA; iNOS and GFAP levels in hippocampus and cerebellum: VPA > VPA+PEA; IкBα and NF-kB levels: VPA < VPA+PEA; Bax: VPA > VPA+PEA; Bcl-2: VPA < VPA+PEA; BrdU+ cells, DCX+ cells and development of dendritic spines in the dentate gyrus of hippocampus: VPA < VPA+PEA |
| Kerr et al. (2013) (Ireland) | To assess PEA and other ECBs/AEs brain levels and their association with behavior, and ECBs/AEs-related gene expression and enzyme activity | Quantitative brain assessment in animals | 1. 16 saline; 2. 14 VPA | 30 | 1. Brain ECBs/AEs levels (LC-MS/MS); 2. behavior (sociability test); 3. gene expression (real-time PCR); 4. enzyme activity | 1. AEA, 2-AG, PEA and OEA in frontal cortex, hippocampus and cerebellum: VPA vs. saline NS; 2. AEA, OEA and PEA in hippocampus after sociability test: VPA > saline (other ECBs in other brain regions NS); 3. MAGL mRNA in the hippocampus, DAGLα mRNA in the cerebellum, VPA < saline (other brain region NS); PPARα and PPARγ mRNA in frontal cortex and hippocampus, VPA < saline; GPR55 mRNA in frontal cortex and hippocampus, VPA < saline (cerebellum NS); CB1/CB2 receptor mRNA in frontal cortex, hippocampus and cerebellum, VPA vs. saline, NS; 4. MAGL activity in the hippocampus, VPA > saline (FAAH NS) |
| Blanco et al. (2015) (Spain) | To assess ECBs/AEs-related enzyme and receptor activity | Quantitative brain assessment in animals | 1.CTL; 2. C+; 3. PA | 15 (mothers) | 1. Brain ECBs/AEs-related enzyme and receptor activity (immunohistochemistry, immunostaining quantification) | 1. NeuN: NS; GFAP-positive cells: PA > CTL and C+ in CA1, CA3, and DG regions of dorsal hippocampus; DAGLα expression: PA and C+ > CTL in CA1, C+ > CTL in DG (other regions or groups NS); NAPE-PLD expression: PA < C+ and CTL in CA1 and CA3, PA < C+ in DG (other regions or groups NS); CB1 expression, NS; PPARα-positive cells: PA < C+ and CTL in CA1 and CA3 (DG NS); FAAH expression: C+ > CTL and PA in CA3 (other regions or groups NS) |
| Herrera et al. (2018) (Spain) | To assess the effect of PEA on behavior and neuronal damage | In vivo postnatal exposure in animals | 1. 15 PA+VHI; 2. 13 CTL (VHI); 3. 18 PA+PEA; 4. 17 CTL+PEA | 63 | 1. Behavior (OFT, EPM); 2. neuronal damage (electron microscopy, immunohistochemistry (NeuN, pNF-H/M, MAP-2, GFAP), Western Blot (pNF H/M, MAP-2, GFAP)) | 1. OFT: time spent rearing, PA < CTL, PA < PA+PEA, CTL vs. PA+PEA NS, CTL vs. CTL+PEA NS; time spent grooming, PA > CTL, PA > PA+PEA, CTL vs. PA+PEA NS, CTL vs. CTL+PEA NS; EPM: time spent rearing, PA < CTL, PA < PA+PEA, CTL vs. PA+PEA NS, CTL vs. CTL+PEA NS; time spent grooming, PA > CTL, PA vs. PA+PEA NS, PA+PEA > CTL; time spent HD, PA > CTL, PA > PA+PEA, CTL vs. PA+PEA NS, CTL vs. CTL+PEA NS; 2. pyknotic nucleus in the hippocampal CA1 neurons: PA > CTL, ↓ in PA+PEA; NeuN abnormal neurons in the hippocampal CA1 layer: PA > CTL, ↓ in PA+PEA; hippocampal pNF-H/M reactive area/ protein expression: PA > CTL, ↓in PA+PEA (PA+PEA still > CTL); hippocampal MAP-2reactive area/ protein expression: PA < CTL, ↑ in PA+PEA (PA+PEA still < CTL); GFAP-positive cells and protein expression: NS |
| Cristiano et al. (2018) (Italy) | To assess the effect of PEA on behavior, gene expression, receptor activity, neurotrophins, mitochondrial function, neuroinflammation, and microbiota-gut-brain axis | In vivo postnatal exposure in animals | 1. C57Bl/6J+VHI (control B6); 2. BTBR T+tf/J+VHI (BTBR); 3. BTBR+PEA; 4. BTBR+GW; 5. BTBR+GW+PEA; 6. B6 PPAR-α null (KO)+VHI; 7. KO+PEA | 6–12 per group of experiment | 1. Behavior (MBA, SGT, TST); 2. gene expression, receptor activity, neurotrophins (Western Blot, real-time PCR); 3. mitochondrial function and neuroinflammation (serum parameters, mitochondrial parameters, oxydative stress assay); 4. microbiota-gut-brain axis (intestinal permeability assay (FITC-labeled dextran, faecal microbiota)) | 1. MBA and SGT: BTBR > B6, BTBR+PEA10 ↓, BTBR+PEA30 ↓↓, BTBR+GW+PEA vs. BTBR NS, BTBR+GW vs. BTBR NS, KO+PEA vs. KO NS; TST: BTBR+PEA10 ↑, BTBR+PEA30 ↑↑, BTBR+GW+PEA vs. BTBR NS, BTBR+GW vs. BTBR NS, KO+PEA vs. KO NS; BTBR+PEA30 early (1h) effect NS; B6+PEA vs. B6 NS; 2. PPAR-α mRNA and protein, BDNF protein, TrkB mRNA, CREB protein (CREB mRNA NS): BTBR < B6, BTBR+PEA30 ↑; 3. mitochondrial state 3 respiration and SOD activity: BTBR < B6, BTBR+PEA ↑ (mitochondrial state 4 respiration and oligomycin state 4 respiration NS); FCCP-stimulated respiration, mitochondrial energetic efficiency, ROS: BTBR+PEA ↓; TNFα, IL1-β, IL6 in hippocampus, colon and serum: BTBR > B6, BTBR+PEA ↓; 4. epithelial barrier integrity (Tjp1 and Ocln mRNA levels): BTBR < B6, BTBR+PEA ↑; Microbial community: Firmicutes/Bacteroidetes ratio, BTBR+PEA > BTBR |
| Tomas-Roig et al. (2018) (Germany) | To assess the effect of repeated stress and acute cannabinoid exposure on behavior and ECBs/AEs brain levels | Quantitative brain assessment in animals | 1. 60 STS; 2. 60 CTL; 3. VHI; 4. WIN+VHI; 5. Rim+VHI; 6. Rim+WIN; 7.STS+VHI; 8. STS+WIN+VHI; 9. STS+Rim+VHI; 10. STS+Rim+WIN; 11. CTL+VHI; 12. CTL+WIN+VHI; 13. CTL+Rim+VHI; 14. CTL+Rim+WIN | 120 | 1. Behavior (FOB, OFT); 2. brain ECBs/AEs and related gene expression (LC-APCI-MS, real-time PCR) | 1. Effect of stress: AEA levels in dorsal CPu, STS < CTL; Chrna6 gene expression, STS > CTL; 2. Effect of drugs exposure: OFT, distance travelled, WIN+VHI < VHI, Rim+WIN < Rim+VHI; distance travelled in center, Rim+VHI > WIN+VHI; rearing activity, Rim+WIN > WIN+VHI; PEA and OEA levels in dorsal Cpu, WIN+VHI > others; Fkpb5 expression, Rim+WIN > others; 3. Effect of stress under drug influence: OFT, distance travelled, STS+VHI > CTL+VHI, STS/CTL+WIN+VHI < STS/CTL+Rim+VHI, STS+WIN+VHI < STS+VHI, CTL+Rim+VHI > CTL+VHI; 2-AG levels, CTL+Rim+VHI > CTL+VHI/CTL+Rim+WIN; Chrna6 gene expression: CTL+WIN+VHI/CTL+Rim+VHI/CTL+Rim+WIN > CTL+VHI, STS+Rim+VHI/STS+Rim+WIN > STS+WIN+VHI/CTL groups; Slc6a4 gene expression: Rim+VHI > others |
| Udovin et al. (2020) (Argentina) | To assess the effect of PEA on neuronal damage | In vivo postnatal exposure in animals | 1. 15 PA+VHI; 2. 13 CTL (VHI); 3. 18 PA+PEA; 4. 7 CTL+PEA | 53 | 1. Neuronal damage (electron microscopy, immunohistochemistry (pNF-H/M, MAP-2, GFAP), Western Blot (pNF H/M, MAP-2, GFAP, antiglyceraldehyde-3-phosphate dehydrogenase)) | 1. Striatal pNF-H/M reactive area/protein expression: PA < CTL, ↑ in PA+PEA (PA+PEA still < CTL); striatal MAP-2 reactive area/protein expression: PA < CTL, ↑ in PA+PEA; GFAP-positive cells: PA < CTL, ↑ in PA+PEA (PA+PEA still < CTL; protein expression NS) |
| Study | Study Design | Defined Study Population | Age (Years) | Gender | PEA Measure | Adequate PEA Evaluation | Control | Comparability of Subjects | Other Comorbidity | Excluded/Adjusted for Confounding Factors | Statistical Analyses | Funding or Sponsorship |
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Antonucci et al. (2015) (Italy) | √ Case report | √ Clinical diagnosis | √ 1. 13 years old; 2. 15 years old | √ Male | √ 1. Normast 600 mg, 1/2 tablet twice daily for one week, THEN 1 tablet twice daily (oral administration); 2. Normast 600 mg, once daily (oral administration) | √ 1. 1 month exposure; 2. 3 month exposure | X | NA | √ 1. Atopia; 2. Epilepsy | X | NA | √ |
| Aran et al. (2019) (Israel) | √ Analytic, observational | √ ADOS-2, DSM-5, additional assessments (VABS-II, SCQ lifetime form, CARS2-ST, HSQ-ASD, CBCL-validated Hebrew version, APSI, SRS-2 Hebrew version, CGI-S) | √ Mean (SD) [range]: 1. HC: 11.8 (4.3) [5.5-21]; 2. ASD: 13.1 (4.1) [6–21] | √ Male (%): 1. HC: 79%; 2. ASD: 79% | √ Serum blood levels | √ Single assessment | √ | √ Matched for age, gender and BMI | √ 1. HC, no neuropsychiatric comorbidity other than ADHD | √ 1. Results adjusted for age, gender, BMI, and ADHD; 2. results correlated with anxiety, behavior, epilepsy, and perinatal complication comorbidity | √ t-test, Bonferroni correction, Pearson χ2 test, Pearson correlation, multivariate logistic regression, linear regression | √ |
| Bertolino et al. (2016) (Italy) | √ Case report | √ Clinical diagnosis | √ 10 years old | √ Male | √ co-ultraPEA-Lut 700 mg + 70 mg twice daily (oral administration) | √ 12/14 months | X | NA | √ Tetralogy of Fallot | X | NA | √ |
| Khalaj et al. (2018) (Iran) | √ Double-blind, randomized (parallel), placebo controlled | √ DSM-5, having irritability symptoms of at least moderate severity (scores ≥ 12 on the ABC-C Irritability subscale). | √ Mean (SD) [range]: 1. Risperidone + PEA, 6.84 (2.1) [4–12]; 2. Risperidone + placebo, 7.42 (2.35) [4–12] | √ Male, N (%): 1. Risperidone + PEA, 22 (70.97); 2. Risperidone + placebo, 25 (80.65) | √ 600 mg twice daily (oral administration) | √ 10 weeks | √ | √ Matched for age, sex, weight, ESRS score, ABC-C irritability, lethargy, stereotypy, hyperactivity, and inappropriate speech domains | √ Exclusion criterion | √ Excluded if 1. symptoms not so severe for treatment with risperidone; 2. concomitant psychiatric disorder; 3. preexisting medical condition; 4. severe intellectual disability; 5. alcohol/drug abuse; 6. dyskinesia; 7. antipsychotic medication or behavior treatment within the past 6 months | √ t-test with Levene’s test for equality of variance, Freeman-Halton extension of Fisher’s exact test, Cohen’s d, repeated measures ANOVA, ITT | √ |
| Study | Study Design | Defined Study Population | Age | Gender | PEA Measure | Adequate PEA Evaluation | Control Group | Statistical Analyses | Funding or Sponsorship |
|---|---|---|---|---|---|---|---|---|---|
| Bertolino et al. (2016) (Italy) | √ Analytic, observational, interventional | √ C57/BL6 mice injected SC with VPA (400 mg/kg) on P14 | √ P15-P120 | √ Male | √ co-ultraPEA-LUT 1 mg/kg (oral administration by gavage) | √ 1. 2 weeks for behavior, immunochemistry and Western Blot studies; 2. 3 months for neurogenesis studies | √ SHAM/VPA + vehicle, SHAM+PEA | √ Behavior: one-way ANOVA, Newman–Keuls multiple comparison test; Immunohistochemistry: ANOVA and post hoc Tukey tests with Bonferroni correction for multiple comparisons; all other results: ANOVA and Bonferroni post hoc for multiple comparisons | √ |
| Kerr et al. (2013) (Ireland) | √ Analytic, observational | √ Litters of female Sprague Dawley rats SC injected with VPA (600 mg/kg) at G12.5 | √ P33-P40 | √ Male and female | √ Brain tissue levels | √ Single assessment | √ Saline-treated | √ Shapiro–Wilk test; Levene test; unpaired t-test | √ |
| Blanco et al. (2015) (Spain) | √ Analytic, observational | √ Rats exposed to PA procedures | √ P30 | √ Male | √ Brain tissue levels of PEA-related enzymes and receptors | √ Single assessment | √ CTL, C+ | √ ANOVA, Tukey’s post hoc tests for multiple comparisons, Bonferroni’s correction, Kuskal-Wallis test, Mann–Whitney test | √ |
| Herrera et al. (2018) (Spain) | √ Analytic, observational, interventional | √ Rats exposed to PA procedures | √ P30 | √ Male | √ 10 mg/Kg (SC injection) | √ Single administration (within the 1st h of life) | √ CTL/PA + vehicle, CTL+PEA | √ Shapiro–Wilk test; Levene test; ANOVA; Student’s t-test; Bonferroni’s correction | √ |
| Cristiano et al. (2018) (Italy) | √ Analytic, observational, interventional | √ BTBR T+tf/J (BTBR) mice | √ 3–4 months | √ Male | √ 10 or 30 mg/Kg (IP injection) | √ Daily administration (10 days) | √ B6/BTBR/BTBR+GW/KO + vehicle, BTBR+GW/KO + PEA | √ ANOVA, Bonferroni’s correction | √ |
| Tomas-Roig et al. (2018) (Germany) | √ Analytic, observational, interventional | √ C57BI6/J mice exposed to stress (1 h/day per 21 days) | √ 7-8 weeks | √ Male | √ Brain tissue levels | √ Single assessment | √ CTL (left undisturbed) | √ ANOVA, Brown–Forsythe test, Bonferroni’s correction, Tamhane post hoc test, Student’s t-test | √ |
| Udovin et al. (2020) (Argentina) | √ Analytic, observational, interventional | √ Rats exposed to PA procedures | √ P30 | √ Male | √ 10 mg/Kg (SC injection) | √ Single administration (within the 1st h of life) | √ CTL/PA + vehicle, CTL+PEA | √ Shapiro–Wilk test; Levene test; ANOVA; Student’s t-test; Bonferroni’s correction | √ |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Colizzi, M.; Bortoletto, R.; Costa, R.; Zoccante, L. Palmitoylethanolamide and Its Biobehavioral Correlates in Autism Spectrum Disorder: A Systematic Review of Human and Animal Evidence. Nutrients 2021, 13, 1346. https://doi.org/10.3390/nu13041346
Colizzi M, Bortoletto R, Costa R, Zoccante L. Palmitoylethanolamide and Its Biobehavioral Correlates in Autism Spectrum Disorder: A Systematic Review of Human and Animal Evidence. Nutrients. 2021; 13(4):1346. https://doi.org/10.3390/nu13041346
Chicago/Turabian StyleColizzi, Marco, Riccardo Bortoletto, Rosalia Costa, and Leonardo Zoccante. 2021. "Palmitoylethanolamide and Its Biobehavioral Correlates in Autism Spectrum Disorder: A Systematic Review of Human and Animal Evidence" Nutrients 13, no. 4: 1346. https://doi.org/10.3390/nu13041346
APA StyleColizzi, M., Bortoletto, R., Costa, R., & Zoccante, L. (2021). Palmitoylethanolamide and Its Biobehavioral Correlates in Autism Spectrum Disorder: A Systematic Review of Human and Animal Evidence. Nutrients, 13(4), 1346. https://doi.org/10.3390/nu13041346