
Ketogenic Diet Decreases Alcohol Intake in Adult Male Mice

 , , ,
, , , {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Subjects
2.2. Drugs Treatment
2.3. Apparatus and Procedure
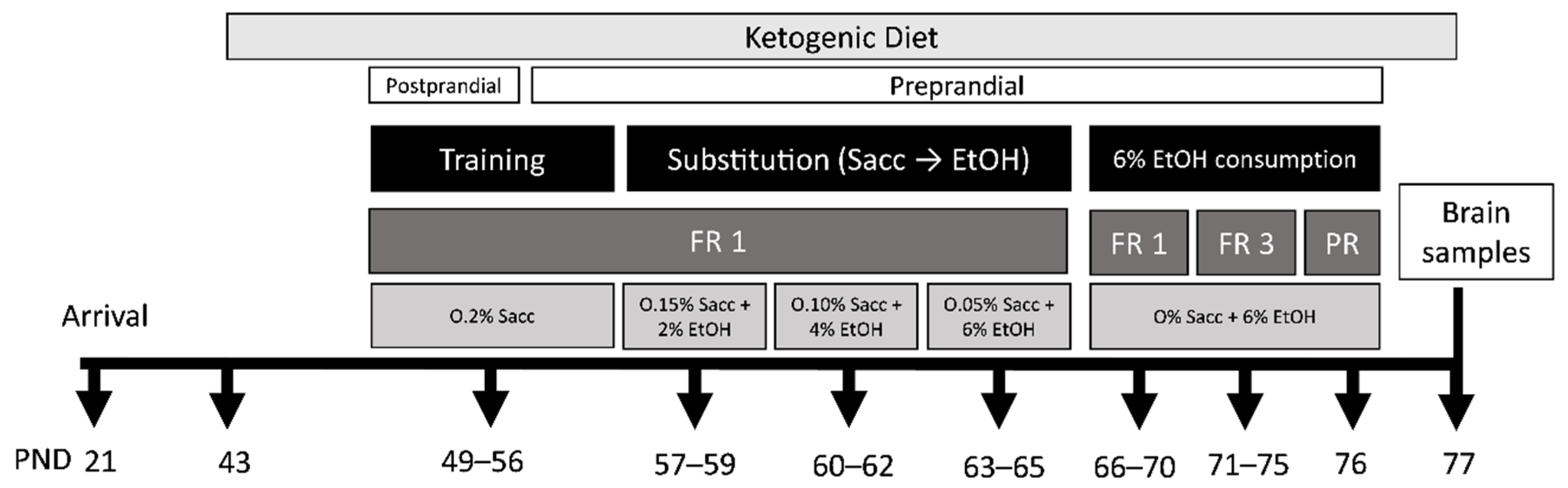
2.3.1. Experimental Design
2.3.2. Feeding Conditions
2.3.3. Ketosis Status: β-Hydroxybutyrate Blood Levels
2.3.4. Oral Ethanol Self-Administration
- Training phase (8 days)In the training phase, animals had to nose-poke into the active wholes to obtain 37 μL of saccharin (0.2% (w/v)). To facilitate learning acquisition, two days before beginning the training, chow was restricted to 1 h/day and water was suspended for 24 h before the first session. Only during the subsequent 3 days, 1 h before initiating the operant session animals had access to food but not to water (postprandial). On the subsequent four days and during the course of the experiment, to avoid EtOH intake due to thirst, water was available anytime and food was available for 1 h after each training session (preprandial).
- Saccharin substitution (9 days)
- 6% Ethanol consumption (11 days)This phase evaluates the number of active nose-poke responses, the 6% EtOH (w/v) intake and motivation to obtain it. First, animals were exposed to 5 days of fixed ratio 1 (FR1) sessions, in which the number of effective responses on the active nose-poke and EtOH consumption (all) was measured. After each session, the remaining fluid in the receptacle was collected and quantified with a micropipette. Following the FR1 sessions, animals were exposed to the fixed ratio 3 (FR3) schedule for 5 days, where they had to respond three times with an active nose poke to obtain one EtOH reinforcement. To set the breaking point for each animal, which is the maximum number of active nose-pokes the animal is capable of accomplishing to obtain one reinforcement, a progressive ratio (PR) session with a 2 h duration was carried out. The response requirement to achieve reinforcements increased corresponding to the series: 1-2-3-5-12-18-27-40-60-90-135-200-300-450-675-1000. The breaking point that the animal achieved was calculated based on this scale, which defines the animal’s motivation toward EtOH consumption.
2.3.5. RNA Isolation, Reverse Transcription, and Quantitative RT-PCR
2.4. Statistics
3. Results
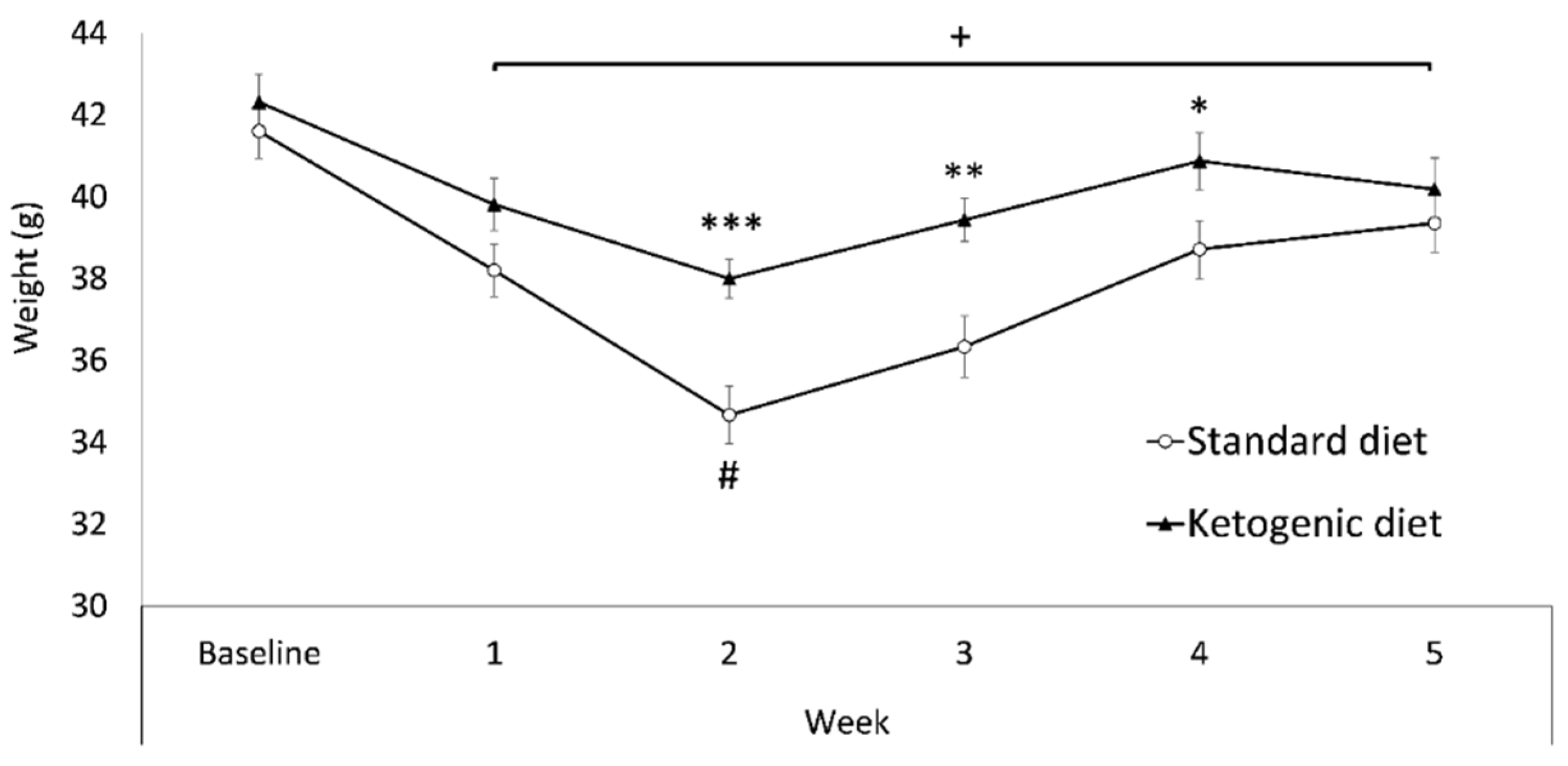
3.1. Increased β-Hydroxybutyrate (βOHB) and Bodyweight in Mice Fed on KD
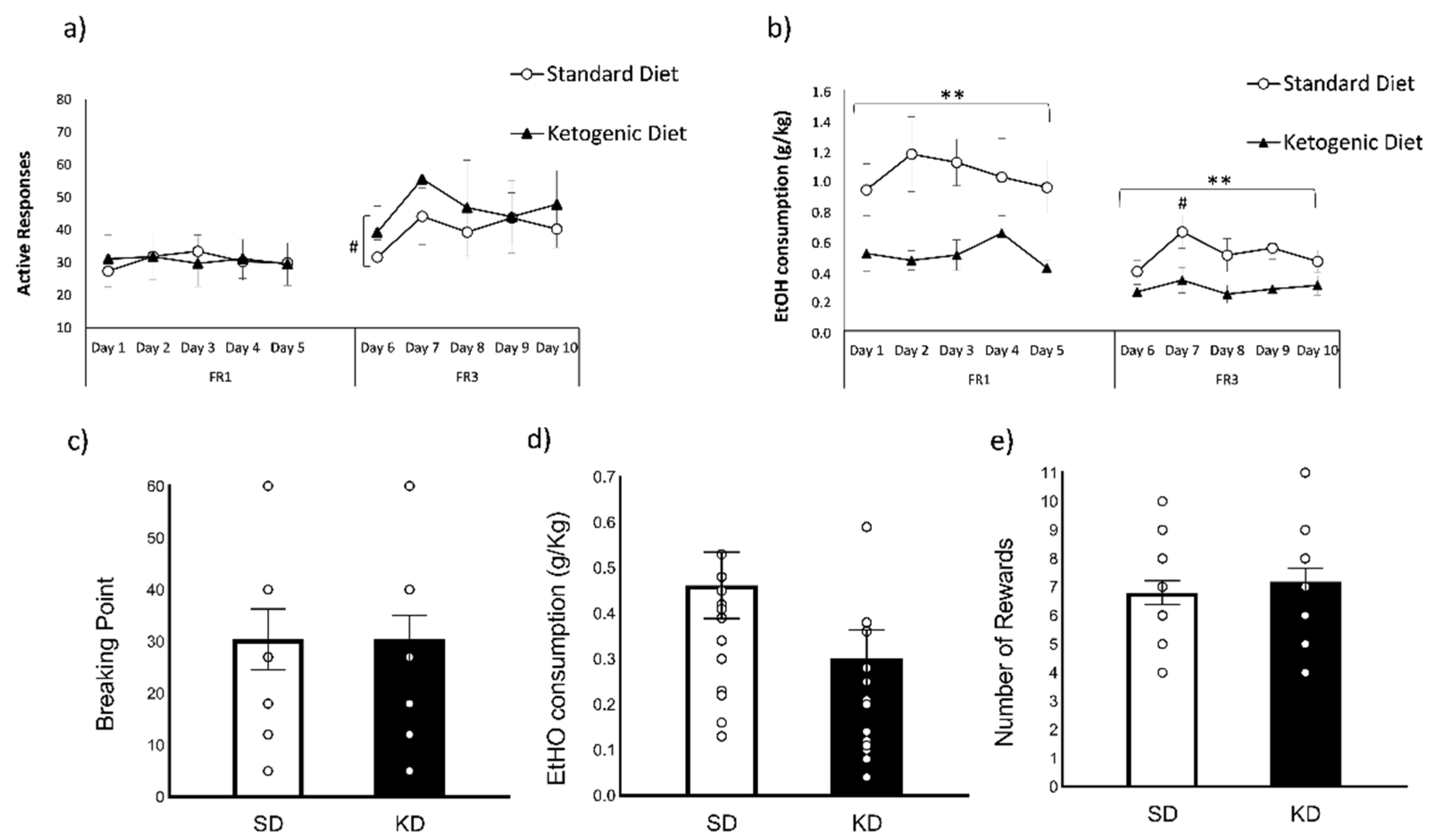
3.2. Ketogenic Diet Decreased Oral Ethanol Self-Administration
3.3. Gene Expression Analyses
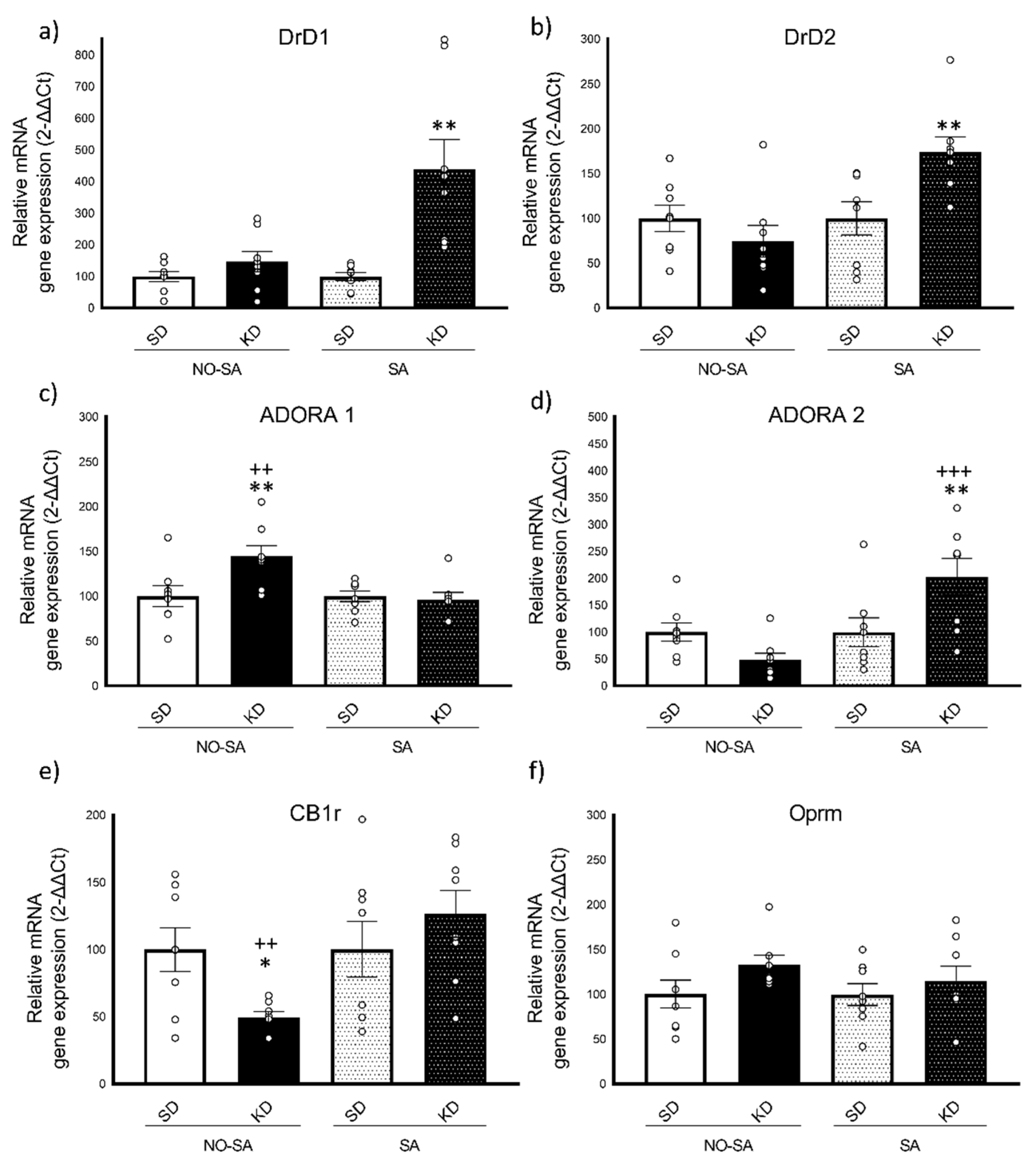
3.3.1. Ketogenic Diet Induced Increased Expression of DrD1 and DrD2 Gene Expression after Ethanol Self-Administration
3.3.2. Opposite Changes in ADORA1 and ADORA2 Gene Expression in Response to Ketogenic Diet and Ethanol Self-Administration
3.3.3. Ketogenic Diet Decreases CB1r Gene Expression
4. Discussion
4.1. Ketogenic Diet Modulates Bodyweight and Increases β-Hydroxybutyrate Blood Levels
4.2. Ketogenic Diet Diminishes Ethanol Self-Administration
4.3. Ketogenic Diet Diminishes Ethanol Intake through the Adenosine-Dopamine Binomial
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Kenny, P.J. Reward mechanisms in obesity: New insights and future directions. Neuron 2011, 69, 664–679. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Garber, A.K.; Lustig, R.H. Is fast food addictive? Curr. Drug Abuse Rev. 2011, 4, 146–162. [Google Scholar] [CrossRef] [PubMed]
- Avena, N.M.; Gold, J.A.; Kroll, C.; Gold, M.S. Further developments in the neurobiology of food and addiction: Update on the state of the science. Nutrition 2012, 28, 341–343. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Blanco-Gandía, M.C.; Aracil-Fernández, A.; Montagud-Romero, S.; Aguilar, M.A.; Manzanares, J.; Miñarro, J.; Rodríguez-Arias, M. Changes in gene expression and sensitivity of cocaine reward produced by a continuous fat diet. Psychopharmacology 2017, 234, 2337–2352. [Google Scholar] [CrossRef]
- Blanco-Gandia, M.; Cantacorps, L.; Aracil-Fernández, A.; Romero, S.M.; Aguilar, M.A.; Manzanares, J.; Valverde, O.; Miñarro, J.; Rodríguez-Arias, M. Effects of bingeing on fat during adolescence on the reinforcing effects of cocaine in adult male mice. Neuropharmacology 2017, 113, 31–44. [Google Scholar] [CrossRef] [Green Version]
- Blanco-Gandia, M.; Ledesma, J.C.; Aracil-Fernández, A.; Navarrete, F.; Romero, S.M.; Aguilar, M.A.; Manzanares, J.; Miñarro, J.; Rodríguez-Arias, M. The rewarding effects of ethanol are modulated by binge eating of a high-fat diet during adolescence. Neuropharmacology 2017, 121, 219–230. [Google Scholar] [CrossRef]
- Blanco-Gandía, M.C.; Miñarro, J.; Aguilar, M.A.; Rodríguez-Arias, M. Increased ethanol consumption after interruption of fat bingeing. PLoS ONE 2018, 13, e0194431. [Google Scholar] [CrossRef] [Green Version]
- Puhl, M.D.; Cason, A.M.; Wojnicki, F.H.; Corwin, R.L.; Grigson, P.S. A history of bingeing on fat enhances cocaine seeking and taking. Behav. Neurosci. 2011, 125, 930. [Google Scholar] [CrossRef] [Green Version]
- Ródenas-González, F.; del Carmen Blanco-Gandía, M.; Pascual, M.; Molari, I.; Guerri, C.; López, J.M.; Rodríguez-Arias, M. A limited and intermittent access to a high-fat diet modulates the effects of cocaine-induced reinstatement in the conditioned place preference in male and female mice. Psychopharmacology 2021, 1–13. [Google Scholar] [CrossRef]
- Politi, K.; Shemer-Meiri, L.; Shuper, A.; Aharoni, S. The ketogenic diet 2011: How it works. Epilepsy Res. Treat. 2011, 2011. [Google Scholar] [CrossRef]
- Masino, S.A.; Rho, J.M. Mechanisms of ketogenic diet action. Epilepsia 2010, 51, 85. [Google Scholar] [CrossRef]
- Hartman, A.L.; Gasior, M.; Vining, E.P.; Rogawski, M.A. The neuropharmacology of the ketogenic diet. Pediatr. Neurol. 2007, 36, 281–292. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cotter, D.G.; d’Avignon, D.A.; Wentz, A.E.; Weber, M.L.; Crawford, P.A. Obligate role for ketone body oxidation in neonatal metabolic homeostasis. J. Biol. Chem. 2011, 286, 6902–6910. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cunnane, S.C.; Courchesne-Loyer, A.; St-Pierre, V.; Vandenberghe, C.; Pierotti, T.; Fortier, M.; Croteau, E.; Castellano, C. Can ketones compensate for deteriorating brain glucose uptake during aging? Implications for the risk and treatment of Alzheimer’s disease. Ann. N. Y. Acad. Sci. 2016, 1367, 12–20. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Breckenridge, W.C.; Kuksis, A. Molecular weight distributions of milk fat triglycerides from seven species. J. Lipid Res. 1967, 8, 473–478. [Google Scholar] [CrossRef]
- Nehlig, A. Age-dependent pathways of brain energy metabolism: The suckling rat, a natural model of the ketogenic diet. Epilepsy Res. 1999, 37, 211–221. [Google Scholar] [CrossRef]
- Gasior, M.; Rogawski, M.A.; Hartman, A.L. Neuroprotective and disease-modifying effects of the ketogenic diet. Behav. Pharmacol. 2006, 17, 431. [Google Scholar] [CrossRef] [Green Version]
- Nehlig, A. Brain uptake and metabolism of ketone bodies in animal models. Prostaglandins Leukot Essent Fat. Acids. 2004, 70, 265–275. [Google Scholar] [CrossRef]
- Morris, A.A.M. Cerebral ketone body metabolism. J. Inherit. Metab. Dis. 2005, 28, 109–121. [Google Scholar] [CrossRef]
- Maalouf, M.; Rho, J.M.; Mattson, M.P. The neuroprotective properties of calorie restriction, the ketogenic diet, and ketone bodies. Brain Res. Rev. 2009, 59, 293–315. [Google Scholar] [CrossRef] [Green Version]
- Neal, E.G.; Chaffe, H.; Schwartz, R.H.; Lawson, M.S.; Edwards, N.; Fitzsimmons, G.; Whitney, A.; Cross, J.H. The ketogenic diet for the treatment of childhood epilepsy: A randomised controlled trial. Lancet Neurol. 2008, 7, 500–506. [Google Scholar] [CrossRef]
- Kessler, S.K.; Neal, E.G.; Camfield, C.S.; Kossoff, E.H. Dietary therapies for epilepsy: Future research. Epilepsy Behav. 2011, 22, 17–22. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kashiwaya, Y.; Takeshima, T.; Mori, N.; Nakashima, K.; Clarke, K.; Veech, R.L. d-β-Hydroxybutyrate protects neurons in models of Alzheimer’s and Parkinson’s disease. Proc. Natl. Acad. Sci. USA 2000, 97, 5440–5444. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Henderson, S.T.; Vogel, J.L.; Barr, L.J.; Garvin, F.; Jones, J.J.; Costantini, L.C. Study of the ketogenic agent AC-1202 in mild to moderate Alzheimer’s disease: A randomized, double-blind, placebo-controlled, multicenter trial. Nutr. Metab. 2009, 6, 31. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yao, J.; Chen, S.; Mao, Z.; Cadenas, E.; Brinton, R.D. 2-Deoxy-D-glucose treatment induces ketogenesis, sustains mitochondrial function, and reduces pathology in female mouse model of Alzheimer’s disease. PLoS ONE 2011, 6, e21788. [Google Scholar] [CrossRef] [PubMed]
- Tieu, K.; Perier, C.; Caspersen, C.; Teismann, P.; Wu, D.-C.; Yan, S.-D.; Naini, A.; Vila, M.; Jackson-Lewis, V.; Ramasamy, R.; et al. D-β-Hydroxybutyrate rescues mitochondrial respiration and mitigates features of Parkinson disease. J. Clin. Investig. 2003, 112, 892–901. [Google Scholar] [CrossRef] [Green Version]
- Yang, X.; Cheng, B. Neuroprotective and anti-inflammatory activities of ketogenic diet on MPTP-induced neurotoxicity. J. Mol. Neurosci. 2010, 42, 145–153. [Google Scholar] [CrossRef]
- Seyfried, T.N.; Marsh, J.; Shelton, L.M.; Huysentruyt, L.C.; Mukherjee, P. Is the restricted ketogenic diet a viable alternative to the standard of care for managing malignant brain cancer? Epilepsy Res. 2012, 100, 310–326. [Google Scholar] [CrossRef] [Green Version]
- Schmidt, M.; Pfetzer, N.; Schwab, M.; Strauss, I.; Kämmerer, U. Effects of a ketogenic diet on the quality of life in 16 patients with advanced cancer: A pilot trial. Nutr. Metab. 2011, 8, 54. [Google Scholar] [CrossRef] [Green Version]
- Evangeliou, A.; Vlachonikolis, I.; Mihailidou, H.; Spilioti, M.; Skarpalezou, A.; Makaronas, N.; Prokopiou, A.; Christodoulou, P.; Liapi-Adamidou, G.; Helidonis, E.; et al. Application of a ketogenic diet in children with autistic behavior: Pilot study. J. Child Neurol. 2003, 18, 113–118. [Google Scholar] [CrossRef]
- Masino, S.A.; Kawamura, M., Jr.; Plotkin, L.M.; Svedova, J.; DiMario, F.J., Jr.; Eigsti, I.M. The relationship between the neuromodulator adenosine and behavioral symptoms of autism. Neurosci. Lett. 2011, 500, 1–5. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhao, Z.; Lange, D.J.; Voustianiouk, A.; MacGrogan, D.; Ho, L.; Suh, J.; Humala, N.; Thiyagarajan, M.; Wang, J.; Pasinetti, G.M. A ketogenic diet as a potential novel therapeutic intervention in amyotrophic lateral sclerosis. BMC Neurosci. 2006, 7, 29. [Google Scholar] [CrossRef] [Green Version]
- Waldbaum, S.; Patel, M. Mitochondrial dysfunction and oxidative stress: A contributing link to acquired epilepsy? J. Bioenerg. Biomembr. 2010, 42, 449–455. [Google Scholar] [CrossRef] [Green Version]
- Roth, J.; Szulc, A.L.; Danoff, A. Energy, evolution, and human diseases: An overview. Am. J. Clin. Nutr. 2011, 93, 875–883. [Google Scholar] [CrossRef] [PubMed]
- Schiff, M.; Bénit, P.; Coulibaly, A.; Loublier, S.; El-Khoury, R.; Rustin, P. Mitochondrial response to controlled nutrition in health and disease. Nutr. Rev. 2011, 69, 65–75. [Google Scholar] [CrossRef]
- Masino, S.A.; Kawamura, M., Jr.; Ruskin, D.N.; Geiger, J.D.; Boison, D. Purines and neuronal excitability: Links to the ketogenic diet. Epilepsy Res. 2012, 100, 229–238. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lusardi, T.A.; Akula, K.K.; Coffman, S.Q.; Ruskin, D.N.; Masino, S.A.; Boison, D. Ketogenic diet prevents epileptogenesis and disease progression in adult mice and rats. Neuropharmacology 2015, 99, 500–509. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kawamura, M.; Ruskin, D.N.; Masino, S.A. Metabolic autocrine regulation of neurons involves cooperation among pannexin hemichannels, adenosine receptors, and KATP channels. J. Neurosci. 2010, 30, 3886–3895. [Google Scholar] [CrossRef]
- Masino, S.A.; Kawamura, M., Jr.; Wasser, C.A.; Pomeroy, L.T.; Ruskin, D.N. Adenosine, ketogenic diet and epilepsy: The emerging therapeutic relationship between metabolism and brain activity. Curr. Neuropharmacol. 2009, 7, 257–268. [Google Scholar] [CrossRef]
- Masino, S.A.; Li, T.; Theofilas, P.; Sandau, U.S.; Ruskin, D.N.; Fredholm, B.B.; Geiger, J.D.; Aronica, E.; Boison, D. A ketogenic diet suppresses seizures in mice through adenosine A 1 receptors. J. Clin. Investig. 2011, 121, 2679–2683. [Google Scholar] [CrossRef] [Green Version]
- Ferré, S.; Fuxe, K.; Fredholm, B.B.; Morelli, M.; Popoli, P. Adenosine–dopamine receptor–receptor interactions as an integrative mechanism in the basal ganglia. Trends Neurosci. 1997, 20, 482–487. [Google Scholar] [CrossRef]
- Franco, R.; Ferré, S.; Agnati, L.; Torvinen, M.; Ginés, S.; Hillion, J.; Casadó, V.; Lledó, P.M.; Zoli, M.; Lluis, C.; et al. Evidence for adenosine/dopamine receptor interactions: Indications for heteromerization. Neuropsychopharmacology 2000, 23, 50–59. [Google Scholar] [CrossRef]
- Kaplan, G.B.; Sears, M.T. Adenosine receptor agonists attenuate and adenosine receptor antagonists exacerbate opiate withdrawal signs. Psychopharmacology 1996, 123, 64–70. [Google Scholar] [CrossRef]
- Berrendero, F.; Castañé, A.; Ledent, C.; Parmentier, M.; Maldonado, R.; Valverde, O. Increase of morphine withdrawal in mice lacking A2a receptors and no changes in CB1/A2a double knockout mice. Eur. J. Neurosci. 2003, 17, 315–324. [Google Scholar] [CrossRef] [PubMed]
- Soria, G.; Castané, A.; Ledent, C.; Parmentier, M.; Maldonado, R.; Valverde, O. The lack of A 2A adenosine receptors diminishes the reinforcing efficacy of cocaine. Neuropsychopharmacology 2006, 31, 978–987. [Google Scholar] [CrossRef] [PubMed]
- Listos, J.; Talarek, S.; Fidecka, S. Involvement of adenosine receptor agonists on the development of hypersensitivity to acute dose of morphine during morphine withdrawal period. Pharmacol. Rep. 2008, 60, 679. [Google Scholar] [CrossRef]
- Marcellino, D.; Roberts, D.; Navarro, G.; Filip, M.; Agnati, L.; Lluís, C.; Franco, R.; Fuxe, K. Increase in A2A receptors in the nucleus accumbens after extended cocaine self-administration and its disappearance after cocaine withdrawal. Brain Res. 2007, 1143, 208–220. [Google Scholar] [CrossRef]
- Martinez, L.A.; Lees, M.E.; Ruskin, D.N.; Masino, S.A. A ketogenic diet diminishes behavioral responses to cocaine in young adult male and female rats. Neuropharmacology 2019, 149, 27–34. [Google Scholar] [CrossRef]
- Dencker, D.; Molander, A.; Thomsen, M.; Schlumberger, C.; Wortwein, G.; Weikop, P.; Benveniste, H.; Volkow, N.D.; Fink-Jensen, A. Ketogenic diet suppresses alcohol withdrawal syndrome in rats. Alcohol. Clin. Exp. Res. 2018, 42, 270–277. [Google Scholar] [CrossRef]
- Bornebusch, A.B.; Mason, G.F.; Tonetto, S.; Damsgaard, J.; Gjedde, A.; Fink-Jensen, A.; Thomsen, M. Effects of ketogenic diet and ketone monoester supplement on acute alcohol withdrawal symptoms in male mice. Psychopharmacology 2021, 238, 833–844. [Google Scholar] [CrossRef]
- Wiers, C.E.; Vendruscolo, L.F.; van der Veen, J.-W.; Manza, P.; Shokri-Kojori, E.; Kroll, D.S.; Feldman, D.E.; McPherson, K.L.; Biesecker, C.L.; Zhang, R.; et al. Ketogenic diet reduces alcohol withdrawal symptoms in humans and alcohol intake in rodents. Sci. Adv. 2021, 7, eabf6780. [Google Scholar] [CrossRef]
- Fritz, B.M.; Muñoz, B.; Yin, F.; Bauchle, C.; Atwood, B.K. A high-fat, high-sugar ‘Western’ diet alters dorsal striatal glutamate, opioid, and dopamine transmission in mice. Neuroscience 2018, 372, 1–15. [Google Scholar] [CrossRef] [PubMed]
- Barson, J.R.; Morganstern, I.; Leibowitz, S.F. Neurobiology of consummatory behavior: Mechanisms underlying overeating and drug use. ILAR J. 2012, 53, 35–58. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kawahara, Y.; Kaneko, F.; Yamada, M.; Kishikawa, Y.; Kawahara, H.; Nishi, A. Food reward-sensitive interaction of ghrelin and opioid receptor pathways in mesolimbic dopamine system. Neuropharmacology 2013, 67, 395–402. [Google Scholar] [CrossRef]
- Navarrete, F.; Rubio, G.; Manzanares, J. Effects of naltrexone plus topiramate on ethanol self-administration and tyrosine hydroxylase gene expression changes. Addict. Biol. 2014, 19, 862–873. [Google Scholar] [CrossRef]
- Samson, H.H.; Tolliver, G.A.; Pfeffer, A.O.; Sadeghi, K.G.; Mills, F.G. Oral ethanol reinforcement in the rat: Effect of the partial inverse benzodiazepine agonist RO15-4513. Pharmacol. Biochem. Behav. 1987, 27, 517–519. [Google Scholar] [CrossRef]
- de Macedo, I.C.; de Freitas, J.S.; da Silva Torres, I.L. The influence of palatable diets in reward system activation: A mini review. Adv. Pharmacol. Sci. 2016, 2016, 7238679. [Google Scholar] [CrossRef] [Green Version]
- Blanco-Gandía, M.C.; Miñarro, J.; Rodríguez-Arias, M. Common Neural Mechanisms of Palatable Food Intake and Drug Abuse: Knowledge Obtained with Animal Models. Curr. Pharm. Des. 2020, 26, 2372–2384. [Google Scholar] [CrossRef]
- Chen, Y.; Ouyang, X.; Hoque, R.; Garcia-Martinez, I.; Yousaf, M.N.; Tonack, S.; Offermanns, S.; Dubuquoy, L.; Louvet, A.; Mathurin, P.; et al. β-Hydroxybutyrate protects from alcohol-induced liver injury via a Hcar2-cAMP dependent pathway. J. Hepatol. 2018, 69, 687–696. [Google Scholar] [CrossRef]
- Newman, J.C.; Verdin, E. Ketone bodies as signaling metabolites. Trends Endocrinol. Metab. 2014, 25, 42–52. [Google Scholar] [CrossRef] [Green Version]
- Jiang, Y.; Yang, Y.; Wang, S.; Ding, Y.; Guo, Y.; Zhang, M.-M.; Wen, S.-Q.; Ding, M.-P. Ketogenic diet protects against epileptogenesis as well as neuronal loss in amygdaloid-kindling seizures. Neurosci. Lett. 2012, 508, 22–26. [Google Scholar] [CrossRef]
- Ribeiro, L.C.; Chittó, A.L.; Müller, A.P.; Rocha, J.K.; Da Silva, M.C.; Quincozes-Santos, A.; Nardin, P.; Rotta, L.; Ziegler, D.R.; Gonçalves, C.-A.; et al. Ketogenic diet-fed rats have increased fat mass and phosphoenolpyruvate carboxykinase activity. Mol. Nutr. Food Res. 2008, 52, 1365–1371. [Google Scholar] [CrossRef]
- Srivastava, S.; Baxa, U.; Niu, G.; Chen, X.L.; Veech, R. A ketogenic diet increases brown adipose tissue mitochondrial proteins and UCP1 levels in mice. IUBMB Life 2013, 65, 58–66. [Google Scholar] [CrossRef] [PubMed]
- Panlilio, L.V.; Goldberg, S.R. Self-administration of drugs in animals and humans as a model and an investigative tool. Addiction 2007, 102, 1863–1870. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Richardson, N.R.; Roberts, D.C. Progressive ratio schedules in drug self-administration studies in rats: A method to evaluate reinforcing efficacy. J. Neurosci. Methods 1996, 66, 1–11. [Google Scholar] [CrossRef]
- Samson, H.H.; Czachowski, C.L. Behavioral measures of alcohol self-administration and intake control: Rodent models. Int. Rev. Neurobiol. 2003, 54, 107–143. [Google Scholar] [CrossRef] [PubMed]
- Sanchis-Segura, C.; Spanagel, R. Behavioural assessment of drug reinforcement and addictive features in rodents: An overview. Addict. Biol. 2006, 11, 2–38. [Google Scholar] [CrossRef]
- McGuire, L.C.; Cruickshank, A.M.; Munro, P.T. Alcoholic ketoacidosis. Emerg. Med. J. 2006, 23, 417–420. [Google Scholar] [CrossRef] [PubMed]
- Alhadeff, A.L.; Goldstein, N.; Park, O.; Klima, M.L.; Vargas, A.; Betley, J.N. Natural and drug rewards engage distinct pathways that converge on coordinated hypothalamic and reward circuits. Neuron 2019, 103, 891–908. [Google Scholar] [CrossRef]
- Gibson, A.A.; Seimon, R.V.; Lee, C.M.; Ayre, J.; Franklin, J.; Markovic, T.P.; Caterson, I.D.; Sainsbury, A. Do ketogenic diets really suppress appetite? A systematic review and meta-analysis. Obes. Rev. 2015, 16, 64–76. [Google Scholar] [CrossRef] [Green Version]
- Kwiterovich, P.O., Jr.; Vining, E.P.; Pyzik, P.; Skolasky, R., Jr.; Freeman, J.M. Effect of a high-fat ketogenic diet on plasma levels of lipids, lipoproteins, and apolipoproteins in children. JAMA 2003, 290, 912–920. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kinzig, K.P.; Taylor, R.J. Maintenance on a ketogenic diet: Voluntary exercise, adiposity and neuroendocrine effects. Int. J. Obes. 2009, 33, 824–830. [Google Scholar] [CrossRef] [Green Version]
- Vining, E.P. Clinical efficacy of the ketogenic diet. Epilepsy Res. 1999, 37, 181–190. [Google Scholar] [CrossRef]
- Kang, H.C.; Chung, D.E.; Kim, D.W.; Kim, H.D. Early- and late-onset complications of the ketogenic diet for intractable epilepsy. Epilepsia 2004, 45, 1116–1123. [Google Scholar] [CrossRef]
- Noakes, M.; Foster, P.R.; Keogh, J.B.; James, A.P.; Mamo, J.C.; Clifton, P.M. Comparison of isocaloric very low carbohydrate/high saturated fat and high carbohydrate/low saturated fat diets on body composition and cardiovascular risk. Nutr. Metab. 2006, 3, 7. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nuttall, F.Q.; Almokayyad, R.M.; Gannon, M.C. Comparison of a carbohydrate-free diet vs. fasting on plasma glucose, insulin and glucagon in type 2 diabetes. Metabolism 2015, 64, 253–262. [Google Scholar] [CrossRef]
- Furth, S.L.; Casey, J.C.; Pyzik, P.L.; Neu, A.M.; Docimo, S.G.; Vining, E.P.; Freeman, J.M.; Fivush, B.A. Risk factors for urolithiasis in children on the ketogenic diet. Pediatr. Nephrol. 2000, 15, 125–128. [Google Scholar] [CrossRef]
- Jeynes, K.D.; Gibson, E.L. The importance of nutrition in aiding recovery from substance use disorders: A review. Drug Alcohol. Depend. 2017, 179, 229–239. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kesl, S.L.; Poff, A.M.; Ward, N.P.; Fiorelli, T.N.; Ari, C.; Van Putten, A.J.; Sherwood, J.W.; Arnold, P.; D’Agostino, D.P. Effects of exogenous ketone supplementation on blood ketone, glucose, triglyceride, and lipoprotein levels in Sprague-Dawley rats. Nutr. Metab. 2016, 13, 9. [Google Scholar] [CrossRef] [Green Version]
- Kashiwaya, Y.; Bergman, C.; Lee, J.H.; Wan, R.; King, M.T.; Mughal, M.R.; Okun, E.; Clarke, K.; Mattson, M.P.; Veech, R.L. A ketone ester diet exhibits anxiolytic and cognition-sparing properties, and lessens amyloid and tau pathologies in a mouse model of Alzheimer’s disease. Neurobiol. Aging 2013, 34, 1530–1539. [Google Scholar] [CrossRef] [Green Version]
- Fuxe, K.; Marcellino, D.; Borroto-Escuela, D.O.; Guescini, M.; Fernandez-Duenas, V.; Tanganelli, S.; Rivera, A.; Ciruela, F.; Agnati, L.F. Adenosine–dopamine interactions in the pathophysiology and treatment of CNS disorders. CNS Neurosci. Ther. 2010, 16, 18–42. [Google Scholar] [CrossRef] [PubMed]
- Weerts, E.M.; Griffiths, R.R. The adenosine receptor antagonist CGS15943 reinstates cocaine-seeking behavior and maintains self-administration in baboons. Psychopharmacology 2003, 168, 155–163. [Google Scholar] [CrossRef] [PubMed]
- Knapp, C.M.; Foye, M.M.; Cottam, N.; Ciraulo, D.A.; Kornetsky, C. Adenosine agonists CGS 21680 and NECA inhibit the initiation of cocaine self-administration. Pharmacol. Biochem. Behav. 2001, 68, 797–803. [Google Scholar] [CrossRef]
- Filip, M.; Frankowska, M.; Zaniewska, M.; Przegaliński, E.; Műller, C.E.; Agnati, L.; Franco, R.; Roberts, D.; Fuxe, K. Involvement of adenosine A2A and dopamine receptors in the locomotor and sensitizing effects of cocaine. Brain Res. 2006, 1077, 67–80. [Google Scholar] [CrossRef]
- Chen, J.-F.; Beilstein, M.; Xu, Y.-H.; Turner, T.; Moratalla, R.; Standaert, D.; Aloyo, V.; Fink, J.; Schwarzschild, M. Selective attenuation of psychostimulant-induced behavioral responses in mice lacking A2A adenosine receptors. Neuroscience 2000, 97, 195–204. [Google Scholar] [CrossRef]
- Feltmann, K.; Borroto-Escuela, D.O.; Rüegg, J.; Pinton, L.; Sergio, T.D.O.; Narváez, M.; Jimenez-Beristain, A.; Ekström, T.J.; Fuxe, K.; Steensland, P. Effects of Long-Term Alcohol Drinking on the Dopamine D2 Receptor: Gene Expression and Heteroreceptor Complexes in the Striatum in Rats. Alcohol. Clin. Exp. Res. 2018, 42, 338–351. [Google Scholar] [CrossRef] [Green Version]
- Di Bonaventura, M.V.M.; Cifani, C.; Lambertucci, C.; Volpini, R.; Cristalli, G.; Froldi, R.; Massi, M. Effects of A 2A adenosine receptor blockade or stimulation on alcohol intake in alcohol-preferring rats. Psychopharmacology 2012, 219, 945–957. [Google Scholar] [CrossRef]
- Nam, H.W.; Hinton, D.J.; Kang, N.Y.; Kim, T.; Lee, M.R.; Oliveros, A.; Adams, C.; Ruby, C.L.; Choi, D.-S. Adenosine transporter ENT1 regulates the acquisition of goal-directed behavior and ethanol drinking through A2A receptor in the dorsomedial striatum. J. Neurosci. 2013, 33, 4329–4338. [Google Scholar] [CrossRef] [Green Version]
- Church, W.H.; Adams, R.E.; Wyss, L.S. Ketogenic diet alters dopaminergic activity in the mouse cortex. Neurosci. Lett. 2014, 571, 1–4. [Google Scholar] [CrossRef]
- Enoch, M.A. The role of GABAA receptors in the development of alcoholism. Pharmacol. Biochem. Behav. 2008, 90, 95–104. [Google Scholar] [CrossRef] [Green Version]
- Gano, L.B.; Patel, M.; Rho, J.M. Ketogenic diets, mitochondria, and neurological diseases. J. Lipid Res. 2014, 55, 2211–2228. [Google Scholar] [CrossRef] [Green Version]
- Mironov, S.L.; Richter, D.W. Intracellular signalling pathways modulate KATP channels in inspiratory brainstem neurones and their hypoxic activation: Involvement of metabotropic receptors, G-proteins and cytoskeleton. Brain Res. 2000, 853, 60–67. [Google Scholar] [CrossRef]
- Koob, G.F. A role for GABA mechanisms in the motivational effects of alcohol. Biochem. Pharmacol. 2004, 68, 1515–1525. [Google Scholar] [CrossRef] [PubMed]
- Janak, P.H.; Gill, T.M. Comparison of the effects of allopregnanolone with direct GABAergic agonists on ethanol self-administration with and without concurrently available sucrose. Alcohol 2003, 30, 1–7. [Google Scholar] [CrossRef]
- Colombo, G.; Vacca, G.; Serra, S.; Brunetti, G.; Carai, M.A.; Gessa, G.L. Baclofen suppresses motivation to consume alcohol in rats. Psychopharmacology 2003, 167, 221–224. [Google Scholar] [CrossRef]
- Harrold, J.A.; Elliott, J.C.; King, P.J.; Widdowson, P.S.; Williams, G. Down-regulation of cannabinoid-1 (CB-1) receptors in specific extrahypothalamic regions of rats with dietary obesity: A role for endogenous cannabinoids in driving appetite for palatable food? Brain Res. 2002, 952, 232–238. [Google Scholar] [CrossRef]
- Hájos, N.; Freund, T.F. Distinct cannabinoid sensitive receptors regulate hippocampal excitation and inhibition. Chem. Phys. Lipids 2002, 121, 73–82. [Google Scholar] [CrossRef]
- Berghuis, P.; Rajnicek, A.M.; Morozov, Y.M.; Ross, R.A.; Mulder, J.; Urbán, G.M.; Monory, K.; Marsicano, G.; Matteoli, M.; Canty, A.; et al. Hardwiring the brain: Endocannabinoids shape neuronal connectivity. Science 2007, 316, 1212–1216. [Google Scholar] [CrossRef] [Green Version]
- Peciña, S.; Smith, K.S. Hedonic and motivational roles of opioids in food reward: Implications for overeating disorders. Pharmacol. Biochem. Behav. 2010, 97, 34–46. [Google Scholar] [CrossRef] [PubMed]
- Gosnell, B.A.; Levine, A.S. Reward systems and food intake: Role of opioids. Int. J. Obes. 2009, 33, 54–58. [Google Scholar] [CrossRef] [PubMed] [Green Version]

Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Blanco-Gandía, M.d.C.; Ródenas-González, F.; Pascual, M.; Reguilón, M.D.; Guerri, C.; Miñarro, J.; Rodríguez-Arias, M. Ketogenic Diet Decreases Alcohol Intake in Adult Male Mice. Nutrients 2021, 13, 2167. https://doi.org/10.3390/nu13072167
Blanco-Gandía MdC, Ródenas-González F, Pascual M, Reguilón MD, Guerri C, Miñarro J, Rodríguez-Arias M. Ketogenic Diet Decreases Alcohol Intake in Adult Male Mice. Nutrients. 2021; 13(7):2167. https://doi.org/10.3390/nu13072167
Chicago/Turabian StyleBlanco-Gandía, María del Carmen, Francisco Ródenas-González, María Pascual, Marina Daiana Reguilón, Consuelo Guerri, José Miñarro, and Marta Rodríguez-Arias. 2021. "Ketogenic Diet Decreases Alcohol Intake in Adult Male Mice" Nutrients 13, no. 7: 2167. https://doi.org/10.3390/nu13072167