
Beneficial Effects of Exogenous Ketogenic Supplements on Aging Processes and Age-Related Neurodegenerative Diseases




Abstract
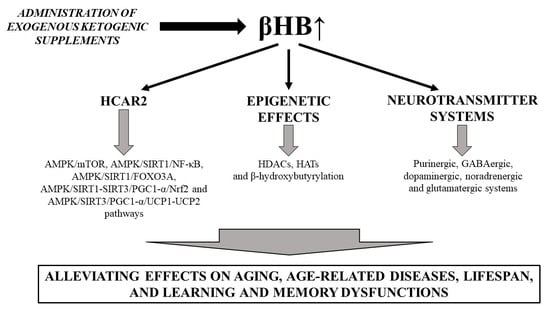
:
1. Introduction
2. Main Features of Aging Processes
2.1. Nutrient Sensing Pathways
2.2. Telomere Shortening and Genome Instability
2.3. Epigenetic Alterations
2.4. Mitochondrial Dysfunction
2.5. Altered Intercellular Communication: Increased Inflammatory Processes
2.6. Cellular Senescence
2.7. Loss of Proteostasis and Stem Cell Exhaustion
2.8. Effects of Senotherapeutic Drugs on Aging Hallmarks and Neurodegenerative Diseases: Main Signaling Pathways
3. Alleviating Effects of Ketosis on Lifespan, Aging and Age-Related Neurodegenerative Diseases
3.1. Ketosis-Evoked Neuroprotective Effects and Downstream Signaling Pathways
3.2. Beneficial Effects of EKSs-Evoked Ketosis (βHB) on Lifespan, Aging, Age-Related Diseases, as Well as Learning and Memory Dysfunctions
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Abbreviations
References
- Campisi, J.; Kapahi, P.; Lithgow, G.J.; Melov, S.; Newman, J.C.; Verdin, E. From discoveries in ageing research to therapeutics for healthy ageing. Nature 2019, 571, 183–192. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, Z.; Zhang, Z.; Ren, Y.; Wang, Y.; Fang, J.; Yue, H.; Ma, S.; Guan, F. Aging and age-related diseases: From mechanisms to therapeutic strategies. Biogerontology 2021, 22, 165–187. [Google Scholar] [CrossRef] [PubMed]
- Sen, A.; Capelli, V.; Husain, M. Cognition and dementia in older patients with epilepsy. Brain 2018, 141, 1592–1608. [Google Scholar] [CrossRef]
- United Nations, Department of Economic and Social Affairs, Population Division. World Population Ageing 2019: Highlights; United Nations: New York, NY, USA, 2019; ISBN 978-92-1-148325-3. [Google Scholar]
- Drygalski, K.; Fereniec, E.; Koryciński, K.; Chomentowski, A.; Kiełczewska, A.; Odrzygóźdź, C.; Modzelewska, B. Resveratrol and Alzheimer’s disease. From molecular pathophysiology to clinical trials. Exp. Gerontol. 2018, 113, 36–47. [Google Scholar] [CrossRef]
- Yang, C.; Zhang, W.; Dong, X.; Fu, C.; Yuan, J.; Xu, M.; Liang, Z.; Qiu, C.; Xu, C. A natural product solution to aging and aging-associated diseases. Pharmacol. Ther. 2020, 216, 107673. [Google Scholar] [CrossRef] [PubMed]
- De Magalhães, J.P.; Stevens, M.; Thornton, D. The Business of Anti-Aging Science. Trends Biotechnol. 2017, 35, 1062–1073. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Anisimov, V.N.; Zabezhinski, M.A.; Popovich, I.G.; Piskunova, T.S.; Semenchenko, A.V.; Tyndyk, M.L.; Yurova, M.N.; Rosenfeld, S.V.; Blagosklonny, M.V. Rapamycin increases lifespan and inhibits spontaneous tumorigenesis in inbred female mice. Cell Cycle 2011, 10, 4230–4236. [Google Scholar] [CrossRef] [PubMed]
- Bitto, A.; Ito, T.K.; Pineda, V.V.; LeTexier, N.J.; Huang, H.Z.; Sutlief, E.; Tung, H.; Vizzini, N.; Chen, B.; Smith, K.; et al. Transient rapamycin treatment can increase lifespan and healthspan in middle-aged mice. Elife 2016, 5, e16351. [Google Scholar] [CrossRef]
- Mannick, J.B.; Morris, M.; Hockey, H.P.; Roma, G.; Beibel, M.; Kulmatycki, K.; Watkins, M.; Shavlakadze, T.; Zhou, W.; Quinn, D.; et al. TORC1 inhibition enhances immune function and reduces infections in the elderly. Sci. Transl. Med. 2018, 10, eaaq1564. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bannister, C.A.; Holden, S.E.; Jenkins-Jones, S.; Morgan, C.L.; Halcox, J.P.; Schernthaner, G.; Mukherjee, J.; Currie, C.J. Can people with type 2 diabetes live longer than those without? A comparison of mortality in people initiated with metformin or sulphonylurea monotherapy and matched, non-diabetic controls. Diabetes Obes. Metab. 2014, 16, 1165–1173. [Google Scholar] [CrossRef] [Green Version]
- Barzilai, N.; Crandall, J.P.; Kritchevsky, S.B.; Espeland, M.A. Metformin as a Tool to Target Aging. Cell Metab. 2016, 23, 1060–1065. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bonkowski, M.S.; Sinclair, D.A. Slowing ageing by design: The rise of NAD+ and sirtuin-activating compounds. Nat. Rev. Mol. Cell Biol. 2016, 17, 679–690. [Google Scholar] [CrossRef]
- Kane, A.E.; Sinclair, D.A. Sirtuins and NAD+ in the Development and Treatment of Metabolic and Cardiovascular Diseases. Circ. Res. 2018, 123, 868–885. [Google Scholar] [CrossRef] [PubMed]
- Kirkland, J.L.; Tchkonia, T.; Zhu, Y.; Niedernhofer, L.J.; Robbins, P.D. The Clinical Potential of Senolytic Drugs. J. Am. Geriatr. Soc. 2017, 65, 2297–2301. [Google Scholar] [CrossRef] [PubMed]
- Brownlow, M.L.; Jung, S.H.; Moore, R.J.; Bechmann, N.; Jankord, R. Nutritional Ketosis Affects Metabolism and Behavior in Sprague-Dawley Rats in Both Control and Chronic Stress Environments. Front. Mol. Neurosci. 2017, 10, 129. [Google Scholar] [CrossRef] [Green Version]
- Brunengraber, H. Potential of ketone body esters for parenteral and oral nutrition. Nutrition 1997, 13, 233–235. [Google Scholar] [CrossRef]
- Clarke, K.; Tchabanenko, K.; Pawlosky, R.; Carter, E.; Knight, N.S.; Murray, A.J.; Cochlin, L.E.; King, M.T.; Wong, A.W.; Roberts, A.; et al. Oral 28-day and developmental toxicity studies of (R)-3-hydroxybutyl (R)-3-hydroxybutyrate. Regul. Toxicol. Pharmacol. 2012, 63, 196–208. [Google Scholar] [CrossRef] [Green Version]
- Clarke, K.; Tchabanenko, K.; Pawlosky, R.; Carter, E.; Todd King, M.; Musa-Veloso, K.; Ho, M.; Roberts, A.; Robertson, J.; Vanitallie, T.B.; et al. Kinetics, safety and tolerability of (R)-3-hydroxybutyl (R)-3-hydroxybutyrate in healthy adult subjects. Regul. Toxicol. Pharmacol. 2012, 63, 401–408. [Google Scholar] [CrossRef] [Green Version]
- Schönfeld, P.; Wojtczak, L. Short- and medium-chain fatty acids in energy metabolism: The cellular perspective. J. Lipid Res. 2016, 57, 943–954. [Google Scholar] [CrossRef] [Green Version]
- Kovács, Z.; Brunner, B.; D’Agostino, D.P.; Ari, C. Age- and Sex-Dependent Modulation of Exogenous Ketone Supplement-Evoked Effects on Blood Glucose and Ketone Body Levels in Wistar Albino Glaxo Rijswijk Rats. Front. Neurosci. 2021, 14, 618422. [Google Scholar] [CrossRef]
- Achanta, L.B.; Rae, C.D. β-Hydroxybutyrate in the Brain: One Molecule, Multiple Mechanisms. Neurochem. Res. 2017, 42, 35–49. [Google Scholar] [CrossRef]
- Koppel, S.J.; Swerdlow, R.H. Neuroketotherapeutics: A modern review of a century-old therapy. Neurochem. Int. 2018, 117, 114–125. [Google Scholar] [CrossRef] [PubMed]
- Newman, J.C.; Verdin, E. Ketone bodies as signaling metabolites. Trends Endocrinol. Metab. 2014, 25, 42–52. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Soto-Mota, A.; Norwitz, N.G.; Clarke, K. Why a d-β-hydroxybutyrate monoester? Biochem. Soc. Trans. 2020, 48, 51–59. [Google Scholar] [CrossRef] [Green Version]
- Ari, C.; Kovács, Z.; Juhasz, G.; Murdun, C.; Goldhagen, C.R.; Koutnik, A.P.; Poff, A.M.; Kesl, S.L.; D’Agostino, D.P. Exogenous Ketone Supplements Reduce Anxiety-Related Behavior in Sprague-Dawley and Wistar Albino Glaxo/Rijswijk Rats. Front. Mol. Neurosci. 2016, 9, 137. [Google Scholar] [CrossRef] [Green Version]
- Kesl, S.L.; Poff, A.M.; Ward, N.P.; Fiorelli, T.N.; Ari, C.; Van Putten, A.J.; Sherwood, J.W.; Arnold, P.; D’Agostino, D.P. Effects of exogenous ketone supplementation on blood ketone, glucose, triglyceride, and lipoprotein levels in Sprague-Dawley rats. Nutr. Metab. 2016, 13, 9. [Google Scholar] [CrossRef] [Green Version]
- Myette-Côté, É.; Neudorf, H.; Rafiei, H.; Clarke, K.; Little, J.P. Prior ingestion of exogenous ketone monoester attenuates the glycaemic response to an oral glucose tolerance test in healthy young individuals. J. Physiol. 2018, 596, 1385–1395. [Google Scholar] [CrossRef] [PubMed]
- Stubbs, B.J.; Cox, P.J.; Evans, R.D.; Santer, P.; Miller, J.J.; Faull, O.K.; Magor-Elliott, S.; Hiyama, S.; Stirling, M.; Clarke, K. On the Metabolism of Exogenous Ketones in Humans. Front. Physiol. 2017, 8, 848. [Google Scholar] [CrossRef]
- Hashim, S.A.; VanItallie, T.B. Ketone body therapy: From the ketogenic diet to the oral administration of ketone ester. J. Lipid Res. 2014, 55, 1818–1826. [Google Scholar] [CrossRef] [Green Version]
- McDonald, T.J.; Cervenka, M.C. Lessons learned from recent clinical trials of ketogenic diet therapies in adults. Curr. Opin. Clin. Nutr. Metab. Care 2019, 22, 418–424. [Google Scholar] [CrossRef]
- Fortier, M.; Castellano, C.A.; St-Pierre, V.; Myette-Côté, É.; Langlois, F.; Roy, M.; Morin, M.C.; Bocti, C.; Fulop, T.; Godin, J.P.; et al. A ketogenic drink improves cognition in mild cognitive impairment: Results of a 6-month RCT. Alzheimers Dement. 2021, 17, 543–552. [Google Scholar] [CrossRef]
- Soto-Mota, A.; Norwitz, N.G.; Evans, R.; Clarke, K.; Barber, T.M. Exogenous ketosis in patients with type 2 diabetes: Safety, tolerability and effect on glycaemic control. Endocrinol. Diabetes Metab. 2021, e00264. [Google Scholar] [CrossRef]
- Camberos-Luna, L.; Massieu, L. Therapeutic strategies for ketosis induction and their potential efficacy for the treatment of acute brain injury and neurodegenerative diseases. Neurochem. Int. 2020, 133, 104614. [Google Scholar] [CrossRef]
- Han, Y.M.; Ramprasath, T.; Zou, M.H. β-hydroxybutyrate and its metabolic effects on age-associated pathology. Exp. Mol. Med. 2020, 52, 548–555. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kovács, Z.; D’Agostino, D.P.; Dobolyi, A.; Ari, C. Adenosine A1 Receptor Antagonism Abolished the Anti-seizure Effects of Exogenous Ketone Supplementation in Wistar Albino Glaxo Rijswijk Rats. Front. Mol. Neurosci. 2017, 10, 235. [Google Scholar] [CrossRef] [Green Version]
- Branco, A.F.; Ferreira, A.; Simões, R.F.; Magalhães-Novais, S.; Zehowski, C.; Cope, E.; Silva, A.M.; Pereira, D.; Sardão, V.A.; Cunha-Oliveira, T. Ketogenic diets: From cancer to mitochondrial diseases and beyond. Eur. J. Clin. Investig. 2016, 46, 285–298. [Google Scholar] [CrossRef] [Green Version]
- Kim, D.Y.; Simeone, K.A.; Simeone, T.A.; Pandya, J.D.; Wilke, J.C.; Ahn, Y.; Geddes, J.W.; Sullivan, P.G.; Rho, J.M. Ketone bodies mediate antiseizure effects through mitochondrial permeability transition. Ann. Neurol. 2015, 78, 77–87. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kovács, Z.; D’Agostino, D.P.; Diamond, D.; Kindy, M.S.; Rogers, C.; Ari, C. Therapeutic Potential of Exogenous Ketone Supplement Induced Ketosis in the Treatment of Psychiatric Disorders: Review of Current Literature. Front. Psychiatry 2019, 10, 363. [Google Scholar] [CrossRef]
- Berk, B.A.; Law, T.H.; Packer, R.M.A.; Wessmann, A.; Bathen-Nöthen, A.; Jokinen, T.S.; Knebel, A.; Tipold, A.; Pelligand, L.; Meads, Z.; et al. A multicenter randomized controlled trial of medium-chain triglyceride dietary supplementation on epilepsy in dogs. J. Vet. Intern. Med. 2020, 34, 1248–1259. [Google Scholar] [CrossRef] [Green Version]
- Ciarlone, S.L.; Grieco, J.C.; D’Agostino, D.P.; Weeber, E.J. Ketone ester supplementation attenuates seizure activity, and improves behavior and hippocampal synaptic plasticity in an Angelman syndrome mouse model. Neurobiol. Dis. 2016, 96, 38–46. [Google Scholar] [CrossRef] [Green Version]
- D’Agostino, D.P.; Pilla, R.; Held, H.E.; Landon, C.S.; Puchowicz, M.; Brunengraber, H.; Ari, C.; Arnold, P.; Dean, J.B. Therapeutic ketosis with ketone ester delays central nervous system oxygen toxicity seizures in rats. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2013, 304, 829–836. [Google Scholar] [CrossRef] [Green Version]
- Kashiwaya, Y.; Bergman, C.; Lee, J.H.; Wan, R.; King, M.T.; Mughal, M.R.; Okun, E.; Clarke, K.; Mattson, M.P.; Veech, R.L. A ketone ester diet exhibits anxiolytic and cognition-sparing properties, and lessens amyloid and tau pathologies in a mouse model of Alzheimer’s disease. Neurobiol. Aging 2013, 34, 1530–1539. [Google Scholar] [CrossRef] [Green Version]
- Kovács, Z.; D’Agostino, D.P.; Ari, C. Anxiolytic Effect of Exogenous Ketone Supplementation Is Abolished by Adenosine A1 Receptor Inhibition in Wistar Albino Glaxo/Rijswijk Rats. Front. Behav. Neurosci. 2018, 12, 29. [Google Scholar] [CrossRef] [Green Version]
- Ari, C.; Zippert, M.; D’Agostino, D.P. Neuroregeneration improved by ketones. FASEB J. 2018, 32, 545–549. [Google Scholar] [CrossRef]
- Ari, C.; Pilla, R.; D’Agostino, D. Nutritional/metabolic therapies in animal models of amyotrophic lateral sclerosis, Alzheimer’s disease, and seizures. In Bioactive Nutraceuticals and Dietary Supplements in Neurological and Brain Disease; Ross Watson, R., Preedy, V.R., Eds.; Academic Press: New York, NY, USA, 2015; pp. 449–459. ISBN 978-0-12-411462-3. [Google Scholar]
- Newport, M.T.; VanItallie, T.B.; Kashiwaya, Y.; King, M.T.; Veech, R.L. A new way to produce hyperketonemia: Use of ketone ester in a case of Alzheimer’s disease. Alzheimers Dement. 2015, 11, 99–103. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tefera, T.W.; Wong, Y.; Barkl-Luke, M.E.; Ngo, S.T.; Thomas, N.K.; McDonald, T.S.; Borges, K. Triheptanoin Protects Motor Neurons and Delays the Onset of Motor Symptoms in a Mouse Model of Amyotrophic Lateral Sclerosis. PLoS ONE 2016, 11, e0161816. [Google Scholar] [CrossRef] [Green Version]
- Maalouf, M.; Rho, J.M.; Mattson, M.P. The neuroprotective properties of calorie restriction, the ketogenic diet, and ketone bodies. Brain Res. Rev. 2009, 59, 293–315. [Google Scholar] [CrossRef] [Green Version]
- Wu, Y.; Gong, Y.; Luan, Y.; Li, Y.; Liu, J.; Yue, Z.; Yuan, B.; Sun, J.; Xie, C.; Li, L.; et al. BHBA treatment improves cognitive function by targeting pleiotropic mechanisms in transgenic mouse model of Alzheimer’s disease. FASEB J. 2020, 34, 1412–1429. [Google Scholar] [CrossRef] [Green Version]
- Roberts, M.N.; Wallace, M.A.; Tomilov, A.A.; Zhou, Z.; Marcotte, G.R.; Tran, D.; Perez, G.; Gutierrez-Casado, E.; Koike, S.; Knotts, T.A.; et al. A Ketogenic Diet Extends Longevity and Healthspan in Adult Mice. Cell Metab. 2017, 26, 539–546. [Google Scholar] [CrossRef] [Green Version]
- Veech, R.L.; Bradshaw, P.C.; Clarke, K.; Curtis, W.; Pawlosky, R.; King, M.T. Ketone bodies mimic the life span extending properties of caloric restriction. IUBMB Life 2017, 69, 305–314. [Google Scholar] [CrossRef]
- Newman, J.C.; Verdin, E. β-hydroxybutyrate: Much more than a metabolite. Diabetes Res. Clin. Pract. 2014, 106, 173–181. [Google Scholar] [CrossRef] [Green Version]
- Rahman, M.; Muhammad, S.; Khan, M.A.; Chen, H.; Ridder, D.A.; Müller-Fielitz, H.; Pokorná, B.; Vollbrandt, T.; Stölting, I.; Nadrowitz, R.; et al. The β-hydroxybutyrate receptor HCA2 activates a neuroprotective subset of macrophages. Nat. Commun. 2014, 5, 3944. [Google Scholar] [CrossRef]
- Fu, S.P.; Wang, J.F.; Xue, W.J.; Liu, H.M.; Liu, B.R.; Zeng, Y.L.; Li, S.N.; Huang, B.X.; Lv, Q.K.; Wang, W.; et al. Anti-inflammatory effects of BHBA in both in vivo and in vitro Parkinson’s disease models are mediated by GPR109A-dependent mechanisms. J. Neuroinflamm. 2015, 12, 9. [Google Scholar] [CrossRef] [Green Version]
- Rezq, S.; Abdel-Rahman, A.A. Central GPR109A Activation Mediates Glutamate-Dependent Pressor Response in Conscious Rats. J. Pharmacol. Exp. Ther. 2016, 356, 456–465. [Google Scholar] [CrossRef] [Green Version]
- Graff, E.C.; Fang, H.; Wanders, D.; Judd, R.L. Anti-inflammatory effects of the hydroxycarboxylic acid receptor 2. Metabolism 2016, 65, 102–113. [Google Scholar] [CrossRef]
- Wakade, C.; Chong, R.; Bradley, E.; Thomas, B.; Morgan, J. Upregulation of GPR109A in Parkinson’s disease. PLoS ONE 2014, 9, e109818. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Han, Y.M.; Bedarida, T.; Ding, Y.; Somba, B.K.; Lu, Q.; Wang, Q.; Song, P.; Zou, M.H. β-Hydroxybutyrate Prevents Vascular Senescence through hnRNP A1-Mediated Upregulation of Oct4. Mol. Cell 2018, 71, 1064–1078. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Edwards, C.; Canfield, J.; Copes, N.; Rehan, M.; Lipps, D.; Bradshaw, P.C. D-beta-hydroxybutyrate extends lifespan in C. elegans. Aging 2014, 6, 621–644. [Google Scholar] [CrossRef] [Green Version]
- Trevisan, K.; Cristina-Pereira, R.; Silva-Amaral, D.; Aversi-Ferreira, T.A. Theories of Aging and the Prevalence of Alzheimer’s Disease. Biomed. Res. Int. 2019, 2019, 9171424. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ascherio, A.; Schwarzschild, M.A. The epidemiology of Parkinson’s disease: Risk factors and prevention. Lancet Neurol. 2016, 15, 1257–1272. [Google Scholar] [CrossRef]
- Hou, Y.; Dan, X.; Babbar, M.; Wei, Y.; Hasselbalch, S.G.; Croteau, D.L.; Bohr, V.A. Ageing as a risk factor for neurodegenerative disease. Nat. Rev. Neurol. 2019, 15, 565–581. [Google Scholar] [CrossRef] [PubMed]
- Pandya, V.A.; Patani, R. Decoding the relationship between ageing and amyotrophic lateral sclerosis: A cellular perspective. Brain 2020, 143, 1057–1072. [Google Scholar] [CrossRef]
- Broughton, S.; Partridge, L. Insulin/IGF-like signalling, the central nervous system and aging. Biochem. J. 2009, 418, 1–12. [Google Scholar] [CrossRef] [Green Version]
- Klement, R.J.; Fink, M.K. Dietary and pharmacological modification of the insulin/IGF-1 system: Exploiting the full repertoire against cancer. Oncogenesis 2016, 5, e193. [Google Scholar] [CrossRef] [Green Version]
- Martin, B.; Mattson, M.P.; Maudsley, S. Caloric restriction and intermittent fasting: Two potential diets for successful brain aging. Ageing Res. Rev. 2006, 5, 332–353. [Google Scholar] [CrossRef] [Green Version]
- Santos, J.; Leitão-Correia, F.; Sousa, M.J.; Leão, C. Dietary Restriction and Nutrient Balance in Aging. Oxid. Med. Cell Longev. 2016, 2016, 4010357. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- López-Otín, C.; Blasco, M.A.; Partridge, L.; Serrano, M.; Kroemer, G. The hallmarks of aging. Cell 2013, 153, 1194–1217. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nugent, S.; Tremblay, S.; Chen, K.W.; Ayutyanont, N.; Roontiva, A.; Castellano, C.A.; Fortier, M.; Roy, M.; Courchesne-Loyer, A.; Bocti, C.; et al. Brain glucose and acetoacetate metabolism: A comparison of young and older adults. Neurobiol. Aging 2014, 35, 1386–1395. [Google Scholar] [CrossRef] [PubMed]
- Schultz, M.B.; Sinclair, D.A. When stem cells grow old: Phenotypes and mechanisms of stem cell aging. Development 2016, 143, 3–14. [Google Scholar] [CrossRef] [Green Version]
- Mathew, R.; Pal Bhadra, M.; Bhadra, U. Insulin/insulin-like growth factor-1 signalling (IIS) based regulation of lifespan across species. Biogerontology 2017, 18, 35–53. [Google Scholar] [CrossRef]
- Cohen, E.; Bieschke, J.; Perciavalle, R.M.; Kelly, J.W.; Dillin, A. Opposing activities protect against age-onset proteotoxicity. Science 2006, 313, 1604–1610. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Kim, S.G.; Blenis, J. Rapamycin: One drug, many effects. Cell Metab. 2014, 19, 373–379. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Saxton, R.A.; Sabatini, D.M. mTOR Signaling in Growth, Metabolism, and Disease. Cell 2017, 168, 960–976. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Johnson, S.C.; Rabinovitch, P.S.; Kaeberlein, M. mTOR is a key modulator of ageing and age-related disease. Nature 2013, 493, 338–345. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Menzies, F.M.; Fleming, A.; Caricasole, A.; Bento, C.F.; Andrews, S.P.; Ashkenazi, A.; Füllgrabe, J.; Jackson, A.; Jimenez Sanchez, M.; Karabiyik, C.; et al. Autophagy and Neurodegeneration: Pathogenic Mechanisms and Therapeutic Opportunities. Neuron 2017, 93, 1015–1034. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sen, P.; Shah, P.P.; Nativio, R.; Berger, S.L. Epigenetic Mechanisms of Longevity and Aging. Cell 2016, 166, 822–839. [Google Scholar] [CrossRef] [Green Version]
- Carling, D. The AMP-activated protein kinase cascade—A unifying system for energy control. Trends Biochem. Sci. 2004, 29, 18–24. [Google Scholar] [CrossRef]
- Steinberg, G.R.; Kemp, B.E. AMPK in Health and Disease. Physiol. Rev. 2009, 89, 1025–1078. [Google Scholar] [CrossRef]
- Chung, M.M.; Nicol, C.J.; Cheng, Y.C.; Lin, K.H.; Chen, Y.L.; Pei, D.; Lin, C.H.; Shih, Y.N.; Yen, C.H.; Chen, S.J.; et al. Metformin activation of AMPK suppresses AGE-induced inflammatory response in hNSCs. Exp. Cell Res. 2017, 352, 75–83. [Google Scholar] [CrossRef]
- Salminen, A.; Hyttinen, J.M.; Kaarniranta, K. AMP-activated protein kinase inhibits NF-κB signaling and inflammation: Impact on healthspan and lifespan. J. Mol. Med. 2011, 89, 667–676. [Google Scholar] [CrossRef] [Green Version]
- Cantó, C.; Gerhart-Hines, Z.; Feige, J.N.; Lagouge, M.; Noriega, L.; Milne, J.C.; Elliott, P.J.; Puigserver, P.; Auwerx, J. AMPK regulates energy expenditure by modulating NAD+ metabolism and SIRT1 activity. Nature 2009, 458, 1056–1060. [Google Scholar] [CrossRef]
- Kapahi, P.; Kaeberlein, M.; Hansen, M. Dietary restriction and lifespan: Lessons from invertebrate models. Ageing Res. Rev. 2017, 39, 3–14. [Google Scholar] [CrossRef]
- He, W.; Newman, J.C.; Wang, M.Z.; Ho, L.; Verdin, E. Mitochondrial sirtuins: Regulators of protein acylation and metabolism. Trends Endocrinol. Metab. 2012, 23, 467–476. [Google Scholar] [CrossRef]
- Gillum, M.P.; Kotas, M.E.; Erion, D.M.; Kursawe, R.; Chatterjee, P.; Nead, K.T.; Muise, E.S.; Hsiao, J.J.; Frederick, D.W.; Yonemitsu, S.; et al. SirT1 regulates adipose tissue inflammation. Diabetes 2011, 60, 3235–3245. [Google Scholar] [CrossRef] [Green Version]
- Rodgers, J.T.; Lerin, C.; Haas, W.; Gygi, S.P.; Spiegelman, B.M.; Puigserver, P. Nutrient control of glucose homeostasis through a complex of PGC-1alpha and SIRT1. Nature 2005, 434, 113–118. [Google Scholar] [CrossRef]
- Imai, S.; Guarente, L. NAD+ and sirtuins in aging and disease. Trends Cell Biol. 2014, 24, 464–471. [Google Scholar] [CrossRef] [PubMed]
- Morigi, M.; Perico, L.; Benigni, A. Sirtuins in Renal Health and Disease. J. Am. Soc. Nephrol. 2018, 29, 1799–1809. [Google Scholar] [CrossRef]
- Wątroba, M.; Dudek, I.; Skoda, M.; Stangret, A.; Rzodkiewicz, P.; Szukiewicz, D. Sirtuins, epigenetics and longevity. Ageing Res. Rev. 2017, 40, 11–19. [Google Scholar] [CrossRef]
- Cho, S.H.; Chen, J.A.; Sayed, F.; Ward, M.E.; Gao, F.; Nguyen, T.A.; Krabbe, G.; Sohn, P.D.; Lo, I.; Minami, S.; et al. SIRT1 deficiency in microglia contributes to cognitive decline in aging and neurodegeneration via epigenetic regulation of IL-1β. J. Neurosci. 2015, 35, 807–818. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Patel, N.V.; Gordon, M.N.; Connor, K.E.; Good, R.A.; Engelman, R.W.; Mason, J.; Morgan, D.G.; Morgan, T.E.; Finch, C.E. Caloric restriction attenuates Abeta-deposition in Alzheimer transgenic models. Neurobiol. Aging 2005, 26, 995–1000. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Fivecoat, H.; Ho, L.; Pan, Y.; Ling, E.; Pasinetti, G.M. The role of Sirt1: At the crossroad between promotion of longevity and protection against Alzheimer’s disease neuropathology. Biochim. Biophys. Acta 2010, 1804, 1690–1694. [Google Scholar] [CrossRef]
- Kim, D.; Nguyen, M.D.; Dobbin, M.M.; Fischer, A.; Sananbenesi, F.; Rodgers, J.T.; Delalle, I.; Baur, J.A.; Sui, G.; Armour, S.M.; et al. SIRT1 deacetylase protects against neurodegeneration in models for Alzheimer’s disease and amyotrophic lateral sclerosis. EMBO J. 2007, 26, 3169–3179. [Google Scholar] [CrossRef] [PubMed]
- Marambaud, P.; Zhao, H.; Davies, P. Resveratrol promotes clearance of Alzheimer’s disease amyloid-beta peptides. J. Biol. Chem. 2005, 280, 37377–37382. [Google Scholar] [CrossRef] [Green Version]
- Luchsinger, J.A.; Tang, M.X.; Shea, S.; Mayeux, R. Caloric intake and the risk of Alzheimer disease. Arch. Neurol. 2002, 59, 1258–1263. [Google Scholar] [CrossRef] [PubMed]
- Julien, C.; Tremblay, C.; Emond, V.; Lebbadi, M.; Salem, N., Jr.; Bennett, D.A.; Calon, F. Sirtuin 1 reduction parallels the accumulation of tau in Alzheimer disease. J. Neuropathol. Exp. Neurol. 2009, 68, 48–58. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Oosterhof, N.; Dekens, D.W.; Lawerman, T.F.; van Dijk, M. Yet another role for SIRT1: Reduction of α-synuclein aggregation in stressed neurons. J. Neurosci. 2012, 32, 6413–6414. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Araki, T.; Sasaki, Y.; Milbrandt, J. Increased nuclear NAD biosynthesis and SIRT1 activation prevent axonal degeneration. Science 2004, 305, 1010–1013. [Google Scholar] [CrossRef] [Green Version]
- Virgili, M.; Contestabile, A. Partial neuroprotection of in vivo excitotoxic brain damage by chronic administration of the red wine antioxidant agent, trans-resveratrol in rats. Neurosci. Lett. 2000, 281, 123–126. [Google Scholar] [CrossRef]
- Brunet, A.; Sweeney, L.B.; Sturgill, J.F.; Chua, K.F.; Greer, P.L.; Lin, Y.; Tran, H.; Ross, S.E.; Mostoslavsky, R.; Cohen, H.Y.; et al. Stress-dependent regulation of FOXO transcription factors by the SIRT1 deacetylase. Science 2004, 303, 2011–2015. [Google Scholar] [CrossRef] [Green Version]
- Zhao, L.; Cao, J.; Hu, K.; He, X.; Yun, D.; Tong, T.; Han, L. Sirtuins and their Biological Relevance in Aging and Age-Related Diseases. Aging Dis. 2020, 11, 927–945. [Google Scholar] [CrossRef]
- Vaziri, H.; Dessain, S.K.; Ng Eaton, E.; Imai, S.I.; Frye, R.A.; Pandita, T.K.; Guarente, L.; Weinberg, R.A. hSIR2(SIRT1) functions as an NAD-dependent p53 deacetylase. Cell 2001, 107, 149–159. [Google Scholar] [CrossRef] [Green Version]
- Prozorovski, T.; Schulze-Topphoff, U.; Glumm, R.; Baumgart, J.; Schröter, F.; Ninnemann, O.; Siegert, E.; Bendix, I.; Brüstle, O.; Nitsch, R.; et al. Sirt1 contributes critically to the redox-dependent fate of neural progenitors. Nat. Cell Biol. 2008, 10, 385–394. [Google Scholar] [CrossRef] [PubMed]
- Chang, H.C.; Guarente, L. SIRT1 mediates central circadian control in the SCN by a mechanism that decays with aging. Cell 2013, 153, 1448–1460. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yaku, K.; Okabe, K.; Nakagawa, T. NAD metabolism: Implications in aging and longevity. Ageing Res. Rev. 2018, 47, 1–17. [Google Scholar] [CrossRef] [PubMed]
- Blüher, M.; Kahn, B.B.; Kahn, C.R. Extended longevity in mice lacking the insulin receptor in adipose tissue. Science 2003, 299, 572–574. [Google Scholar] [CrossRef] [Green Version]
- Kenyon, C.J. The genetics of ageing. Nature 2010, 464, 504–512. [Google Scholar] [CrossRef] [PubMed]
- Greer, E.L.; Brunet, A. Different dietary restriction regimens extend lifespan by both independent and overlapping genetic pathways in C. elegans. Aging Cell 2009, 8, 113–127. [Google Scholar] [CrossRef] [Green Version]
- Zhu, Y.; Liu, X.; Ding, X.; Wang, F.; Geng, X. Telomere and its role in the aging pathways: Telomere shortening, cell senescence and mitochondria dysfunction. Biogerontology 2019, 20, 1–16. [Google Scholar] [CrossRef]
- Herrmann, M.; Pusceddu, I.; März, W.; Herrmann, W. Telomere biology and age-related diseases. Clin. Chem. Lab. Med. 2018, 56, 1210–1222. [Google Scholar] [CrossRef]
- Sahin, E.; DePinho, R.A. Axis of ageing: Telomeres, p53 and mitochondria. Nat. Rev. Mol. Cell Biol. 2012, 13, 397–404. [Google Scholar] [CrossRef] [Green Version]
- Bernardes de Jesus, B.; Vera, E.; Schneeberger, K.; Tejera, A.M.; Ayuso, E.; Bosch, F.; Blasco, M.A. Telomerase gene therapy in adult and old mice delays aging and increases longevity without increasing cancer. EMBO Mol. Med. 2012, 4, 691–704. [Google Scholar] [CrossRef] [PubMed]
- Shay, J.W. Role of Telomeres and Telomerase in Aging and Cancer. Cancer Discov. 2016, 6, 584–593. [Google Scholar] [CrossRef] [Green Version]
- Palacios, J.A.; Herranz, D.; De Bonis, M.L.; Velasco, S.; Serrano, M.; Blasco, M.A. SIRT1 contributes to telomere maintenance and augments global homologous recombination. J. Cell Biol. 2010, 191, 1299–1313. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tiwari, V.; Wilson, D.M., 3rd. DNA Damage and Associated DNA Repair Defects in Disease and Premature Aging. Am. J. Hum. Genet. 2019, 105, 237–257. [Google Scholar] [CrossRef] [Green Version]
- Foo, M.X.R.; Ong, P.F.; Dreesen, O. Premature aging syndromes: From patients to mechanism. J. Dermatol. Sci. 2019, 96, 58–65. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hoeijmakers, J.H. DNA damage, aging, and cancer. N. Engl. J. Med. 2009, 361, 1475–1485. [Google Scholar] [CrossRef] [PubMed]
- Kauppila, T.E.S.; Bratic, A.; Jensen, M.B.; Baggio, F.; Partridge, L.; Jasper, H.; Grönke, S.; Larsson, N.G. Mutations of mitochondrial DNA are not major contributors to aging of fruit flies. Proc. Natl. Acad. Sci. USA 2018, 115, 9620–9629. [Google Scholar] [CrossRef] [Green Version]
- Thanan, R.; Oikawa, S.; Hiraku, Y.; Ohnishi, S.; Ma, N.; Pinlaor, S.; Yongvanit, P.; Kawanishi, S.; Murata, M. Oxidative stress and its significant roles in neurodegenerative diseases and cancer. Int. J. Mol. Sci. 2014, 16, 193–217. [Google Scholar] [CrossRef] [Green Version]
- Feinberg, A.P.; Tycko, B. The history of cancer epigenetics. Nat. Rev. Cancer 2004, 4, 143–153. [Google Scholar] [CrossRef]
- Kane, A.E.; Sinclair, D.A. Epigenetic changes during aging and their reprogramming potential. Crit. Rev. Biochem. Mol. Biol. 2019, 54, 61–83. [Google Scholar] [CrossRef]
- Burzynski, S.R. Aging: Gene silencing or gene activation? Med. Hypotheses 2005, 64, 201–208. [Google Scholar] [CrossRef]
- Ben-Avraham, D.; Muzumdar, R.H.; Atzmon, G. Epigenetic genome-wide association methylation in aging and longevity. Epigenomics 2012, 4, 503–509. [Google Scholar] [CrossRef] [Green Version]
- Benayoun, B.A.; Pollina, E.A.; Brunet, A. Epigenetic regulation of ageing: Linking environmental inputs to genomic stability. Nat. Rev. Mol. Cell Biol. 2015, 16, 593–610. [Google Scholar] [CrossRef] [Green Version]
- Hernandez, D.G.; Nalls, M.A.; Gibbs, J.R.; Arepalli, S.; van der Brug, M.; Chong, S.; Moore, M.; Longo, D.L.; Cookson, M.R.; Traynor, B.J.; et al. Distinct DNA methylation changes highly correlated with chronological age in the human brain. Hum. Mol. Genet. 2011, 20, 1164–1172. [Google Scholar] [CrossRef] [Green Version]
- Waki, T.; Tamura, G.; Sato, M.; Motoyama, T. Age-related methylation of tumor suppressor and tumor-related genes: An analysis of autopsy samples. Oncogene 2003, 22, 4128–4133. [Google Scholar] [CrossRef] [Green Version]
- Casillas, M.A., Jr.; Lopatina, N.; Andrews, L.G.; Tollefsbol, T.O. Transcriptional control of the DNA methyltransferases is altered in aging and neoplastically-transformed human fibroblasts. Mol. Cell. Biochem. 2003, 252, 33–43. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Liu, L.; Tollefsbol, T.O. Glucose restriction can extend normal cell lifespan and impair precancerous cell growth through epigenetic control of hTERT and p16 expression. FASEB J. 2010, 24, 1442–1453. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wakeling, L.A.; Ions, L.J.; Ford, D. Could Sirt1-mediated epigenetic effects contribute to the longevity response to dietary restriction and be mimicked by other dietary interventions? Age 2009, 31, 327–341. [Google Scholar] [CrossRef] [Green Version]
- Alageel, A.; Tomasi, J.; Tersigni, C.; Brietzke, E.; Zuckerman, H.; Subramaniapillai, M.; Lee, Y.; Iacobucci, M.; Rosenblat, J.D.; Mansur, R.B.; et al. Evidence supporting a mechanistic role of sirtuins in mood and metabolic disorders. Prog. Neuropsychopharmacol. Biol. Psychiatry 2018, 86, 95–101. [Google Scholar] [CrossRef]
- De Ruijter, A.J.; van Gennip, A.H.; Caron, H.N.; Kemp, S.; van Kuilenburg, A.B. Histone deacetylases (HDACs): Characterization of the classical HDAC family. Biochem. J. 2003, 370, 737–749. [Google Scholar] [CrossRef] [PubMed]
- Satoh, A.; Brace, C.S.; Rensing, N.; Cliften, P.; Wozniak, D.F.; Herzog, E.D.; Yamada, K.A.; Imai, S. Sirt1 extends life span and delays aging in mice through the regulation of Nk2 homeobox 1 in the DMH and LH. Cell Metab. 2013, 18, 416–430. [Google Scholar] [CrossRef] [Green Version]
- Siebold, A.P.; Banerjee, R.; Tie, F.; Kiss, D.L.; Moskowitz, J.; Harte, P.J. Polycomb Repressive Complex 2 and Trithorax modulate Drosophila longevity and stress resistance. Proc. Natl. Acad. Sci. USA 2010, 107, 169–174. [Google Scholar] [CrossRef] [Green Version]
- Pasyukova, E.G.; Vaiserman, A.M. HDAC inhibitors: A new promising drug class in anti-aging research. Mech. Ageing Dev. 2017, 166, 6–15. [Google Scholar] [CrossRef] [PubMed]
- Sharma, S.; Taliyan, R. Targeting histone deacetylases: A novel approach in Parkinson’s disease. Parkinsons Dis. 2015, 2015, 303294. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yoo, Y.E.; Ko, C.P. Treatment with trichostatin A initiated after disease onset delays disease progression and increases survival in a mouse model of amyotrophic lateral sclerosis. Exp. Neurol. 2011, 231, 147–159. [Google Scholar] [CrossRef] [PubMed]
- Ricobaraza, A.; Cuadrado-Tejedor, M.; Marco, S.; Pérez-Otaño, I.; García-Osta, A. Phenylbutyrate rescues dendritic spine loss associated with memory deficits in a mouse model of Alzheimer disease. Hippocampus 2012, 22, 1040–1050. [Google Scholar] [CrossRef]
- Wiley, J.C.; Pettan-Brewer, C.; Ladiges, W.C. Phenylbutyric acid reduces amyloid plaques and rescues cognitive behavior in AD transgenic mice. Aging Cell 2011, 10, 418–428. [Google Scholar] [CrossRef]
- Harrison, I.F.; Crum, W.R.; Vernon, A.C.; Dexter, D.T. Neurorestoration induced by the HDAC inhibitor sodium valproate in the lactacystin model of Parkinson’s is associated with histone acetylation and up-regulation of neurotrophic factors. Br. J. Pharmacol. 2015, 172, 4200–4215. [Google Scholar] [CrossRef] [Green Version]
- Grillari, J.; Grillari-Voglauer, R. Novel modulators of senescence, aging, and longevity: Small non-coding RNAs enter the stage. Exp. Gerontol. 2010, 45, 302–311. [Google Scholar] [CrossRef] [Green Version]
- Nelson, P.T.; Wang, W.X.; Rajeev, B.W. MicroRNAs (miRNAs) in neurodegenerative diseases. Brain Pathol. 2008, 18, 130–138. [Google Scholar] [CrossRef]
- Rodriguez-Ortiz, C.J.; Baglietto-Vargas, D.; Martinez-Coria, H.; LaFerla, F.M.; Kitazawa, M. Upregulation of miR-181 decreases c-Fos and SIRT-1 in the hippocampus of 3xTg-AD mice. J. Alzheimers Dis. 2014, 42, 1229–1238. [Google Scholar] [CrossRef]
- Mao, K.; Zhang, G. The role of PARP1 in neurodegenerative diseases and aging. FEBS J 2021. [Google Scholar] [CrossRef]
- Martire, S.; Mosca, L.; d’Erme, M. PARP-1 involvement in neurodegeneration: A focus on Alzheimer’s and Parkinson’s diseases. Mech. Ageing Dev. 2015, 146-148, 53–64. [Google Scholar] [CrossRef] [PubMed]
- Narne, P.; Pandey, V.; Simhadri, P.K.; Phanithi, P.B. Poly(ADP-ribose)polymerase-1 hyperactivation in neurodegenerative diseases: The death knell tolls for neurons. Semin. Cell Dev. Biol. 2017, 63, 154–166. [Google Scholar] [CrossRef] [PubMed]
- Beneke, S.; Cohausz, O.; Malanga, M.; Boukamp, P.; Althaus, F.; Bürkle, A. Rapid regulation of telomere length is mediated by poly(ADP-ribose) polymerase-1. Nucleic Acids Res. 2008, 36, 6309–6317. [Google Scholar] [CrossRef] [PubMed]
- Ye, T.J.; Lu, Y.L.; Yan, X.F.; Hu, X.D.; Wang, X.L. High mobility group box-1 release from H2O2-injured hepatocytes due to sirt1 functional inhibition. World J. Gastroenterol. 2019, 25, 5434–5450. [Google Scholar] [CrossRef]
- Kam, T.I.; Mao, X.; Park, H.; Chou, S.C.; Karuppagounder, S.S.; Umanah, G.E.; Yun, S.P.; Brahmachari, S.; Panicker, N.; Chen, R.; et al. Poly(ADP-ribose) drives pathologic α-synuclein neurodegeneration in Parkinson’s disease. Science 2018, 362, eaat8407. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rulten, S.L.; Rotheray, A.; Green, R.L.; Grundy, G.J.; Moore, D.A.; Gómez-Herreros, F.; Hafezparast, M.; Caldecott, K.W. PARP-1 dependent recruitment of the amyotrophic lateral sclerosis-associated protein FUS/TLS to sites of oxidative DNA damage. Nucleic Acids Res. 2014, 42, 307–314. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chini, C.C.S.; Tarragó, M.G.; Chini, E.N. NAD and the aging process: Role in life, death and everything in between. Mol. Cell. Endocrinol. 2017, 455, 62–74. [Google Scholar] [CrossRef]
- Xie, Z.; Zhang, D.; Chung, D.; Tang, Z.; Huang, H.; Dai, L.; Qi, S.; Li, J.; Colak, G.; Chen, Y.; et al. Metabolic Regulation of Gene Expression by Histone Lysine β-Hydroxybutyrylation. Mol. Cell 2016, 62, 194–206. [Google Scholar] [CrossRef] [Green Version]
- Green, D.R.; Galluzzi, L.; Kroemer, G. Mitochondria and the autophagy-inflammation-cell death axis in organismal aging. Science 2011, 333, 1109–1112. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Moehle, E.A.; Shen, K.; Dillin, A. Mitochondrial proteostasis in the context of cellular and organismal health and aging. J. Biol. Chem. 2019, 294, 5396–5407. [Google Scholar] [CrossRef] [Green Version]
- Hekimi, S.; Lapointe, J.; Wen, Y. Taking a “good” look at free radicals in the aging process. Trends Cell Biol. 2011, 21, 569–576. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lipinski, M.M.; Zheng, B.; Lu, T.; Yan, Z.; Py, B.F.; Ng, A.; Xavier, R.J.; Li, C.; Yankner, B.A.; Scherzer, C.R.; et al. Genome-wide analysis reveals mechanisms modulating autophagy in normal brain aging and in Alzheimer’s disease. Proc. Natl. Acad. Sci. USA 2010, 107, 14164–14169. [Google Scholar] [CrossRef] [Green Version]
- Winslow, A.R.; Chen, C.W.; Corrochano, S.; Acevedo-Arozena, A.; Gordon, D.E.; Peden, A.A.; Lichtenberg, M.; Menzies, F.M.; Ravikumar, B.; Imarisio, S.; et al. α-Synuclein impairs macroautophagy: Implications for Parkinson’s disease. J. Cell Biol. 2010, 190, 1023–1037. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Youle, R.J.; Narendra, D.P. Mechanisms of mitophagy. Nat. Rev. Mol. Cell. Biol. 2011, 12, 9–14. [Google Scholar] [CrossRef]
- Gottlieb, R.A.; Mentzer, R.M. Autophagy during cardiac stress: Joys and frustrations of autophagy. Annu. Rev. Physiol. 2010, 72, 45–59. [Google Scholar] [CrossRef] [Green Version]
- Madeo, F.; Tavernarakis, N.; Kroemer, G. Can autophagy promote longevity? Nat. Cell Biol. 2010, 12, 842–846. [Google Scholar] [CrossRef]
- Zhou, R.; Yazdi, A.S.; Menu, P.; Tschopp, J. A role for mitochondria in NLRP3 inflammasome activation. Nature 2011, 469, 221–225. [Google Scholar] [CrossRef]
- Herzig, S.; Shaw, R.J. AMPK: Guardian of metabolism and mitochondrial homeostasis. Nat. Rev. Mol. Cell Biol. 2018, 19, 121–135. [Google Scholar] [CrossRef] [Green Version]
- Lee, I.H.; Cao, L.; Mostoslavsky, R.; Lombard, D.B.; Liu, J.; Bruns, N.E.; Tsokos, M.; Alt, F.W.; Finkel, T. A role for the NAD-dependent deacetylase Sirt1 in the regulation of autophagy. Proc. Natl. Acad. Sci. USA 2008, 105, 3374–3379. [Google Scholar] [CrossRef] [Green Version]
- McCarty, M.F.; DiNicolantonio, J.J.; O’Keefe, J.H. Ketosis may promote brain macroautophagy by activating Sirt1 and hypoxia-inducible factor-1. Med. Hypotheses 2015, 85, 631–639. [Google Scholar] [CrossRef] [PubMed]
- Qiu, X.; Brown, K.; Hirschey, M.D.; Verdin, E.; Chen, D. Calorie restriction reduces oxidative stress by SIRT3-mediated SOD2 activation. Cell Metab. 2010, 12, 662–667. [Google Scholar] [CrossRef] [Green Version]
- Hafner, A.V.; Dai, J.; Gomes, A.P.; Xiao, C.Y.; Palmeira, C.M.; Rosenzweig, A.; Sinclair, D.A. Regulation of the mPTP by SIRT3-mediated deacetylation of CypD at lysine 166 suppresses age-related cardiac hypertrophy. Aging 2010, 2, 914–923. [Google Scholar] [CrossRef] [Green Version]
- Halling, J.F.; Pilegaard, H. PGC-1α-mediated regulation of mitochondrial function and physiological implications. Appl. Physiol. Nutr. Metab. 2020, 45, 927–936. [Google Scholar] [CrossRef] [PubMed]
- Austin, S.; St-Pierre, J. PGC1α and mitochondrial metabolism--emerging concepts and relevance in ageing and neurodegenerative disorders. J. Cell Sci. 2012, 125, 4963–4971. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cantó, C.; Auwerx, J. PGC-1alpha, SIRT1 and AMPK, an energy sensing network that controls energy expenditure. Curr. Opin. Lipidol. 2009, 20, 98–105. [Google Scholar] [CrossRef] [Green Version]
- Puigserver, P.; Spiegelman, B.M. Peroxisome proliferator-activated receptor-gamma coactivator 1 alpha (PGC-1α): Transcriptional coactivator and metabolic regulator. Endocr. Rev. 2003, 24, 78–90. [Google Scholar] [CrossRef] [Green Version]
- Scirpo, R.; Fiorotto, R.; Villani, A.; Amenduni, M.; Spirli, C.; Strazzabosco, M. Stimulation of nuclear receptor peroxisome proliferator-activated receptor-γ limits NF-κB-dependent inflammation in mouse cystic fibrosis biliary epithelium. Hepatology 2015, 62, 1551–1562. [Google Scholar] [CrossRef] [Green Version]
- Tyagi, S.; Gupta, P.; Saini, A.S.; Kaushal, C.; Sharma, S. The peroxisome proliferator-activated receptor: A family of nuclear receptors role in various diseases. J. Adv. Pharm. Technol. Res. 2011, 2, 236–240. [Google Scholar] [CrossRef]
- Qin, W.; Yang, T.; Ho, L.; Zhao, Z.; Wang, J.; Chen, L.; Zhao, W.; Thiyagarajan, M.; MacGrogan, D.; Rodgers, J.T.; et al. Neuronal SIRT1 activation as a novel mechanism underlying the prevention of Alzheimer disease amyloid neuropathology by calorie restriction. J. Biol. Chem. 2006, 281, 21745–21754. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Qin, W.; Haroutunian, V.; Katsel, P.; Cardozo, C.P.; Ho, L.; Buxbaum, J.D.; Pasinetti, G.M. PGC-1alpha expression decreases in the Alzheimer disease brain as a function of dementia. Arch. Neurol. 2009, 66, 352–361. [Google Scholar] [CrossRef] [PubMed]
- Eisele, P.S.; Salatino, S.; Sobek, J.; Hottiger, M.O.; Handschin, C. The peroxisome proliferator-activated receptor γ coactivator 1α/β (PGC-1) coactivators repress the transcriptional activity of NF-κB in skeletal muscle cells. J. Biol. Chem. 2013, 288, 2246–2260. [Google Scholar] [CrossRef] [Green Version]
- Mookerjee, S.A.; Divakaruni, A.S.; Jastroch, M.; Brand, M.D. Mitochondrial uncoupling and lifespan. Mech. Ageing Dev. 2010, 131, 463–472. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Franceschi, C.; Garagnani, P.; Parini, P.; Giuliani, C.; Santoro, A. Inflammaging: A new immune-metabolic viewpoint for age-related diseases. Nat. Rev. Endocrinol. 2018, 14, 576–590. [Google Scholar] [CrossRef]
- Josephson, A.M.; Bradaschia-Correa, V.; Lee, S.; Leclerc, K.; Patel, K.S.; Muinos Lopez, E.; Litwa, H.P.; Neibart, S.S.; Kadiyala, M.; Wong, M.Z.; et al. Age-related inflammation triggers skeletal stem/progenitor cell dysfunction. Proc. Natl. Acad. Sci. USA 2019, 116, 6995–7004. [Google Scholar] [CrossRef] [Green Version]
- Bauernfeind, F.; Ablasser, A.; Bartok, E.; Kim, S.; Schmid-Burgk, J.; Cavlar, T.; Hornung, V. Inflammasomes: Current understanding and open questions. Cell. Mol. Life Sci. 2011, 68, 765–783. [Google Scholar] [CrossRef]
- Salminen, A.; Kaarniranta, K.; Kauppinen, A. Inflammaging: Disturbed interplay between autophagy and inflammasomes. Aging 2012, 4, 166–175. [Google Scholar] [CrossRef] [Green Version]
- Martinez-Vicente, M.; Cuervo, A.M. Autophagy and neurodegeneration: When the cleaning crew goes on strike. Lancet Neurol. 2007, 6, 352–361. [Google Scholar] [CrossRef]
- Nixon, R.A.; Yang, D.S. Autophagy failure in Alzheimer’s disease--locating the primary defect. Neurobiol. Dis. 2011, 43, 38–45. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Masters, S.L.; O’Neill, L.A. Disease-associated amyloid and misfolded protein aggregates activate the inflammasome. Trends Mol. Med. 2011, 17, 276–282. [Google Scholar] [CrossRef]
- Salminen, A.; Huuskonen, J.; Ojala, J.; Kauppinen, A.; Kaarniranta, K.; Suuronen, T. Activation of innate immunity system during aging: NF-kB signaling is the molecular culprit of inflamm-aging. Ageing Res. Rev. 2008, 7, 83–105. [Google Scholar] [CrossRef]
- Zhou, R.; Tardivel, A.; Thorens, B.; Choi, I.; Tschopp, J. Thioredoxin-interacting protein links oxidative stress to inflammasome activation. Nat. Immunol. 2010, 11, 136–140. [Google Scholar] [CrossRef] [PubMed]
- Levy, M.; Thaiss, C.A.; Elinav, E. Taming the inflammasome. Nat. Med. 2015, 21, 213–215. [Google Scholar] [CrossRef]
- Patel, M.N.; Carroll, R.G.; Galván-Peña, S.; Mills, E.L.; Olden, R.; Triantafilou, M.; Wolf, A.I.; Bryant, C.E.; Triantafilou, K.; Masters, S.L. Inflammasome Priming in Sterile Inflammatory Disease. Trends Mol. Med. 2017, 23, 165–180. [Google Scholar] [CrossRef] [PubMed]
- Mihaylova, M.M.; Shaw, R.J. The AMPK signalling pathway coordinates cell growth, autophagy and metabolism. Nat. Cell Biol. 2011, 13, 1016–1023. [Google Scholar] [CrossRef] [PubMed]
- Abdullah, A.; Mohd Murshid, N.; Makpol, S. Antioxidant Modulation of mTOR and Sirtuin Pathways in Age-Related Neurodegenerative Diseases. Mol. Neurobiol. 2020, 57, 5193–5207. [Google Scholar] [CrossRef]
- Osorio, F.G.; Bárcena, C.; Soria-Valles, C.; Ramsay, A.J.; de Carlos, F.; Cobo, J.; Fueyo, A.; Freije, J.M.; López-Otín, C. Nuclear lamina defects cause ATM-dependent NF-κB activation and link accelerated aging to a systemic inflammatory response. Genes Dev. 2012, 26, 2311–2324. [Google Scholar] [CrossRef] [Green Version]
- Amaya-Montoya, M.; Pérez-Londoño, A.; Guatibonza-García, V.; Vargas-Villanueva, A.; Mendivil, C.O. Cellular Senescence as a Therapeutic Target for Age-Related Diseases: A Review. Adv. Ther. 2020, 37, 1407–1424. [Google Scholar] [CrossRef] [Green Version]
- Borghesan, M.; Hoogaars, W.M.H.; Varela-Eirin, M.; Talma, N.; Demaria, M. A Senescence-Centric View of Aging: Implications for Longevity and Disease. Trends Cell Biol. 2020, 30, 777–791. [Google Scholar] [CrossRef]
- Campisi, J. Aging, cellular senescence, and cancer. Annu. Rev. Physiol. 2013, 75, 685–705. [Google Scholar] [CrossRef] [Green Version]
- Di Micco, R.; Krizhanovsky, V.; Baker, D.; d’Adda di Fagagna, F. Cellular senescence in ageing: From mechanisms to therapeutic opportunities. Nat. Rev. Mol. Cell Biol. 2021, 22, 75–95. [Google Scholar] [CrossRef] [PubMed]
- He, S.; Sharpless, N.E. Senescence in Health and Disease. Cell 2017, 169, 1000–1011. [Google Scholar] [CrossRef] [PubMed]
- Tran, D.; Bergholz, J.; Zhang, H.; He, H.; Wang, Y.; Zhang, Y.; Li, Q.; Kirkland, J.L.; Xiao, Z.X. Insulin-like growth factor-1 regulates the SIRT1-p53 pathway in cellular senescence. Aging Cell 2014, 13, 669–678. [Google Scholar] [CrossRef]
- Young, A.R.; Narita, M.; Ferreira, M.; Kirschner, K.; Sadaie, M.; Darot, J.F.; Tavaré, S.; Arakawa, S.; Shimizu, S.; Watt, F.M.; et al. Autophagy mediates the mitotic senescence transition. Genes Dev. 2009, 23, 798–803. [Google Scholar] [CrossRef] [Green Version]
- Chen, C.; Zhou, M.; Ge, Y.; Wang, X. SIRT1 and aging related signaling pathways. Mech. Ageing Dev. 2020, 187, 111215. [Google Scholar] [CrossRef]
- Salmenperä, P.; Karhemo, P.R.; Räsänen, K.; Laakkonen, P.; Vaheri, A. Fibroblast spheroids as a model to study sustained fibroblast quiescence and their crosstalk with tumor cells. Exp. Cell Res. 2016, 345, 17–24. [Google Scholar] [CrossRef] [Green Version]
- Cai, J.; Weiss, M.L.; Rao, M.S. In search of “stemness”. Exp. Hematol. 2004, 32, 585–598. [Google Scholar] [CrossRef]
- Kim, K.H.; Chen, C.C.; Monzon, R.I.; Lau, L.F. Matricellular protein CCN1 promotes regression of liver fibrosis through induction of cellular senescence in hepatic myofibroblasts. Mol. Cell Biol. 2013, 33, 2078–2090. [Google Scholar] [CrossRef] [Green Version]
- Collado, M.; Serrano, M. Senescence in tumours: Evidence from mice and humans. Nat. Rev. Cancer 2010, 10, 51–57. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chinta, S.J.; Woods, G.; Demaria, M.; Rane, A.; Zou, Y.; McQuade, A.; Rajagopalan, S.; Limbad, C.; Madden, D.T.; Campisi, J.; et al. Cellular Senescence Is Induced by the Environmental Neurotoxin Paraquat and Contributes to Neuropathology Linked to Parkinson’s Disease. Cell Rep. 2018, 22, 930–940. [Google Scholar] [CrossRef] [Green Version]
- Finkel, T.; Serrano, M.; Blasco, M.A. The common biology of cancer and ageing. Nature 2007, 448, 767–774. [Google Scholar] [CrossRef] [Green Version]
- Wang, J.C.; Bennett, M. Aging and atherosclerosis: Mechanisms, functional consequences, and potential therapeutics for cellular senescence. Circ. Res. 2012, 111, 245–259. [Google Scholar] [CrossRef] [Green Version]
- Cao, L.; Li, W.; Kim, S.; Brodie, S.G.; Deng, C.X. Senescence, aging, and malignant transformation mediated by p53 in mice lacking the Brca1 full-length isoform. Genes Dev. 2003, 17, 201–213. [Google Scholar] [CrossRef] [Green Version]
- Jones, R.G.; Plas, D.R.; Kubek, S.; Buzzai, M.; Mu, J.; Xu, Y.; Birnbaum, M.J.; Thompson, C.B. AMP-activated protein kinase induces a p53-dependent metabolic checkpoint. Mol. Cell 2005, 18, 283–293. [Google Scholar] [CrossRef] [PubMed]
- Balch, W.E.; Morimoto, R.I.; Dillin, A.; Kelly, J.W. Adapting proteostasis for disease intervention. Science 2008, 319, 916–919. [Google Scholar] [CrossRef] [Green Version]
- Klaips, C.L.; Jayaraj, G.G.; Hartl, F.U. Pathways of cellular proteostasis in aging and disease. J. Cell Biol. 2018, 217, 51–63. [Google Scholar] [CrossRef] [PubMed]
- Lapierre, L.R.; De Magalhaes Filho, C.D.; McQuary, P.R.; Chu, C.C.; Visvikis, O.; Chang, J.T.; Gelino, S.; Ong, B.; Davis, A.E.; Irazoqui, J.E.; et al. The TFEB orthologue HLH-30 regulates autophagy and modulates longevity in Caenorhabditis elegans. Nat. Commun. 2013, 4, 2267. [Google Scholar] [CrossRef] [Green Version]
- Basisty, N.; Meyer, J.G.; Schilling, B. Protein Turnover in Aging and Longevity. Proteomics 2018, 18, e1700108. [Google Scholar] [CrossRef] [Green Version]
- Wong, S.Q.; Kumar, A.V.; Mills, J.; Lapierre, L.R. Autophagy in aging and longevity. Hum. Genet. 2020, 139, 277–290. [Google Scholar] [CrossRef] [PubMed]
- Sorrentino, V.; Romani, M.; Mouchiroud, L.; Beck, J.S.; Zhang, H.; D’Amico, D.; Moullan, N.; Potenza, F.; Schmid, A.W.; Rietsch, S.; et al. Enhancing mitochondrial proteostasis reduces amyloid-β proteotoxicity. Nature 2017, 552, 187–193. [Google Scholar] [CrossRef]
- Fang, E.F.; Hou, Y.; Palikaras, K.; Adriaanse, B.A.; Kerr, J.S.; Yang, B.; Lautrup, S.; Hasan-Olive, M.M.; Caponio, D.; Dan, X.; et al. Mitophagy inhibits amyloid-β and tau pathology and reverses cognitive deficits in models of Alzheimer’s disease. Nat. Neurosci. 2019, 22, 401–412. [Google Scholar] [CrossRef] [PubMed]
- Ryu, D.; Mouchiroud, L.; Andreux, P.A.; Katsyuba, E.; Moullan, N.; Nicolet-Dit-Félix, A.A.; Williams, E.G.; Jha, P.; Lo Sasso, G.; Huzard, D.; et al. Urolithin A induces mitophagy and prolongs lifespan in C. elegans and increases muscle function in rodents. Nat. Med. 2016, 22, 879–888. [Google Scholar] [CrossRef]
- Ou, X.; Lee, M.R.; Huang, X.; Messina-Graham, S.; Broxmeyer, H.E. SIRT1 positively regulates autophagy and mitochondria function in embryonic stem cells under oxidative stress. Stem Cells 2014, 32, 1183–1194. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Goodell, M.A.; Rando, T.A. Stem cells and healthy aging. Science 2015, 350, 1199–1204. [Google Scholar] [CrossRef] [PubMed]
- Keyes, B.E.; Fuchs, E. Stem cells: Aging and transcriptional fingerprints. J. Cell. Biol. 2018, 217, 79–92. [Google Scholar] [CrossRef] [Green Version]
- Zhang, P.; Kishimoto, Y.; Grammatikakis, I.; Gottimukkala, K.; Cutler, R.G.; Zhang, S.; Abdelmohsen, K.; Bohr, V.A.; Misra Sen, J.; Gorospe, M.; et al. Senolytic therapy alleviates Aβ-associated oligodendrocyte progenitor cell senescence and cognitive deficits in an Alzheimer’s disease model. Nat. Neurosci. 2019, 22, 719–728. [Google Scholar] [CrossRef]
- Ehninger, D.; Neff, F.; Xie, K. Longevity, aging and rapamycin. Cell. Mol. Life Sci. 2014, 71, 4325–4346. [Google Scholar] [CrossRef] [Green Version]
- Kaeberlein, M.; Galvan, V. Rapamycin and Alzheimer’s disease: Time for a clinical trial? Sci. Transl. Med. 2019, 11, eaar4289. [Google Scholar] [CrossRef] [PubMed]
- Sharma, M.; Gupta, Y.K. Chronic treatment with trans resveratrol prevents intracerebroventricular streptozotocin induced cognitive impairment and oxidative stress in rats. Life Sci. 2002, 71, 2489–2498. [Google Scholar] [CrossRef]
- Lu, M.; Su, C.; Qiao, C.; Bian, Y.; Ding, J.; Hu, G. Metformin Prevents Dopaminergic Neuron Death in MPTP/P-Induced Mouse Model of Parkinson’s Disease via Autophagy and Mitochondrial ROS Clearance. Int. J. Neuropsychopharmacol. 2016, 19, pyw047. [Google Scholar] [CrossRef] [PubMed]
- Foretz, M.; Guigas, B.; Bertrand, L.; Pollak, M.; Viollet, B. Metformin: From mechanisms of action to therapies. Cell Metab. 2014, 20, 953–966. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kulkarni, A.S.; Gubbi, S.; Barzilai, N. Benefits of Metformin in Attenuating the Hallmarks of Aging. Cell Metab. 2020, 32, 15–30. [Google Scholar] [CrossRef]
- Najafi, M.; Cheki, M.; Rezapoor, S.; Geraily, G.; Motevaseli, E.; Carnovale, C.; Clementi, E.; Shirazi, A. Metformin: Prevention of genomic instability and cancer: A review. Mutat. Res. Genet. Toxicol. Environ. Mutagen. 2018, 827, 1–8. [Google Scholar] [CrossRef]
- Aatsinki, S.M.; Buler, M.; Salomäki, H.; Koulu, M.; Pavek, P.; Hakkola, J. Metformin induces PGC-1α expression and selectively affects hepatic PGC-1α functions. Br. J. Pharmacol. 2014, 171, 2351–2363. [Google Scholar] [CrossRef] [Green Version]
- Prasad, S.; Sajja, R.K.; Kaisar, M.A.; Park, J.H.; Villalba, H.; Liles, T.; Abbruscato, T.; Cucullo, L. Role of Nrf2 and protective effects of Metformin against tobacco smoke-induced cerebrovascular toxicity. Redox Biol. 2017, 12, 58–69. [Google Scholar] [CrossRef]
- Cuyàs, E.; Verdura, S.; Llorach-Parés, L.; Fernández-Arroyo, S.; Joven, J.; Martin-Castillo, B.; Bosch-Barrera, J.; Brunet, J.; Nonell-Canals, A.; Sanchez-Martinez, M.; et al. Metformin Is a Direct SIRT1-Activating Compound: Computational Modeling and Experimental Validation. Front. Endocrinol. 2018, 9, 657. [Google Scholar] [CrossRef] [Green Version]
- Moiseeva, O.; Deschênes-Simard, X.; St-Germain, E.; Igelmann, S.; Huot, G.; Cadar, A.E.; Bourdeau, V.; Pollak, M.N.; Ferbeyre, G. Metformin inhibits the senescence-associated secretory phenotype by interfering with IKK/NF-κB activation. Aging Cell 2013, 12, 489–498. [Google Scholar] [CrossRef]
- Tizazu, A.M.; Nyunt, M.S.Z.; Cexus, O.; Suku, K.; Mok, E.; Xian, C.H.; Chong, J.; Tan, C.; How, W.; Hubert, S.; et al. Metformin Monotherapy Downregulates Diabetes-Associated Inflammatory Status and Impacts on Mortality. Front. Physiol. 2019, 10, 572. [Google Scholar] [CrossRef] [Green Version]
- Wang, C.; Liu, C.; Gao, K.; Zhao, H.; Zhou, Z.; Shen, Z.; Guo, Y.; Li, Z.; Yao, T.; Mei, X. Metformin preconditioning provide neuroprotection through enhancement of autophagy and suppression of inflammation and apoptosis after spinal cord injury. Biochem. Biophys. Res. Commun. 2016, 477, 534–540. [Google Scholar] [CrossRef] [PubMed]
- Markowicz-Piasecka, M.; Sikora, J.; Szydłowska, A.; Skupień, A.; Mikiciuk-Olasik, E.; Huttunen, K.M. Metformin—A Future Therapy for Neurodegenerative Diseases: Theme: Drug Discovery, Development and Delivery in Alzheimer’s Disease Guest Editor: Davide Brambilla. Pharm. Res. 2017, 34, 2614–2627. [Google Scholar] [CrossRef]
- Fang, J.; Yang, J.; Wu, X.; Zhang, G.; Li, T.; Wang, X.; Zhang, H.; Wang, C.C.; Liu, G.H.; Wang, L. Metformin alleviates human cellular aging by upregulating the endoplasmic reticulum glutathione peroxidase 7. Aging Cell 2018, 17, e12765. [Google Scholar] [CrossRef]
- Bridgeman, S.C.; Ellison, G.C.; Melton, P.E.; Newsholme, P.; Mamotte, C.D.S. Epigenetic effects of metformin: From molecular mechanisms to clinical implications. Diabetes Obes. Metab. 2018, 20, 1553–1562. [Google Scholar] [CrossRef] [Green Version]
- Noren Hooten, N.; Martin-Montalvo, A.; Dluzen, D.F.; Zhang, Y.; Bernier, M.; Zonderman, A.B.; Becker, K.G.; Gorospe, M.; de Cabo, R.; Evans, M.K. Metformin-mediated increase in DICER1 regulates microRNA expression and cellular senescence. Aging Cell 2016, 15, 572–581. [Google Scholar] [CrossRef] [PubMed]
- De Zegher, F.; Díaz, M.; Ibáñez, L. Association between Long Telomere Length and Insulin Sensitization in Adolescent Girls with Hyperinsulinemic Androgen Excess. JAMA Pediatr. 2015, 169, 787–788. [Google Scholar] [CrossRef] [Green Version]
- Diman, A.; Boros, J.; Poulain, F.; Rodriguez, J.; Purnelle, M.; Episkopou, H.; Bertrand, L.; Francaux, M.; Deldicque, L.; Decottignies, A. Nuclear respiratory factor 1 and endurance exercise promote human telomere transcription. Sci. Adv. 2016, 2, e1600031. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bhullar, K.S.; Hubbard, B.P. Lifespan and healthspan extension by resveratrol. Biochim. Biophys. Acta 2015, 1852, 1209–1218. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Porquet, D.; Casadesús, G.; Bayod, S.; Vicente, A.; Canudas, A.M.; Vilaplana, J.; Pelegrí, C.; Sanfeliu, C.; Camins, A.; Pallàs, M.; et al. Dietary resveratrol prevents Alzheimer’s markers and increases life span in SAMP8. Age 2013, 35, 1851–1865. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mancuso, R.; del Valle, J.; Modol, L.; Martinez, A.; Granado-Serrano, A.B.; Ramirez-Núñez, O.; Pallás, M.; Portero-Otin, M.; Osta, R.; Navarro, X. Resveratrol improves motoneuron function and extends survival in SOD1(G93A) ALS mice. Neurotherapeutics 2014, 11, 419–432. [Google Scholar] [CrossRef] [Green Version]
- Price, N.L.; Gomes, A.P.; Ling, A.J.; Duarte, F.V.; Martin-Montalvo, A.; North, B.J.; Agarwal, B.; Ye, L.; Ramadori, G.; Teodoro, J.S.; et al. SIRT1 is required for AMPK activation and the beneficial effects of resveratrol on mitochondrial function. Cell Metab. 2012, 15, 675–690. [Google Scholar] [CrossRef] [Green Version]
- Morris, B.J. Seven sirtuins for seven deadly diseases of aging. Free Radic. Biol. Med. 2013, 56, 133–171. [Google Scholar] [CrossRef]
- Choi, H.I.; Kim, H.J.; Park, J.S.; Kim, I.J.; Bae, E.H.; Ma, S.K.; Kim, S.W. PGC-1α attenuates hydrogen peroxide-induced apoptotic cell death by upregulating Nrf-2 via GSK3β inactivation mediated by activated p38 in HK-2 Cells. Sci. Rep. 2017, 7, 4319. [Google Scholar] [CrossRef] [Green Version]
- Huang, K.; Gao, X.; Wei, W. The crosstalk between Sirt1 and Keap1/Nrf2/ARE anti-oxidative pathway forms a positive feedback loop to inhibit FN and TGF-β1 expressions in rat glomerular mesangial cells. Exp. Cell Res. 2017, 361, 63–72. [Google Scholar] [CrossRef] [PubMed]
- Joo, M.S.; Kim, W.D.; Lee, K.Y.; Kim, J.H.; Koo, J.H.; Kim, S.G. AMPK Facilitates Nuclear Accumulation of Nrf2 by Phosphorylating at Serine 550. Mol. Cell Biol. 2016, 36, 1931–1942. [Google Scholar] [CrossRef] [Green Version]
- Mercken, E.M.; Mitchell, S.J.; Martin-Montalvo, A.; Minor, R.K.; Almeida, M.; Gomes, A.P.; Scheibye-Knudsen, M.; Palacios, H.H.; Licata, J.J.; Zhang, Y.; et al. SRT2104 extends survival of male mice on a standard diet and preserves bone and muscle mass. Aging Cell 2014, 13, 787–796. [Google Scholar] [CrossRef] [PubMed]
- Carullo, G.; Cappello, A.R.; Frattaruolo, L.; Badolato, M.; Armentano, B.; Aiello, F. Quercetin and derivatives: Useful tools in inflammation and pain management. Future Med. Chem. 2017, 9, 79–93. [Google Scholar] [CrossRef]
- Feng, X.; Sureda, A.; Jafari, S.; Memariani, Z.; Tewari, D.; Annunziata, G.; Barrea, L.; Hassan, S.T.S.; Šmejkal, K.; Malaník, M.; et al. Berberine in Cardiovascular and Metabolic Diseases: From Mechanisms to Therapeutics. Theranostics 2019, 9, 1923–1951. [Google Scholar] [CrossRef] [PubMed]
- Sarker, M.R.; Franks, S.F. Efficacy of curcumin for age-associated cognitive decline: A narrative review of preclinical and clinical studies. Geroscience 2018, 40, 73–95. [Google Scholar] [CrossRef]
- Sato, K.; Kashiwaya, Y.; Keon, C.A.; Tsuchiya, N.; King, M.T.; Radda, G.K.; Chance, B.; Clarke, K.; Veech, R.L. Insulin, ketone bodies, and mitochondrial energy transduction. FASEB J. 1995, 9, 651–658. [Google Scholar] [CrossRef] [PubMed]
- Sharma, A.K.; Rani, E.; Waheed, A.; Rajput, S.K. Pharmacoresistant Epilepsy: A Current Update on Non-Conventional Pharmacological and Non-Pharmacological Interventions. J. Epilepsy Res. 2015, 5, 1–8. [Google Scholar] [CrossRef] [Green Version]
- VanItallie, T.B.; Nufert, T.H. Ketones: Metabolism’s ugly duckling. Nutr. Rev. 2003, 61, 327–341. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Almeida, C.G.; de Mendonça, A.; Cunha, R.A.; Ribeiro, J.A. Adenosine promotes neuronal recovery from reactive oxygen species induced lesion in rat hippocampal slices. Neurosci. Lett. 2003, 339, 127–130. [Google Scholar] [CrossRef]
- Choudhury, H.; Chellappan, D.K.; Sengupta, P.; Pandey, M.; Gorain, B. Adenosine Receptors in Modulation of Central Nervous System Disorders. Curr. Pharm. Des. 2019, 25, 2808–2827. [Google Scholar] [CrossRef] [PubMed]
- McNally, M.A.; Hartman, A.L. Ketone bodies in epilepsy. J. Neurochem. 2012, 121, 28–35. [Google Scholar] [CrossRef] [Green Version]
- Juge, N.; Gray, J.A.; Omote, H.; Miyaji, T.; Inoue, T.; Hara, C.; Uneyama, H.; Edwards, R.H.; Nicoll, R.A.; Moriyama, Y. Metabolic control of vesicular glutamate transport and release. Neuron 2010, 68, 99–112. [Google Scholar] [CrossRef] [Green Version]
- Tieu, K.; Perier, C.; Caspersen, C.; Teismann, P.; Wu, D.C.; Yan, S.D.; Naini, A.; Vila, M.; Jackson-Lewis, V.; Ramasamy, R.; et al. D-beta-hydroxybutyrate rescues mitochondrial respiration and mitigates features of Parkinson disease. J. Clin. Investig. 2003, 112, 892–901. [Google Scholar] [CrossRef] [Green Version]
- Veech, R.L.; Todd King, M.; Pawlosky, R.; Kashiwaya, Y.; Bradshaw, P.C.; Curtis, W. The “great” controlling nucleotide coenzymes. IUBMB Life 2019, 71, 565–579. [Google Scholar] [CrossRef]
- Shimazu, T.; Hirschey, M.D.; Newman, J.; He, W.; Shirakawa, K.; Le Moan, N.; Grueter, C.A.; Lim, H.; Saunders, L.R.; Stevens, R.D.; et al. Suppression of oxidative stress by β-hydroxybutyrate, an endogenous histone deacetylase inhibitor. Science 2013, 339, 211–214. [Google Scholar] [CrossRef] [Green Version]
- Tseng, A.H.; Shieh, S.S.; Wang, D.L. SIRT3 deacetylates FOXO3 to protect mitochondria against oxidative damage. Free Radic. Biol. Med. 2013, 63, 222–234. [Google Scholar] [CrossRef]
- Zhao, L.; Ackerman, S.L. Endoplasmic reticulum stress in health and disease. Curr. Opin. Cell Biol. 2006, 18, 444–452. [Google Scholar] [CrossRef] [PubMed]
- Sleiman, S.F.; Henry, J.; Al-Haddad, R.; El Hayek, L.; Abou Haidar, E.; Stringer, T.; Ulja, D.; Karuppagounder, S.S.; Holson, E.B.; Ratan, R.R.; et al. Exercise promotes the expression of brain derived neurotrophic factor (BDNF) through the action of the ketone body β-hydroxybutyrate. Elife 2016, 5, e15092. [Google Scholar] [CrossRef]
- Manning, B.D.; Cantley, L.C. AKT/PKB signaling: Navigating downstream. Cell 2007, 129, 1261–1274. [Google Scholar] [CrossRef] [Green Version]
- Xu, D.; Lian, D.; Wu, J.; Liu, Y.; Zhu, M.; Sun, J.; He, D.; Li, L. Brain-derived neurotrophic factor reduces inflammation and hippocampal apoptosis in experimental Streptococcus pneumoniae meningitis. J. Neuroinflamm. 2017, 14, 156. [Google Scholar] [CrossRef] [Green Version]
- Marosi, K.; Kim, S.W.; Moehl, K.; Scheibye-Knudsen, M.; Cheng, A.; Cutler, R.; Camandola, S.; Mattson, M.P. 3-Hydroxybutyrate regulates energy metabolism and induces BDNF expression in cerebral cortical neurons. J. Neurochem. 2016, 139, 769–781. [Google Scholar] [CrossRef] [PubMed]
- Lau, D.; Bengtson, C.P.; Buchthal, B.; Bading, H. BDNF Reduces Toxic Extrasynaptic NMDA Receptor Signaling via Synaptic NMDA Receptors and Nuclear-Calcium-Induced Transcription of inhba/Activin A. Cell Rep. 2015, 12, 1353–1366. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mattson, M.P.; Lovell, M.A.; Furukawa, K.; Markesbery, W.R. Neurotrophic factors attenuate glutamate-induced accumulation of peroxides, elevation of intracellular Ca2+ concentration, and neurotoxicity and increase antioxidant enzyme activities in hippocampal neurons. J. Neurochem. 1995, 65, 1740–1751. [Google Scholar] [CrossRef]
- Menzies, K.J.; Zhang, H.; Katsyuba, E.; Auwerx, J. Protein acetylation in metabolism—Metabolites and cofactors. Nat. Rev. Endocrinol. 2016, 12, 43–60. [Google Scholar] [CrossRef] [PubMed]
- Guil, S.; Long, J.C.; Cáceres, J.F. hnRNP A1 relocalization to the stress granules reflects a role in the stress response. Mol. Cell. Biol. 2006, 26, 5744–5758. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jean-Philippe, J.; Paz, S.; Caputi, M. hnRNP A1: The Swiss army knife of gene expression. Int. J. Mol. Sci. 2013, 14, 18999–19024. [Google Scholar] [CrossRef] [Green Version]
- Parodi, B.; Rossi, S.; Morando, S.; Cordano, C.; Bragoni, A.; Motta, C.; Usai, C.; Wipke, B.T.; Scannevin, R.H.; Mancardi, G.L.; et al. Fumarates modulate microglia activation through a novel HCAR2 signaling pathway and rescue synaptic dysregulation in inflamed CNS. Acta Neuropathol. 2015, 130, 279–295. [Google Scholar] [CrossRef] [Green Version]
- Maalouf, M.; Sullivan, P.G.; Davis, L.; Kim, D.Y.; Rho, J.M. Ketones inhibit mitochondrial production of reactive oxygen species production following glutamate excitotoxicity by increasing NADH oxidation. Neuroscience 2007, 145, 256–264. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pawlosky, R.J.; Kemper, M.F.; Kashiwaya, Y.; King, M.T.; Mattson, M.P.; Veech, R.L. Effects of a dietary ketone ester on hippocampal glycolytic and tricarboxylic acid cycle intermediates and amino acids in a 3xTgAD mouse model of Alzheimer’s disease. J. Neurochem. 2017, 141, 195–207. [Google Scholar] [CrossRef]
- Offermanns, S.; Schwaninger, M. Nutritional or pharmacological activation of HCA(2) ameliorates neuroinflammation. Trends Mol. Med. 2015, 21, 245–255. [Google Scholar] [CrossRef]
- Shabab, T.; Khanabdali, R.; Moghadamtousi, S.Z.; Kadir, H.A.; Mohan, G. Neuroinflammation pathways: A general review. Int. J. Neurosci. 2017, 127, 624–633. [Google Scholar] [CrossRef]
- Norwitz, N.G.; Hu, M.T.; Clarke, K. The Mechanisms by Which the Ketone Body D-β-Hydroxybutyrate May Improve the Multiple Cellular Pathologies of Parkinson’s disease. Front. Nutr. 2019, 6, 63. [Google Scholar] [CrossRef]
- Hasan-Olive, M.M.; Lauritzen, K.H.; Ali, M.; Rasmussen, L.J.; Storm-Mathisen, J.; Bergersen, L.H. A Ketogenic Diet Improves Mitochondrial Biogenesis and Bioenergetics via the PGC1α-SIRT3-UCP2 Axis. Neurochem. Res. 2019, 44, 22–37. [Google Scholar] [CrossRef] [PubMed]
- Zou, X.H.; Li, H.M.; Wang, S.; Leski, M.; Yao, Y.C.; Yang, X.D.; Huang, Q.J.; Chen, G.Q. The effect of 3-hydroxybutyrate methyl ester on learning and memory in mice. Biomaterials 2009, 30, 1532–1541. [Google Scholar] [CrossRef]
- Scheibye-Knudsen, M.; Mitchell, S.J.; Fang, E.F.; Iyama, T.; Ward, T.; Wang, J.; Dunn, C.A.; Singh, N.; Veith, S.; Hasan-Olive, M.M.; et al. A high-fat diet and NAD+ activate Sirt1 to rescue premature aging in cockayne syndrome. Cell Metab. 2014, 20, 840–855. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Houtkooper, R.H.; Auwerx, J. Exploring the therapeutic space around NAD+. J. Cell Biol. 2012, 199, 205–209. [Google Scholar] [CrossRef]
- Kong, X.; Wang, R.; Xue, Y.; Liu, X.; Zhang, H.; Chen, Y.; Fang, F.; Chang, Y. Sirtuin 3, a new target of PGC-1alpha, plays an important role in the suppression of ROS and mitochondrial biogenesis. PLoS ONE 2010, 5, e11707. [Google Scholar] [CrossRef] [Green Version]
- Shi, T.; Wang, F.; Stieren, E.; Tong, Q. SIRT3, a mitochondrial sirtuin deacetylase, regulates mitochondrial function and thermogenesis in brown adipocytes. J. Biol. Chem. 2005, 280, 13560–13567. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cheng, A.; Wan, R.; Yang, J.L.; Kamimura, N.; Son, T.G.; Ouyang, X.; Luo, Y.; Okun, E.; Mattson, M.P. Involvement of PGC-1α in the formation and maintenance of neuronal dendritic spines. Nat. Commun. 2012, 3, 1250. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jeong, E.A.; Jeon, B.T.; Shin, H.J.; Kim, N.; Lee, D.H.; Kim, H.J.; Kang, S.S.; Cho, G.J.; Choi, W.S.; Roh, G.S. Ketogenic diet-induced peroxisome proliferator-activated receptor-γ activation decreases neuroinflammation in the mouse hippocampus after kainic acid-induced seizures. Exp. Neurol. 2011, 232, 195–202. [Google Scholar] [CrossRef] [PubMed]
- Morris, G.; Puri, B.K.; Carvalho, A.; Maes, M.; Berk, M.; Ruusunen, A.; Olive, L. Induced Ketosis as a Treatment for Neuroprogressive Disorders: Food for Thought? Int. J. Neuropsychopharmacol. 2020, 23, 366–384. [Google Scholar] [CrossRef]
- Simeone, T.A.; Matthews, S.A.; Samson, K.K.; Simeone, K.A. Regulation of brain PPARgamma2 contributes to ketogenic diet anti-seizure efficacy. Exp. Neurol. 2017, 287, 54–64. [Google Scholar] [CrossRef] [Green Version]
- Kashiwaya, Y.; Pawlosky, R.; Markis, W.; King, M.T.; Bergman, C.; Srivastava, S.; Murray, A.; Clarke, K.; Veech, R.L. A ketone ester diet increases brain malonyl-CoA and Uncoupling proteins 4 and 5 while decreasing food intake in the normal Wistar Rat. J. Biol. Chem. 2010, 285, 25950–25956. [Google Scholar] [CrossRef] [Green Version]
- Srivastava, S.; Kashiwaya, Y.; King, M.T.; Baxa, U.; Tam, J.; Niu, G.; Chen, X.; Clarke, K.; Veech, R.L. Mitochondrial biogenesis and increased uncoupling protein 1 in brown adipose tissue of mice fed a ketone ester diet. FASEB J. 2012, 26, 2351–2362. [Google Scholar] [CrossRef] [Green Version]
- Bae, H.R.; Kim, D.H.; Park, M.H.; Lee, B.; Kim, M.J.; Lee, E.K.; Chung, K.W.; Kim, S.M.; Im, D.S.; Chung, H.Y. β-Hydroxybutyrate suppresses inflammasome formation by ameliorating endoplasmic reticulum stress via AMPK activation. Oncotarget 2016, 7, 66444–66454. [Google Scholar] [CrossRef] [Green Version]
- Yamanashi, T.; Iwata, M.; Kamiya, N.; Tsunetomi, K.; Kajitani, N.; Wada, N.; Iitsuka, T.; Yamauchi, T.; Miura, A.; Pu, S.; et al. Beta-hydroxybutyrate, an endogenic NLRP3 inflammasome inhibitor, attenuates stress-induced behavioral and inflammatory responses. Sci. Rep. 2017, 7, 7677. [Google Scholar] [CrossRef]
- Youm, Y.H.; Nguyen, K.Y.; Grant, R.W.; Goldberg, E.L.; Bodogai, M.; Kim, D.; D’Agostino, D.; Planavsky, N.; Lupfer, C.; Kanneganti, T.D.; et al. The ketone metabolite β-hydroxybutyrate blocks NLRP3 inflammasome-mediated inflammatory disease. Nat. Med. 2015, 21, 263–269. [Google Scholar] [CrossRef] [Green Version]
- Ari, C.; Murdun, C.; Koutnik, A.P.; Goldhagen, C.R.; Rogers, C.; Park, C.; Bharwani, S.; Diamond, D.M.; Kindy, M.S.; D’Agostino, D.P.; et al. Exogenous Ketones Lower Blood Glucose Level in Rested and Exercised Rodent Models. Nutrients 2019, 11, 2330. [Google Scholar] [CrossRef] [Green Version]
- Edwards, C.; Copes, N.; Bradshaw, P.C. D-ß-hydroxybutyrate: An anti-aging ketone body. Oncotarget 2015, 6, 3477–3478. [Google Scholar] [CrossRef] [PubMed]
- Kenyon, C. The first long-lived mutants: Discovery of the insulin/IGF-1 pathway for ageing. Philos. Trans. R. Soc. Lond. B. Biol. Sci. 2011, 366, 9–16. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Willcox, B.J.; Donlon, T.A.; He, Q.; Chen, R.; Grove, J.S.; Yano, K.; Masaki, K.H.; Willcox, D.C.; Rodriguez, B.; Curb, J.D. FOXO3A genotype is strongly associated with human longevity. Proc. Natl. Acad. Sci. USA 2008, 105, 13987–13992. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hansen, M.; Chandra, A.; Mitic, L.L.; Onken, B.; Driscoll, M.; Kenyon, C. A role for autophagy in the extension of lifespan by dietary restriction in C. elegans. PLoS Genet. 2008, 4, e24. [Google Scholar] [CrossRef] [Green Version]
- Yamada, T.; Zhang, S.J.; Westerblad, H.; Katz, A. β-Hydroxybutyrate inhibits insulin-mediated glucose transport in mouse oxidative muscle. Am. J. Physiol. Endocrinol. Metab. 2010, 299, 364–373. [Google Scholar] [CrossRef]
- Barja, G. Free radicals and aging. Trends Neurosci. 2004, 27, 595–600. [Google Scholar] [CrossRef]
- Newman, J.C.; Covarrubias, A.J.; Zhao, M.; Yu, X.; Gut, P.; Ng, C.P.; Huang, Y.; Haldar, S.; Verdin, E. Ketogenic Diet Reduces Midlife Mortality and Improves Memory in Aging Mice. Cell Metab. 2017, 26, 547–557.e8. [Google Scholar] [CrossRef] [Green Version]
- Simeone, K.A.; Matthews, S.A.; Rho, J.M.; Simeone, T.A. Ketogenic diet treatment increases longevity in Kcna1-null mice, a model of sudden unexpected death in epilepsy. Epilepsia 2016, 57, 178–182. [Google Scholar] [CrossRef] [Green Version]
- Liu, K.; Li, F.; Sun, Q.; Lin, N.; Han, H.; You, K.; Tian, F.; Mao, Z.; Li, T.; Tong, T.; et al. p53 β-hydroxybutyrylation attenuates p53 activity. Cell Death Dis. 2019, 10, 243. [Google Scholar] [CrossRef] [Green Version]
- Habieb, M.E.; Mohamed, M.A.; El Gamal, D.M.; Hawas, A.M.; Mohamed, T.M. Anti-aging effect of DL-β-hydroxybutyrate against hepatic cellular senescence induced by D-galactose or γ-irradiation via autophagic flux stimulation in male rats. Arch. Gerontol. Geriatr. 2021, 92, 104288. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.; Han, L.; Zhao, G.; Shen, H.; Wang, P.; Sun, Z.; Xu, C.; Su, Y.; Li, G.; Tong, T.; et al. hnRNP A1 antagonizes cellular senescence and senescence-associated secretory phenotype via regulation of SIRT1 mRNA stability. Aging Cell 2016, 15, 1063–1073. [Google Scholar] [CrossRef] [PubMed]
- Armada-Moreira, A.; Gomes, J.I.; Pina, C.C.; Savchak, O.K.; Gonçalves-Ribeiro, J.; Rei, N.; Pinto, S.; Morais, T.P.; Martins, R.S.; Ribeiro, F.F.; et al. Going the Extra (Synaptic) Mile: Excitotoxicity as the Road Toward Neurodegenerative Diseases. Front. Cell. Neurosci. 2020, 14, 90. [Google Scholar] [CrossRef]
- Coyle, J.T.; Puttfarcken, P. Oxidative stress, glutamate, and neurodegenerative disorders. Science 1993, 262, 689–695. [Google Scholar] [CrossRef] [PubMed]
- Holper, L.; Ben-Shachar, D.; Mann, J.J. Multivariate meta-analyses of mitochondrial complex I and IV in major depressive disorder, bipolar disorder, schizophrenia, Alzheimer disease, and Parkinson disease. Neuropsychopharmacology 2019, 44, 837–849. [Google Scholar] [CrossRef] [PubMed]
- Mosconi, L.; de Leon, M.; Murray, J.; Lu, J.; Javier, E.; McHugh, P.; Swerdlow, R.H. Reduced mitochondria cytochrome oxidase activity in adult children of mothers with Alzheimer’s disease. J. Alzheimers Dis. 2011, 27, 483–490. [Google Scholar] [CrossRef] [PubMed]
- Lugrin, J.; Rosenblatt-Velin, N.; Parapanov, R.; Liaudet, L. The role of oxidative stress during inflammatory processes. Biol. Chem. 2014, 395, 203–230. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wu, Z.; Yu, J.; Zhu, A.; Nakanishi, H. Nutrients, Microglia Aging, and Brain Aging. Oxid. Med. Cell Longev. 2016, 2016, 7498528. [Google Scholar] [CrossRef] [Green Version]
- Hatanpää, K.; Isaacs, K.R.; Shirao, T.; Brady, D.R.; Rapoport, S.I. Loss of proteins regulating synaptic plasticity in normal aging of the human brain and in Alzheimer disease. J. Neuropathol. Exp. Neurol. 1999, 58, 637–643. [Google Scholar] [CrossRef]
- Rao, J.S.; Kellom, M.; Kim, H.W.; Rapoport, S.I.; Reese, E.A. Neuroinflammation and synaptic loss. Neurochem. Res. 2012, 37, 903–910. [Google Scholar] [CrossRef]
- Scheff, S.W.; Price, D.A.; Schmitt, F.A.; Mufson, E.J. Hippocampal synaptic loss in early Alzheimer’s disease and mild cognitive impairment. Neurobiol. Aging 2006, 27, 1372–1384. [Google Scholar] [CrossRef]
- Appelberg, K.S.; Hovda, D.A.; Prins, M.L. The effects of a ketogenic diet on behavioral outcome after controlled cortical impact injury in the juvenile and adult rat. J. Neurotrauma 2009, 26, 497–506. [Google Scholar] [CrossRef] [Green Version]
- Krikorian, R.; Shidler, M.D.; Dangelo, K.; Couch, S.C.; Benoit, S.C.; Clegg, D.J. Dietary ketosis enhances memory in mild cognitive impairment. Neurobiol. Aging 2012, 33, e19–e27. [Google Scholar] [CrossRef] [Green Version]
- Maalouf, M.; Rho, J.M. Oxidative impairment of hippocampal long-term potentiation involves activation of protein phosphatase 2A and is prevented by ketone bodies. J. Neurosci. Res. 2008, 86, 3322–3330. [Google Scholar] [CrossRef] [Green Version]
- Rusek, M.; Pluta, R.; Ułamek-Kozioł, M.; Czuczwar, S.J. Ketogenic Diet in Alzheimer’s disease. Int. J. Mol. Sci. 2019, 20, 3892. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chatterjee, P.; Fernando, M.; Fernando, B.; Dias, C.B.; Shah, T.; Silva, R.; Williams, S.; Pedrini, S.; Hillebrandt, H.; Goozee, K.; et al. Potential of coconut oil and medium chain triglycerides in the prevention and treatment of Alzheimer’s disease. Mech. Ageing Dev. 2020, 186, 111209. [Google Scholar] [CrossRef]
- Fernando, W.M.; Martins, I.J.; Goozee, K.G.; Brennan, C.S.; Jayasena, V.; Martins, R.N. The role of dietary coconut for the prevention and treatment of Alzheimer’s disease: Potential mechanisms of action. Br. J. Nutr. 2015, 114, 1–14. [Google Scholar] [CrossRef] [PubMed]
- Rebello, C.J.; Keller, J.N.; Liu, A.G.; Johnson, W.D.; Greenway, F.L. Pilot feasibility and safety study examining the effect of medium chain triglyceride supplementation in subjects with mild cognitive impairment: A randomized controlled trial. BBA Clin. 2015, 3, 123–125. [Google Scholar] [CrossRef] [Green Version]
- Ota, M.; Matsuo, J.; Ishida, I.; Hattori, K.; Teraishi, T.; Tonouchi, H.; Ashida, K.; Takahashi, T.; Kunugi, H. Effect of a ketogenic meal on cognitive function in elderly adults: Potential for cognitive enhancement. Psychopharmacology 2016, 233, 3797–3802. [Google Scholar] [CrossRef]
- Pan, Y.; Larson, B.; Araujo, J.A.; Lau, W.; de Rivera, C.; Santana, R.; Gore, A.; Milgram, N.W. Dietary supplementation with medium-chain TAG has long-lasting cognition-enhancing effects in aged dogs. Br. J. Nutr. 2010, 103, 1746–1754. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pawlosky, R.J.; Kashiwaya, Y.; King, M.T.; Veech, R.L. A Dietary Ketone Ester Normalizes Abnormal Behavior in a Mouse Model of Alzheimer’s Disease. Int. J. Mol. Sci. 2020, 21, 1044. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Alexander, G.E.; Chen, K.; Pietrini, P.; Rapoport, S.I.; Reiman, E.M. Longitudinal PET Evaluation of Cerebral Metabolic Decline in Dementia: A Potential Outcome Measure in Alzheimer’s Disease Treatment Studies. Am. J. Psychiatry 2002, 159, 738–745. [Google Scholar] [CrossRef]
- Reiman, E.M.; Chen, K.; Alexander, G.E.; Caselli, R.J.; Bandy, D.; Osborne, D.; Saunders, A.M.; Hardy, J. Functional brain abnormalities in young adults at genetic risk for late-onset Alzheimer’s dementia. Proc. Natl. Acad. Sci. USA 2004, 101, 284–289. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Henderson, S.T.; Vogel, J.L.; Barr, L.J.; Garvin, F.; Jones, J.J.; Costantini, L.C. Study of the ketogenic agent AC-1202 in mild to moderate Alzheimer’s disease: A randomized, double-blind, placebo-controlled, multicenter trial. Nutr. Metab. 2009, 6, 31. [Google Scholar] [CrossRef] [Green Version]
- Reger, M.A.; Henderson, S.T.; Hale, C.; Cholerton, B.; Baker, L.D.; Watson, G.S.; Hyde, K.; Chapman, D.; Craft, S. Effects of beta-hydroxybutyrate on cognition in memory-impaired adults. Neurobiol. Aging 2004, 25, 311–314. [Google Scholar] [CrossRef]
- Kashiwaya, Y.; Takeshima, T.; Mori, N.; Nakashima, K.; Clarke, K.; Veech, R.L. D-beta-hydroxybutyrate protects neurons in models of Alzheimer’s and Parkinson’s disease. Proc. Natl. Acad. Sci. USA 2000, 97, 5440–5444. [Google Scholar] [CrossRef] [Green Version]
- Yin, J.X.; Maalouf, M.; Han, P.; Zhao, M.; Gao, M.; Dharshaun, T.; Ryan, C.; Whitelegge, J.; Wu, J.; Eisenberg, D.; et al. Ketones block amyloid entry and improve cognition in an Alzheimer’s model. Neurobiol. Aging 2016, 39, 25–37. [Google Scholar] [CrossRef]
- Singer, T.P.; Ramsay, R.R.; McKeown, K.; Trevor, A.; Castagnoli, N.E., Jr. Mechanism of the neurotoxicity of 1-methyl-4-phenylpyridinium (MPP+), the toxic bioactivation product of 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine (MPTP). Toxicology 1988, 49, 17–23. [Google Scholar] [CrossRef]
- Vanitallie, T.B.; Nonas, C.; Di Rocco, A.; Boyar, K.; Hyams, K.; Heymsfield, S.B. Treatment of Parkinson disease with diet-induced hyperketonemia: A feasibility study. Neurology 2005, 64, 728–730. [Google Scholar] [CrossRef]
- Ari, C.; Poff, A.M.; Held, H.E.; Landon, C.S.; Goldhagen, C.R.; Mavromates, N.; D’Agostino, D.P. Metabolic therapy with Deanna Protocol supplementation delays disease progression and extends survival in amyotrophic lateral sclerosis (ALS) mouse model. PLoS ONE 2014, 9, e103526. [Google Scholar] [CrossRef]
- Ari, C.; Murdun, C.; Goldhagen, C.; Koutnik, A.P.; Bharwani, S.R.; Diamond, D.M.; Kindy, M.; D’Agostino, D.P.; Kovacs, Z. Exogenous Ketone Supplements Improved Motor Performance in Preclinical Rodent Models. Nutrients 2020, 12, 2459. [Google Scholar] [CrossRef]
- Netzahualcoyotzi, C.; Tapia, R. Degeneration of spinal motor neurons by chronic AMPA-induced excitotoxicity in vivo and protection by energy substrates. Acta Neuropathol. Commun. 2015, 3, 27. [Google Scholar] [CrossRef] [Green Version]
- Zhao, Z.; Lange, D.J.; Voustianiouk, A.; MacGrogan, D.; Ho, L.; Suh, J.; Humala, N.; Thiyagarajan, M.; Wang, J.; Pasinetti, G.M. A ketogenic diet as a potential novel therapeutic intervention in amyotrophic lateral sclerosis. BMC Neurosci. 2006, 7, 29. [Google Scholar] [CrossRef] [Green Version]
- Zhao, W.; Varghese, M.; Vempati, P.; Dzhun, A.; Cheng, A.; Wang, J.; Lange, D.; Bilski, A.; Faravelli, I.; Pasinetti, G.M. Caprylic triglyceride as a novel therapeutic approach to effectively improve the performance and attenuate the symptoms due to the motor neuron loss in ALS disease. PLoS ONE 2012, 7, e49191. [Google Scholar] [CrossRef] [PubMed]
- Abg Abd Wahab, D.Y.; Gau, C.H.; Zakaria, R.; Muthu Karuppan, M.K.; A-Rahbi, B.S.; Abdullah, Z.; Alrafiah, A.; Abdullah, J.M.; Muthuraju, S. Review on Cross Talk between Neurotransmitters and Neuroinflammation in Striatum and Cerebellum in the Mediation of Motor Behaviour. Biomed. Res. Int. 2019, 14, 1767203. [Google Scholar] [CrossRef] [PubMed]
- Blasco, H.; Mavel, S.; Corcia, P.; Gordon, P.H. The glutamate hypothesis in ALS: Pathophysiology and drug development. Curr. Med. Chem. 2014, 21, 3551–3575. [Google Scholar] [CrossRef]
- Brichta, L.; Greengard, P.; Flajolet, M. Advances in the pharmacological treatment of Parkinson’s disease: Targeting neurotransmitter systems. Trends Neurosci. 2013, 36, 543–554. [Google Scholar] [CrossRef]
- Tisch, S.; Silberstein, P.; Limousin-Dowsey, P.; Jahanshahi, M. The basal ganglia: Anatomy, physiology, and pharmacology. Psychiatr. Clin. 2004, 27, 757–799. [Google Scholar] [CrossRef]
- D’Amelio, M.; Puglisi-Allegra, S.; Mercuri, N. The role of dopaminergic midbrain in Alzheimer’s disease: Translating basic science into clinical practice. Pharmacol. Res. 2018, 130, 414–419. [Google Scholar] [CrossRef]
- Huang, D.; Liu, D.; Yin, J.; Qian, T.; Shrestha, S.; Ni, H. Glutamate-glutamine and GABA in brain of normal aged and patients with cognitive impairment. Eur. Radiol. 2017, 27, 2698–2705. [Google Scholar] [CrossRef]
- Stanciu, G.D.; Luca, A.; Rusu, R.N.; Bild, V.; Beschea Chiriac, S.I.; Solcan, C.; Bild, W.; Ababei, D.C. Alzheimer’s Disease Pharmacotherapy in Relation to Cholinergic System Involvement. Biomolecules 2019, 10, 40. [Google Scholar] [CrossRef] [Green Version]
- Ma, S.; Hangya, B.; Leonard, C.S.; Wisden, W.; Gundlach, A.L. Dual-transmitter systems regulating arousal, attention, learning and memory. Neurosci. Biobehav. Rev. 2018, 85, 21–33. [Google Scholar] [CrossRef]
- Choong, C.J.; Sasaki, T.; Hayakawa, H.; Yasuda, T.; Baba, K.; Hirata, Y.; Uesato, S.; Mochizuki, H. A novel histone deacetylase 1 and 2 isoform-specific inhibitor alleviates experimental Parkinson’s disease. Neurobiol. Aging 2016, 37, 103–116. [Google Scholar] [CrossRef] [Green Version]
- D’Mello, S.R. Histone deacetylases as targets for the treatment of human neurodegenerative diseases. Drug News Perspect. 2009, 22, 513–524. [Google Scholar] [CrossRef]
- Feng, H.L.; Leng, Y.; Ma, C.H.; Zhang, J.; Ren, M.; Chuang, D.M. Combined lithium and valproate treatment delays disease onset, reduces neurological deficits and prolongs survival in an amyotrophic lateral sclerosis mouse model. Neuroscience 2008, 155, 567–572. [Google Scholar] [CrossRef] [Green Version]
- Lu, X.; Wang, L.; Yu, C.; Yu, D.; Yu, G. Histone Acetylation Modifiers in the Pathogenesis of Alzheimer’s Disease. Front. Cell. Neurosci. 2015, 9, 226. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chuang, D.M.; Leng, Y.; Marinova, Z.; Kim, H.J.; Chiu, C.T. Multiple roles of HDAC inhibition in neurodegenerative conditions. Trends Neurosci. 2009, 32, 591–601. [Google Scholar] [CrossRef] [Green Version]
- Kazantsev, A.G.; Thompson, L.M. Therapeutic application of histone deacetylase inhibitors for central nervous system disorders. Nat. Rev. Drug Discov. 2008, 7, 854–868. [Google Scholar] [CrossRef] [PubMed]
- Peleg, S.; Sananbenesi, F.; Zovoilis, A.; Burkhardt, S.; Bahari-Javan, S.; Agis-Balboa, R.C.; Cota, P.; Wittnam, J.L.; Gogol-Doering, A.; Opitz, L.; et al. Altered histone acetylation is associated with age-dependent memory impairment in mice. Science 2010, 328, 753–756. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sharma, S.; Taliyan, R.; Ramagiri, S. Histone deacetylase inhibitor, trichostatin A, improves learning and memory in high-fat diet-induced cognitive deficits in mice. J. Mol. Neurosci. 2015, 56, 1–11. [Google Scholar] [CrossRef]
- Peng, S.; Wuu, J.; Mufson, E.J.; Fahnestock, M. Precursor form of brain-derived neurotrophic factor and mature brain-derived neurotrophic factor are decreased in the pre-clinical stages of Alzheimer’s disease. J. Neurochem. 2005, 93, 1412–1421. [Google Scholar] [CrossRef] [PubMed]
- Qin, X.Y.; Cao, C.; Cawley, N.X.; Liu, T.T.; Yuan, J.; Loh, Y.P.; Cheng, Y. Decreased peripheral brain-derived neurotrophic factor levels in Alzheimer’s disease: A meta-analysis study (N = 7277). Mol. Psychiatry 2017, 22, 312–320. [Google Scholar] [CrossRef]
- Valenzuela, P.L.; Castillo-García, A.; Morales, J.S.; de la Villa, P.; Hampel, H.; Emanuele, E.; Lista, S.; Lucia, A. Exercise benefits on Alzheimer’s disease: State-of-the-science. Ageing Res. Rev. 2020, 62, 101108. [Google Scholar] [CrossRef]
- Glass, C.K.; Saijo, K.; Winner, B.; Marchetto, M.C.; Gage, F.H. Mechanisms underlying inflammation in neurodegeneration. Cell 2010, 140, 918–934. [Google Scholar] [CrossRef] [Green Version]
- Swanton, T.; Cook, J.; Beswick, J.A.; Freeman, S.; Lawrence, C.B.; Brough, D. Is Targeting the Inflammasome a Way Forward for Neuroscience Drug Discovery? SLAS Discov. 2018, 23, 991–1017. [Google Scholar] [CrossRef] [Green Version]
- Hyun, D.H.; Lee, M.; Halliwell, B.; Jenner, P. Proteasomal inhibition causes the formation of protein aggregates containing a wide range of proteins, including nitrated proteins. J. Neurochem. 2003, 86, 363–373. [Google Scholar] [CrossRef]
- Lim, J.E.; Song, M.; Jin, J.; Kou, J.; Pattanayak, A.; Lalonde, R.; Fukuchi, K. The effects of MyD88 deficiency on exploratory activity, anxiety, motor coordination, and spatial learning in C57BL/6 and APPswe/PS1dE9 mice. Behav. Brain Res. 2012, 227, 36–42. [Google Scholar] [CrossRef] [Green Version]
- Liu, X.; Wu, Z.; Hayashi, Y.; Nakanishi, H. Age-dependent neuroinflammatory responses and deficits in long-term potentiation in the hippocampus during systemic inflammation. Neuroscience 2012, 216, 133–142. [Google Scholar] [CrossRef] [PubMed]
- Kim, D.Y.; Hao, J.; Liu, R.; Turner, G.; Shi, F.D.; Rho, J.M. Inflammation-mediated memory dysfunction and effects of a ketogenic diet in a murine model of multiple sclerosis. PLoS ONE 2012, 7, e35476. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pallàs, M.; Pizarro, J.G.; Gutierrez-Cuesta, J.; Crespo-Biel, N.; Alvira, D.; Tajes, M.; Yeste-Velasco, M.; Folch, J.; Canudas, A.M.; Sureda, F.X.; et al. Modulation of SIRT1 expression in different neurodegenerative models and human pathologies. Neuroscience 2008, 154, 1388–1397. [Google Scholar] [CrossRef]
- Herskovits, A.Z.; Guarente, L. SIRT1 in neurodevelopment and brain senescence. Neuron 2014, 81, 471–483. [Google Scholar] [CrossRef] [Green Version]
- Corpas, R.; Revilla, S.; Ursulet, S.; Castro-Freire, M.; Kaliman, P.; Petegnief, V.; Giménez-Llort, L.; Sarkis, C.; Pallàs, M.; Sanfeliu, C. SIRT1 Overexpression in Mouse Hippocampus Induces Cognitive Enhancement through Proteostatic and Neurotrophic Mechanisms. Mol. Neurobiol. 2017, 54, 5604–5619. [Google Scholar] [CrossRef]
- Wang, R.; Zhang, Y.; Li, J.; Zhang, C. Resveratrol ameliorates spatial learning memory impairment induced by Aβ1-42 in rats. Neuroscience 2017, 344, 39–47. [Google Scholar] [CrossRef]
- Gao, J.; Wang, W.Y.; Mao, Y.W.; Gräff, J.; Guan, J.S.; Pan, L.; Mak, G.; Kim, D.; Su, S.C.; Tsai, L.H. A novel pathway regulates memory and plasticity via SIRT1 and miR-134. Nature 2010, 466, 1105–1109. [Google Scholar] [CrossRef] [Green Version]
- Michán, S.; Li, Y.; Chou, M.M.; Parrella, E.; Ge, H.; Long, J.M.; Allard, J.S.; Lewis, K.; Miller, M.; Xu, W.; et al. SIRT1 is essential for normal cognitive function and synaptic plasticity. J. Neurosci. 2010, 30, 9695–9707. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Han, S.; Choi, J.R.; Soon Shin, K.; Kang, S.J. Resveratrol upregulated heat shock proteins and extended the survival of G93A-SOD1 mice. Brain Res. 2012, 1483, 112–117. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Zhang, Y.; Tang, L.; Zhang, N.; Fan, D. Protective effects of resveratrol through the up-regulation of SIRT1 expression in the mutant hSOD1-G93A-bearing motor neuron-like cell culture model of amyotrophic lateral sclerosis. Neurosci. Lett. 2011, 503, 250–255. [Google Scholar] [CrossRef] [PubMed]
- Mudò, G.; Mäkelä, J.; Di Liberto, V.; Tselykh, T.V.; Olivieri, M.; Piepponen, P.; Eriksson, O.; Mälkiä, A.; Bonomo, A.; Kairisalo, M.; et al. Transgenic expression and activation of PGC-1α protect dopaminergic neurons in the MPTP mouse model of Parkinson’s disease. Cell. Mol. Life Sci. 2012, 69, 1153–1165. [Google Scholar] [CrossRef]
- Karuppagounder, S.S.; Pinto, J.T.; Xu, H.; Chen, H.L.; Beal, M.F.; Gibson, G.E. Dietary supplementation with resveratrol reduces plaque pathology in a transgenic model of Alzheimer’s disease. Neurochem. Int. 2009, 54, 111–118. [Google Scholar] [CrossRef] [Green Version]
- St-Pierre, J.; Drori, S.; Uldry, M.; Silvaggi, J.M.; Rhee, J.; Jäger, S.; Handschin, C.; Zheng, K.; Lin, J.; Yang, W.; et al. Suppression of reactive oxygen species and neurodegeneration by the PGC-1 transcriptional coactivators. Cell 2006, 127, 397–408. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lin, J.; Wu, P.H.; Tarr, P.T.; Lindenberg, K.S.; St-Pierre, J.; Zhang, C.Y.; Mootha, V.K.; Jäger, S.; Vianna, C.R.; Reznick, R.M.; et al. Defects in adaptive energy metabolism with CNS-linked hyperactivity in PGC-1alpha null mice. Cell 2004, 119, 121–135. [Google Scholar] [CrossRef] [Green Version]
- Wareski, P.; Vaarmann, A.; Choubey, V.; Safiulina, D.; Liiv, J.; Kuum, M.; Kaasik, A. PGC-1{alpha} and PGC-1{beta} regulate mitochondrial density in neurons. J. Biol. Chem. 2009, 284, 21379–21385. [Google Scholar] [CrossRef] [Green Version]
- Zheng, B.; Liao, Z.; Locascio, J.J.; Lesniak, K.A.; Roderick, S.S.; Watt, M.L.; Eklund, A.C.; Zhang-James, Y.; Kim, P.D.; Hauser, M.A.; et al. PGC-1α, a potential therapeutic target for early intervention in Parkinson’s disease. Sci. Transl. Med. 2010, 2, 52ra73. [Google Scholar] [CrossRef] [Green Version]
- Gong, B.; Chen, F.; Pan, Y.; Arrieta-Cruz, I.; Yoshida, Y.; Haroutunian, V.; Pasinetti, G.M. SCFFbx2-E3-ligase-mediated degradation of BACE1 attenuates Alzheimer’s disease amyloidosis and improves synaptic function. Aging Cell 2010, 9, 1018–1031. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kiaei, M.; Kipiani, K.; Chen, J.; Calingasan, N.Y.; Beal, M.F. Peroxisome proliferator-activated receptor-gamma agonist extends survival in transgenic mouse model of amyotrophic lateral sclerosis. Exp. Neurol. 2005, 191, 331–336. [Google Scholar] [CrossRef] [PubMed]
- Zhao, W.; Varghese, M.; Yemul, S.; Pan, Y.; Cheng, A.; Marano, P.; Hassan, S.; Vempati, P.; Chen, F.; Qian, X.; et al. Peroxisome proliferator activator receptor gamma coactivator-1alpha (PGC-1α) improves motor performance and survival in a mouse model of amyotrophic lateral sclerosis. Mol. Neurodegener. 2011, 6, 51. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Agarwal, S.; Yadav, A.; Chaturvedi, R.K. Peroxisome proliferator-activated receptors (PPARs) as therapeutic target in neurodegenerative disorders. Biochem. Biophys. Res. Commun. 2017, 483, 1166–1177. [Google Scholar] [CrossRef] [PubMed]
- D’ANGELO, M.; Castelli, V.; Catanesi, M.; Antonosante, A.; Dominguez-Benot, R.; Ippoliti, R.; Benedetti, E.; Cimini, A. PPARγ and Cognitive Performance. Int. J. Mol. Sci. 2019, 20, 5068. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Im, J.Y.; Lee, K.W.; Woo, J.M.; Junn, E.; Mouradian, M.M. DJ-1 induces thioredoxin 1 expression through the Nrf2 pathway. Hum. Mol. Genet. 2012, 21, 3013–3024. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kensler, T.W.; Wakabayashi, N.; Biswal, S. Cell survival responses to environmental stresses via the Keap1-Nrf2-ARE pathway. Annu. Rev. Pharmacol. Toxicol. 2007, 47, 89–116. [Google Scholar] [CrossRef]
- Kincaid, B.; Bossy-Wetzel, E. Forever young: SIRT3 a shield against mitochondrial meltdown, aging, and neurodegeneration. Front. Aging Neurosci. 2013, 5, 48. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ramesh, S.; Govindarajulu, M.; Lynd, T.; Briggs, G.; Adamek, D.; Jones, E.; Heiner, J.; Majrashi, M.; Moore, T.; Amin, R.; et al. SIRT3 activator Honokiol attenuates β-Amyloid by modulating amyloidogenic pathway. PLoS ONE 2018, 13, e0190350. [Google Scholar] [CrossRef] [Green Version]
- Song, W.; Song, Y.; Kincaid, B.; Bossy, B.; Bossy-Wetzel, E. Mutant SOD1G93A triggers mitochondrial fragmentation in spinal cord motor neurons: Neuroprotection by SIRT3 and PGC-1α. Neurobiol. Dis. 2013, 51, 72–81. [Google Scholar] [CrossRef] [PubMed] [Green Version]



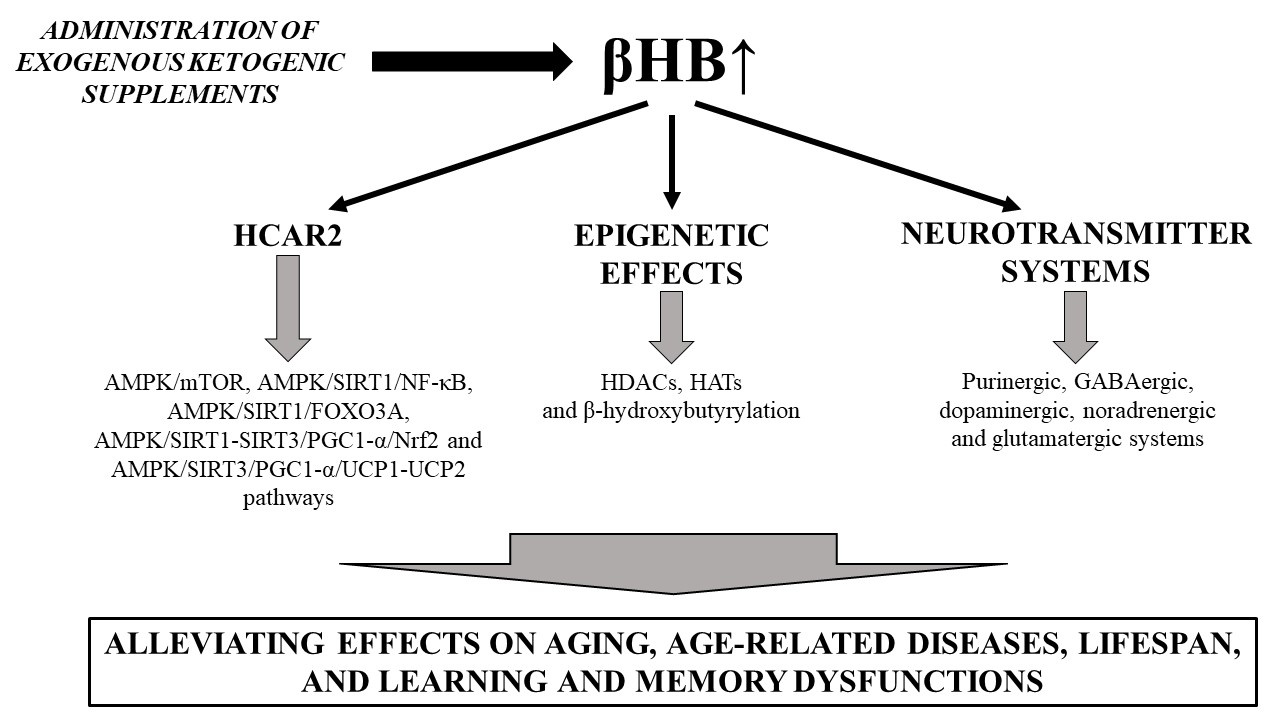{kind=link}
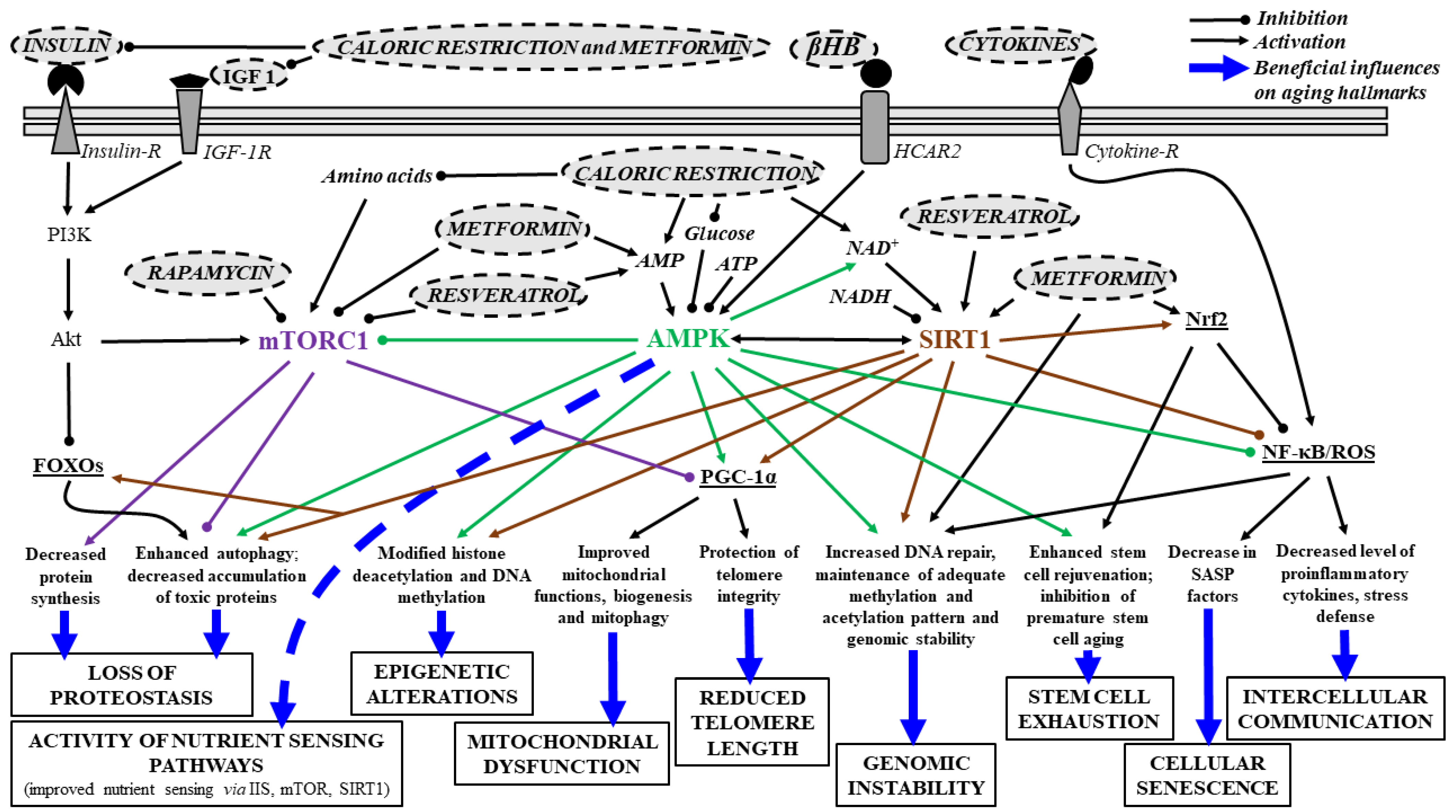{kind=link}
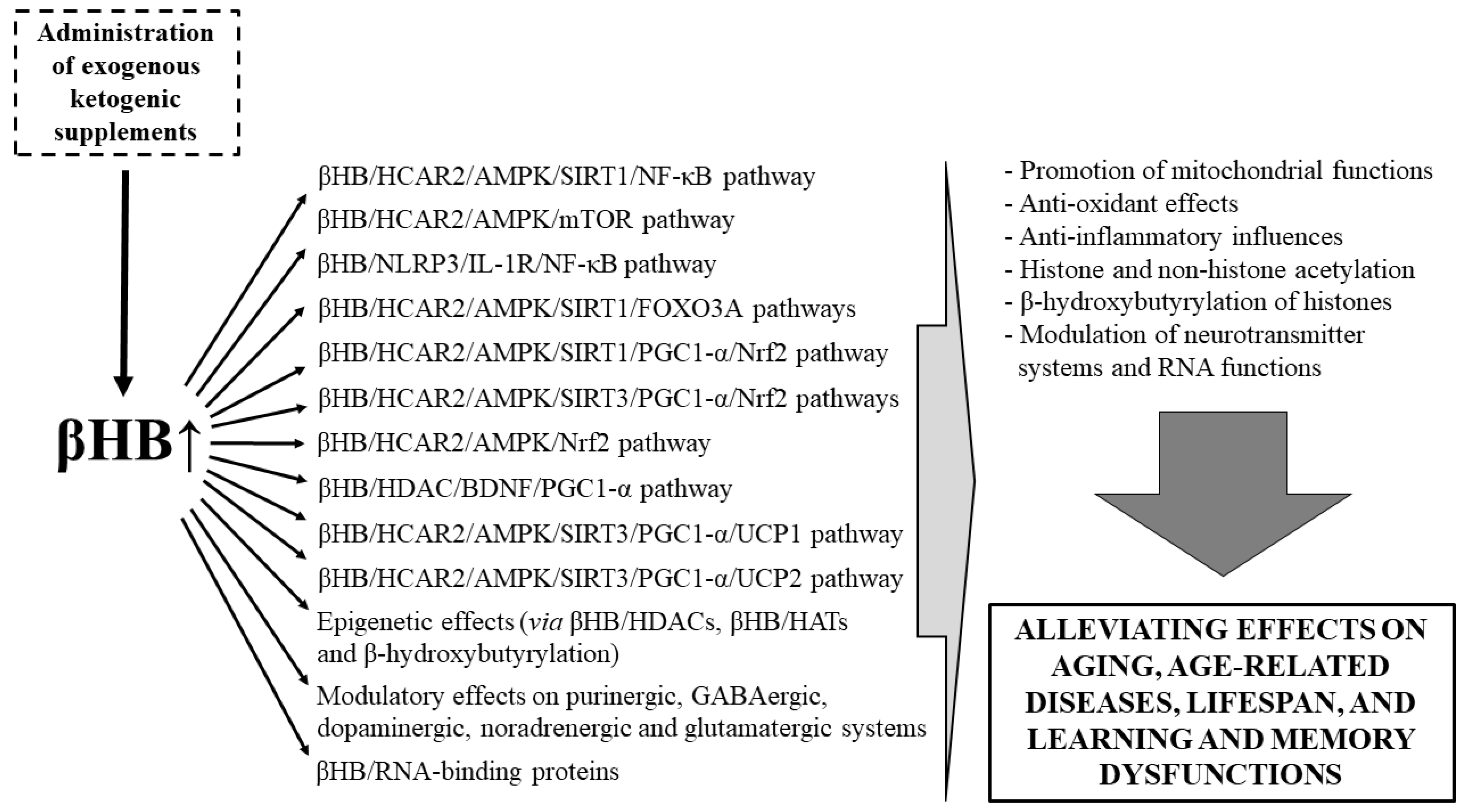{kind=link}
| Name (Components) | Dose and Route of Administration | Treatment Duration | Model Organism (Species) | Significant Increase in Blood βHB Level | Main Findings | Ref. |
|---|---|---|---|---|---|---|
| Beta-hydroxybutyrate (βHB) | ||||||
| βHB (DL-β-Hydroxybutyric acid sodium salt) | 1.5 mmol/kg/day (subcutaneous administration, 0.25 μL/h) | 4 weeks | A mouse model of Alzheimer’s disease (5XFAD) | No data | Improved learning and memory; attenuated Aβ accumulation | [50] |
| βHB + acetoacetate | 600 mg βHB/kg/day + 150 mg acetoacetate/kg/day (subcutaneous injection) | 2 months | A mouse model of Alzheimer’s disease (APPSwInd) | Yes | Improved cognitive performance; reduced Aβ accumulation | [329] |
| βHB | 0.4, 0.8, or 1.6 mmol/kg/day (subcutaneous administration, 1 μL/h) | 28 days | LPS-induced Parkinson’s disease rat model | No data | Beneficial effects on motor dysfunction; protection of dopaminergic neurons | [55] |
| βHB (D-βHB) | 0.4, 0.8, or 1.6 mmol/kg/day (subcutaneous administration, 1 μL/h) | 1 week | MPTP-induced Parkinson’s disease mouse model | Yes | Improved motor performance; decrease in MPTP-induced dopaminergic neurodegeneration | [258] |
| Ketone esters (KEs) | ||||||
| KE (R,S-1,3-butanediol acetoacetate diester: BD-AcAc2; standard rodent chow mixed at 10% BD-AcAc2 by volume and 1% saccharin) | Ad libitum (oral intake) | 8 weeks | A mouse model of Angelman syndrome (UBE3Atm1Alb/J null mutation mice) | Yes | Improved motor coordination, learning and memory | [41] |
| KE (comprised of D-β-hydroxybutyrate and (R)-1,3-butanediol; 125 g KE/1000 g diet) | Animals were fed a 4 to 5 g pellet/animal at approximately 06:00 hours each day (oral intake) | 8 months | A mouse model of Alzheimer’s disease (3xTgAD) | Yes | Improvements in performance on learning and memory tests; decreased Aβ and hyperphosphorylated tau deposition | [43] |
| KE [ketone monoester, (R)-3-hydroxybutyl (R)-3-hydroxybutyrate] + MCT and coconut oil (CO) mixture (4:3) | Normal diet + 28.7 g of the KE thrice daily + 165 mL/day of the MCT/CO mixture (oral intake) | 20 months | A patient with Alzheimer’s disease dementia | Yes | Improving behavior as well as cognitive and daily-activity performance | [47] |
| Medium chain triglycerides (MCTs) | ||||||
| MCT (97% caprylic acid + 3% capric acid; a normal diet supplemented with 5.5% MCT) | Dogs were fed once/day for about one hour; about 200 g supplemented diet/day/animal (oral intake) | 8 months | Aged dogs | Yes | Improvements in learning ability and attention | [322] |
| MCT (the diet was mixed with Deanna protocol/DP at 22% by weight; DP contained 10% MCT high in caprylic acid) | Ad libitum (oral intake) | 6–10 weeks | A mouse model of Amyotrophic lateral sclerosis (SOD1-G93A) | No | Better motor performance, improved (lower) neurological scores and extended survival time | [332] |
| MCT (a diet in which 35% of the calories was derived from triheptanoin) | Ad libitum (oral intake) | 24 weeks | A mouse model of Amyotrophic lateral sclerosis (SOD1-G93A) | Yes | Protection against motor neuron loss; improved motor function | [48] |
| MCT [a diet containing 10% (w/w) caprylic acid] | Ad libitum; about 3 g diet/day was consumed/animal (oral intake) | About 12 weeks | A mouse model of Amyotrophic lateral sclerosis (SOD1-G93A) | Yes | Protection against motor neuron loss; improved motor function | [336] |
| MCT (NeoBee 895, >95% of the fatty acids are caprylic acid; the remainder consists of caproic and capric acids) | 40 mL MCT (oral intake) | Single administration | Adult subjects with Alzheimer’s disease or mild cognitive impairment | Yes | Improvement in cognitive functions (in patients without APOE ε4 allele) | [327] |
| MCT (AC-1202, an MCT composed of glycerin and, almost entirely, caprylic acid, NeoBee 895) | Normal diet + 20 g MCT/day/patient (oral intake) | 3 months | Humans with mild to moderate Alzheimer’s disease | Yes | Improvement in cognitive performance (in patients without APOE ε4 allele) | [326] |
| MCT (50 g Ketogenic meal, Ketonformula containing 20 g of MCTs: 15 g caprylic acid + 5 g capric acid) | 50 g ketogenic meal (oral intake) | Single administration | Humans; elderly, non-demented | Yes | Positive effects on working memory, visual attention, and task switching | [321] |
| MCT (MCT drink: a 12% emulsion of Captex 355, containing 60% caprylic acid and 40% capric acid) | Normal diet + 15 g MCT twice/day/patient in a ketogenic drink (oral intake) | 6 months | Humans; aged participants with mild cognitive impairment | Yes | Improved executive function, memory, and language | [32] |
| MCT (MCT oil, Nestle™) | Normal diet + 56 g MCT/day/patient (oral intake) | 24 weeks | Humans; adults with mild cognitive impairment | Yes | Improved memory | [320] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kovács, Z.; Brunner, B.; Ari, C. Beneficial Effects of Exogenous Ketogenic Supplements on Aging Processes and Age-Related Neurodegenerative Diseases. Nutrients 2021, 13, 2197. https://doi.org/10.3390/nu13072197
Kovács Z, Brunner B, Ari C. Beneficial Effects of Exogenous Ketogenic Supplements on Aging Processes and Age-Related Neurodegenerative Diseases. Nutrients. 2021; 13(7):2197. https://doi.org/10.3390/nu13072197
Chicago/Turabian StyleKovács, Zsolt, Brigitta Brunner, and Csilla Ari. 2021. "Beneficial Effects of Exogenous Ketogenic Supplements on Aging Processes and Age-Related Neurodegenerative Diseases" Nutrients 13, no. 7: 2197. https://doi.org/10.3390/nu13072197
APA StyleKovács, Z., Brunner, B., & Ari, C. (2021). Beneficial Effects of Exogenous Ketogenic Supplements on Aging Processes and Age-Related Neurodegenerative Diseases. Nutrients, 13(7), 2197. https://doi.org/10.3390/nu13072197