
Amino Acid-Induced Impairment of Insulin Signaling and Involvement of G-Protein Coupling Receptor


{kind=link}
{kind=link}
Abstract
:1. Introduction
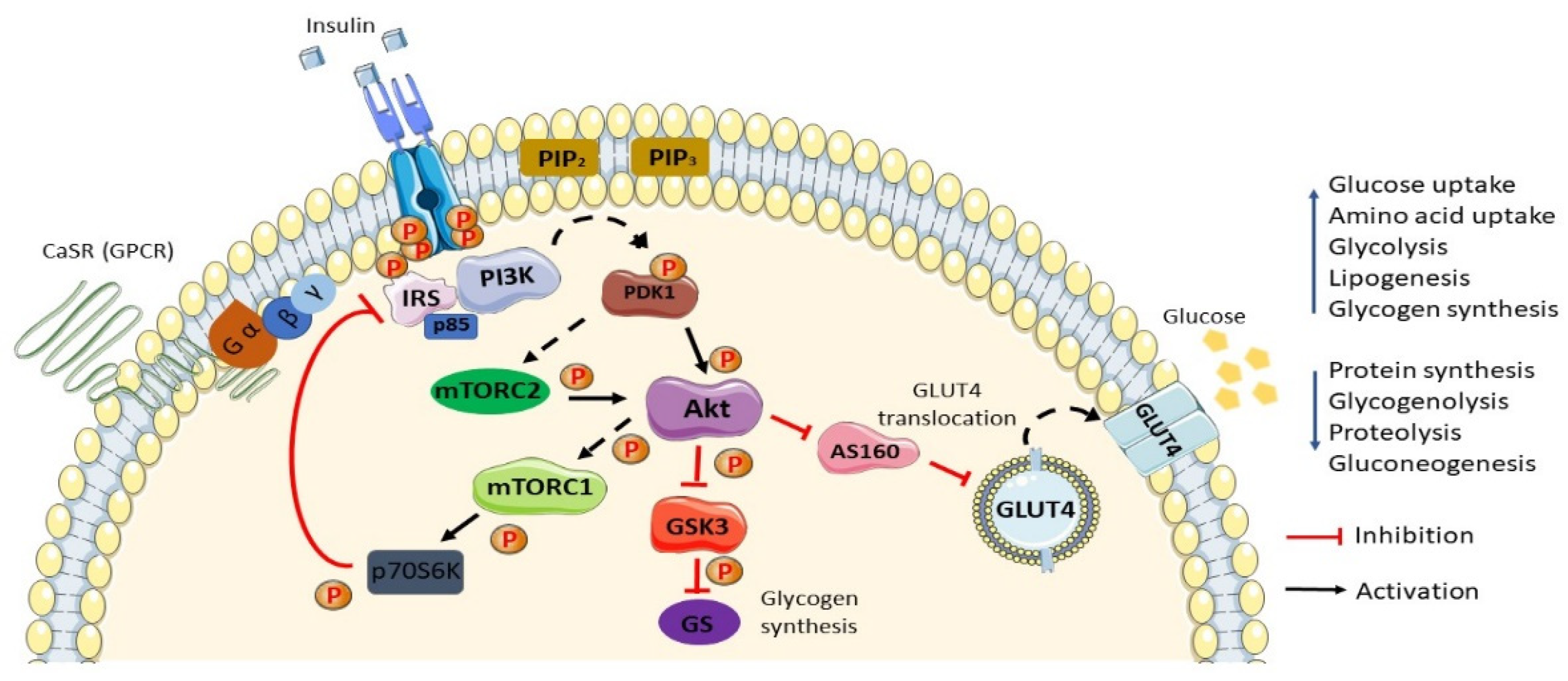
2. Normal Insulin Action/Insulin Action in Normal Settings
3. Insulin Resistance and Type 2 Diabetes Mellitus
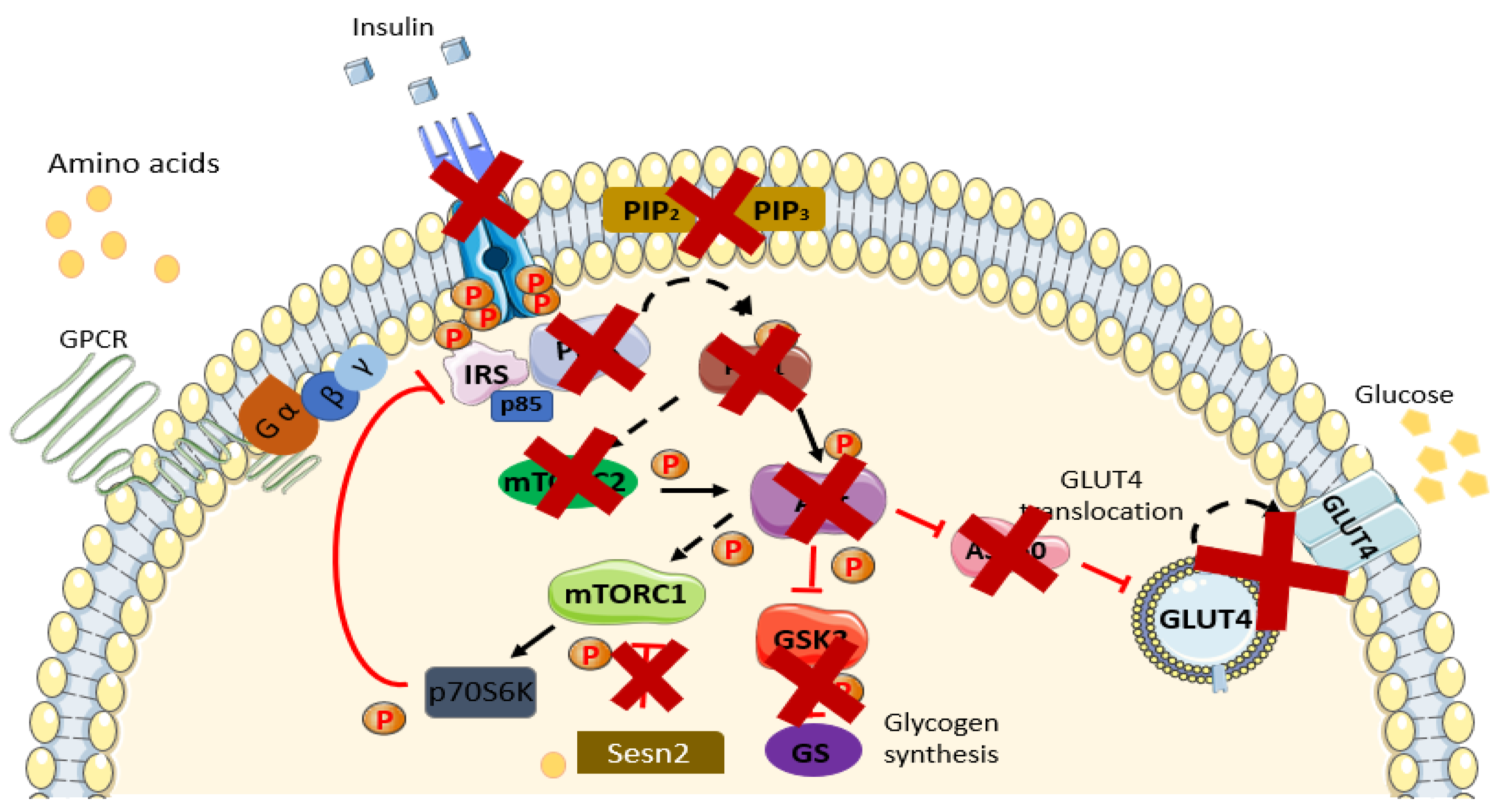
4. Linkage between Amino Acids and IR/T2DM
4.1. Metabolic Dysfunction
4.2. mTOR Pathway
4.3. Gut Microbiome
4.4. 3-Hydroxyisobutyrate (3HIB)
4.5. Inflammation
4.6. Aromatic Amino Acids (AAAs)
5. G-Protein Coupled Receptor Signaling
6. Involvement of G-Protein Coupled Receptor
6.1. GPR142 and GPRC6A
6.2. Umami Taste Receptor T1R1/T1R3
6.3. Calcium-Sensing Receptor (CaSR)
7. Conclusions
Funding
Acknowledgments
Conflicts of Interest
References
- Giesbertz, P.; Daniel, H. Branched-chain amino acids as biomarkers in diabetes. Curr. Opin. Clin. Nutr. Metab. Care 2016, 19, 48–54. [Google Scholar] [CrossRef] [PubMed]
- Karusheva, Y.; Koessler, T.; Strassburger, K.; Markgraf, D.; Mastrototaro, L.; Jelenik, T.; Simon, M.-C.; Pesta, D.; Zaharia, O.-P.; Bódis, K.; et al. Short-term dietary reduction of branched-chain amino acids reduces meal-induced insulin secretion and modifies microbiome composition in type 2 diabetes: A randomized controlled crossover trial. Am. J. Clin. Nutr. 2019, 110, 1098–1107. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nagao, K.; Yamakado, M. The role of amino acid profiles in diabetes risk assessment. Curr. Opin. Clin. Nutr. Metab. Care 2016, 19, 328–335. [Google Scholar] [CrossRef]
- Kawanaka, M.M.; Nishino, K.; Oka, T.; Urata, N.; Nakamura, J.; Suehiro, M.; Kawamoto, H.; Chiba, Y.; Yamada, G. Tyrosine levels are associated with insulin resistance in patients with nonalcoholic fatty liver disease. Hepatic Med. Évid. Res. 2015, 7, 29–35. [Google Scholar] [CrossRef] [Green Version]
- Chen, T.; Ni, Y.; Ma, X.; Bao, Y.; Liu, J.; Huang, F.; Hu, C.; Xie, G.; Zhao, A.; Jia, W.; et al. Branched-chain and aromatic amino acid profiles and diabetes risk in Chinese populations. Sci. Rep. 2016, 6, 20594. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Forouhi, N.; Wareham, N.J. Epidemiology of diabetes. Medicine 2014, 42, 698–702. [Google Scholar] [CrossRef] [Green Version]
- Siddiqui, A.A.; Siddiqui, S.A.; Ahmad, S.; Siddiqui, S.; Ahsan, I.; Sahu, K. Diabetes: Mechanism, pathophysiology and management-A review. Int. J. Drug Dev. Res. 2013, 5, 1–23. [Google Scholar]
- Nucci, L.B.; Toscano, C.M.; Maia, A.L.M.; Fonseca, C.D.; Britto, M.M.B.; Duncan, B.B.; Schmidt, M.I. A nationwide population screening program for diabetes in Brazil. Revista Panamericana Salud Pública 2004, 16, 320–327. [Google Scholar] [CrossRef]
- Catalano, K.J.; Maddux, B.A.; Szary, J.; Youngren, J.F.; Goldfine, I.D.; Schaufele, F. Insulin Resistance Induced by Hyperinsulinemia Coincides with a Persistent Alteration at the Insulin Receptor Tyrosine Kinase Domain. PLoS ONE 2014, 9, e108693. [Google Scholar] [CrossRef] [Green Version]
- Ding, Y.-Y.; Cheng, X.-R.; Li, Z.-Q.; Wu, S.-J.; Yang, Y.; Shi, Y.-H.; Le, G.-W. Effect of dietary oxidized tyrosine products on insulin secretion via the oxidative stress-induced mitochondria damage in mice pancreas. RSC Adv. 2017, 7, 26809–26826. [Google Scholar] [CrossRef] [Green Version]
- Hansen, J.S.; Zhao, X.; Irmler, M.; Liu, X.; Hoene, M.; Scheler, M.; Li, Y.; Beckers, J.; De Angelis, M.H.; Haring, H.-U.; et al. Type 2 diabetes alters metabolic and transcriptional signatures of glucose and amino acid metabolism during exercise and recovery. Diabetologia 2015, 58, 1845–1854. [Google Scholar] [CrossRef] [Green Version]
- Adeva, M.M.; Calviño, J.; Souto, G.; Donapetry, C. Insulin resistance and the metabolism of branched-chain amino acids in humans. Amino Acids 2011, 43, 171–181. [Google Scholar] [CrossRef] [PubMed]
- Chen, X.; Yang, W. Branched-chain amino acids and the association with type 2 diabetes. J. Diabetes Investig. 2015, 6, 369–370. [Google Scholar] [CrossRef] [PubMed]
- Wiklund, P.; Zhang, X.; Pekkala, S.; Autio, R.; Kong, L.; Yang, Y.; Keinänen-Kiukaanniemi, S.; Alen, M.; Cheng, S. Insulin resistance is associated with altered amino acid metabolism and adipose tissue dysfunction in normoglycemic women. Sci. Rep. 2016, 6, 24540. [Google Scholar] [CrossRef]
- Nagata, C.; Nakamura, K.; Wada, K.; Tsuji, M.; Tamai, Y.; Kawachi, T. Branched-chain Amino Acid Intake and the Risk of Diabetes in a Japanese Community: The Takayama Study. Am. J. Epidemiol. 2013, 178, 1226–1232. [Google Scholar] [CrossRef]
- Zhao, X.; Han, Q.; Liu, Y.; Sun, C.; Gang, X.; Wang, G. The Relationship between Branched-Chain Amino Acid Related Metabolomic Signature and Insulin Resistance: A Systematic Review. J. Diabetes Res. 2016, 2016, 1–12. [Google Scholar] [CrossRef]
- Yamada, C.; Kondo, M.; Kishimoto, N.; Shibata, T.; Nagai, Y.; Imanishi, T.; Oroguchi, T.; Ishii, N.; Nishizaki, Y. Association between insulin resistance and plasma amino acid profile in non-diabetic J apanese subjects. J. Diabetes Investig. 2015, 6, 408–415. [Google Scholar] [CrossRef]
- Newgard, C.B.; An, J.; Bain, J.R.; Muehlbauer, M.J.; Stevens, R.D.; Lien, L.F.; Haqq, A.M.; Shah, S.H.; Arlotto, M.; Slentz, C.A.; et al. A Branched-Chain Amino Acid-Related Metabolic Signature that Differentiates Obese and Lean Humans and Contributes to Insulin Resistance. Cell Metab. 2009, 9, 311–326. [Google Scholar] [CrossRef] [Green Version]
- Ghimire, S.; Flury, M.; Scheenstra, E.J.; Miles, C.A. Sampling and degradation of biodegradable plastic and paper mulches in field after tillage incorporation. Sci. Total Environ. 2020, 703, 135577. [Google Scholar] [CrossRef]
- Gar, C.; Rottenkolber, M.; Prehn, C.; Adamski, J.; Seissler, J.; Lechner, A. Serum and plasma amino acids as markers of prediabetes, insulin resistance, and incident diabetes. Crit. Rev. Clin. Lab. Sci. 2017, 55, 21–32. [Google Scholar] [CrossRef]
- Alghamdi, F.; Guo, M.; Abdulkhalek, S.; Crawford, N.; Amith, S.R.; Szewczuk, M.R. A novel insulin receptor-signaling platform and its link to insulin resistance and type 2 diabetes. Cell. Signal. 2014, 26, 1355–1368. [Google Scholar] [CrossRef] [Green Version]
- Liauchonak, I.; Qorri, B.; Dawoud, F.; Riat, Y.; Szewczuk, M.R. Non-Nutritive Sweeteners and Their Implications on the Development of Metabolic Syndrome. Nutrients 2019, 11, 644. [Google Scholar] [CrossRef] [Green Version]
- Liauchonak, I.; Dawoud, F.; Riat, Y.; Qorri, B.; Sambi, M.; Jain, J.; Kalaydina, R.-V.; Mendonza, N.; Bajwa, K.; Szewczuk, M.R. The Biased G-Protein-Coupled Receptor Agonism Bridges the Gap between the Insulin Receptor and the Metabolic Syndrome. Int. J. Mol. Sci. 2018, 19, 575. [Google Scholar] [CrossRef] [Green Version]
- Chaplin, R.; Thach, L.; Hollenberg, M.D.; Cao, Y.; Little, P.J.; Kamato, D. Insights into cellular signalling by G protein coupled receptor transactivation of cell surface protein kinase receptors. J. Cell Commun. Signal. 2017, 11, 117–125. [Google Scholar] [CrossRef] [Green Version]
- Dimitriadis, G.; Mitrou, P.; Lambadiari, V.; Maratou, E.; Raptis, S.A. Insulin effects in muscle and adipose tissue. Diabetes Res. Clin. Pr. 2011, 93, S52–S59. [Google Scholar] [CrossRef]
- Carnagarin, R.; Dharmarajan, A.M.; Dass, C.R. Molecular aspects of glucose homeostasis in skeletal muscle—A focus on the molecular mechanisms of insulin resistance. Mol. Cell. Endocrinol. 2015, 417, 52–62. [Google Scholar] [CrossRef]
- Boucher, J.; Kleinridders, A.; Kahn, C.R. Insulin Receptor Signaling in Normal and Insulin-Resistant States. Cold Spring Harb. Perspect. Biol. 2014, 6, a009191. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Long, Y.C.; Cheng, Z.; Copps, K.D.; White, M.F. Insulin Receptor Substrates Irs1 and Irs2 Coordinate Skeletal Muscle Growth and Metabolism via the Akt and AMPK Pathways. Mol. Cell. Biol. 2010, 31, 430–441. [Google Scholar] [CrossRef] [Green Version]
- Haeusler, R.A.; McGraw, T.E.; Accili, D. Biochemical and cellular properties of insulin receptor signalling. Nat. Rev. Mol. Cell Biol. 2018, 19, 31–44. [Google Scholar] [CrossRef]
- Leto, D.; Saltiel, A.R. Regulation of glucose transport by insulin: Traffic control of GLUT4. Nat. Rev. Mol. Cell Biol. 2012, 13, 383–396. [Google Scholar] [CrossRef]
- Taylor, R. Insulin Resistance and Type 2 Diabetes. Diabetes 2012, 61, 778–779. [Google Scholar] [CrossRef] [Green Version]
- Lu, J.; Xie, G.; Jia, W.; Jia, W. Insulin resistance and the metabolism of branched-chain amino acids. Front. Med. 2013, 7, 53–59. [Google Scholar] [CrossRef]
- Wu, H.; Ballantyne, C.M. Metabolic Inflammation and Insulin Resistance in Obesity. Circ. Res. 2020, 126, 1549–1564. [Google Scholar] [CrossRef]
- Wu, H.; Ballantyne, C.M. Skeletal muscle inflammation and insulin resistance in obesity Find the latest version: Skeletal muscle inflammation and insulin resistance in obesity. J. Clin. Investig. 2017, 127, 43–54. [Google Scholar] [CrossRef]
- Lauterbach, M.A.R.; Wunderlich, F.T. Macrophage function in obesity-induced inflammation and insulin resistance. Pflügers Archiv Eur. J. Physiol. 2017, 469, 385–396. [Google Scholar] [CrossRef] [Green Version]
- Asghar, A.; Sheikh, N. Role of immune cells in obesity induced low grade inflammation and insulin resistance. Cell. Immunol. 2017, 315, 18–26. [Google Scholar] [CrossRef]
- Battiprolu, P.K.; Gillette, T.G.; Wang, Z.; Lavandero, S.; Hill, J.A. Diabetic cardiomyopathy: Mechanisms and therapeutic targets. Drug Discov. Today Dis. Mech. 2010, 7, e135–e143. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, C.-H.; Zhou, B.-G.; Sheng, J.-Q.; Chen, Y.; Cao, Y.-Q.; Chen, C. Molecular mechanisms of hepatic insulin resistance in nonalcoholic fatty liver disease and potential treatment strategies. Pharmacol. Res. 2020, 159, 104984. [Google Scholar] [CrossRef]
- Sergi, D.; Naumovski, N.N.; Heilbronn, L.H.K.; Abeywardena, M.; O’Callaghan, N.; Lionetti, L.; Luscombe-Marsh, N.L.-M. Mitochondrial (Dys)function and Insulin Resistance: From Pathophysiological Molecular Mechanisms to the Impact of Diet. Front. Physiol. 2019, 10, 532. [Google Scholar] [CrossRef] [PubMed]
- Lynch, C.J.; Adams, S. Branched-chain amino acids in metabolic signalling and insulin resistance. Nat. Rev. Endocrinol. 2014, 10, 723–736. [Google Scholar] [CrossRef] [Green Version]
- Akhtar, A.; Sah, S.P. Insulin signaling pathway and related molecules: Role in neurodegeneration and Alzheimer’s disease. Neurochem. Int. 2020, 135, 104707. [Google Scholar] [CrossRef] [PubMed]
- Neis, E.P.J.G.; DeJong, C.H.C.; Rensen, S.S. The Role of Microbial Amino Acid Metabolism in Host Metabolism. Nutrients 2015, 7, 2930–2946. [Google Scholar] [CrossRef] [Green Version]
- Haufe, S.; Engeli, S.; Kaminski, J.; Witt, H.; Rein, D.; Kamlage, B.; Utz, W.; Fuhrmann, J.; Haas, V.; Mähler, A.; et al. Branched-chain amino acid catabolism rather than amino acids plasma concentrations is associated with diet-induced changes in insulin resistance in overweight to obese individuals. Nutr. Metab. Cardiovasc. Dis. 2017, 27, 858–864. [Google Scholar] [CrossRef] [PubMed]
- Liao, X.; Liu, B.; Qu, H.; Zhang, L.; Lu, Y.; Xu, Y.; Lyu, Z.; Zheng, H. A High Level of Circulating Valine Is a Biomarker for Type 2 Diabetes and Associated with the Hypoglycemic Effect of Sitagliptin. Mediat. Inflamm. 2019, 2019, 1–7. [Google Scholar] [CrossRef]
- Jeganathan, S.; Abdullahi, A.; Zargar, S.; Maeda, N.; Riddell, M.C.; Adegoke, O.A.J. Amino acid-induced impairment of insulin sensitivity in healthy and obese rats is reversible. Physiol. Rep. 2014, 2, e12067. [Google Scholar] [CrossRef]
- Chen, Z.; Franco, O.H.; Lamballais, S.; Ikram, M.A.; Schoufour, J.D.; Muka, T.; Voortman, T. Associations of specific dietary protein with longitudinal insulin resistance, prediabetes and type 2 diabetes: The Rotterdam Study. Clin. Nutr. 2020, 39, 242–249. [Google Scholar] [CrossRef]
- Lee, S.-G.; Yim, Y.S.; Lee, Y.-H.; Lee, B.-W.; Kim, H.-S.; Kim, K.-S.; Kim, J.-H. Fasting serum amino acids concentration is associated with insulin resistance and pro-inflammatory cytokines. Diabetes Res. Clin. Pr. 2018, 140, 107–117. [Google Scholar] [CrossRef]
- Hellmuth, C.; Kirchberg, F.F.; Lass, N.; Harder, U.; Peissner, W.; Koletzko, B.; Reinehr, T. Tyrosine Is Associated with Insulin Resistance in Longitudinal Metabolomic Profiling of Obese Children. J. Diabetes Res. 2016, 2016, 1–10. [Google Scholar] [CrossRef] [Green Version]
- Owei, I.; Umekwe, N.; Stentz, F.; Wan, J.; Dagogo-Jack, S. Amino acid signature predictive of incident prediabetes: A case-control study nested within the longitudinal pathobiology of prediabetes in a biracial cohort. Metabolism 2019, 98, 76–83. [Google Scholar] [CrossRef]
- Liu, L.; Feng, R.; Guo, F.; Li, Y.; Jiao, J.; Sun, C. Targeted metabolomic analysis reveals the association between the postprandial change in palmitic acid, branched-chain amino acids and insulin resistance in young obese subjects. Diabetes Res. Clin. Pr. 2015, 108, 84–93. [Google Scholar] [CrossRef]
- Thomas, C.C.; Philipson, L.H. Update on Diabetes Classification. Med. Clin. N. Am. 2015, 99, 1–16. [Google Scholar] [CrossRef]
- Haufe, S.; Witt, H.; Engeli, S.; Kaminski, J.; Utz, W.; Fuhrmann, J.; Rein, D.; Schulz-Menger, J.; Luft, F.; Boschmann, M.; et al. Branched-chain and aromatic amino acids, insulin resistance and liver specific ectopic fat storage in overweight to obese subjects. Nutr. Metab. Cardiovasc. Dis. 2016, 26, 637–642. [Google Scholar] [CrossRef]
- Floegel, A.; Stefan, N.; Yu, Z.; Mühlenbruch, K.; Drogan, D.; Joost, H.-G.; Fritsche, A.; Häring, H.-U.; De Angelis, M.H.; Peters, A.; et al. Identification of Serum Metabolites Associated With Risk of Type 2 Diabetes Using a Targeted Metabolomic Approach. Diabetes 2012, 62, 639–648. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jang, C.; Oh, S.F.; Wada, S.; Rowe, G.; Liu, L.; Chan, M.C.; Rhee, J.; Hoshino, A.; Kim, B.; Ibrahim, A.; et al. A branched-chain amino acid metabolite drives vascular fatty acid transport and causes insulin resistance. Nat. Med. 2016, 22, 421–426. [Google Scholar] [CrossRef] [Green Version]
- Yamakado, M.; Nagao, K.; Imaizumi, A.; Tani, M.; Toda, A.; Tanaka, T.; Jinzu, H.; Miyano, H.; Yamamoto, H.; Daimon, T.; et al. Plasma Free Amino Acid Profiles Predict Four-Year Risk of Developing Diabetes, Metabolic Syndrome, Dyslipidemia and Hypertension in Japanese Population. Sci. Rep. 2015, 5, 11918. [Google Scholar] [CrossRef] [PubMed]
- Xiao, F.; Yu, J.; Guo, Y.; Deng, J.; Li, K.; Du, Y.; Chen, S.; Zhu, J.; Sheng, H.; Guo, F. Effects of individual branched-chain amino acids deprivation on insulin sensitivity and glucose metabolism in mice. Metabolism 2014, 63, 841–850. [Google Scholar] [CrossRef]
- Newsholme, P.; Brennan, L.; Bender, K. Amino Acid Metabolism, -Cell Function, and Diabetes. Diabetes 2006, 55, S39–S47. [Google Scholar] [CrossRef] [Green Version]
- Schriever, S.C.; Deutsch, M.J.; Adamski, J.; Roscher, A.A.; Ensenauer, R. Cellular signaling of amino acids towards mTORC1 activation in impaired human leucine catabolism. J. Nutr. Biochem. 2013, 24, 824–831. [Google Scholar] [CrossRef]
- Manoli, I.; Venditti, C. Disorders of branched chain amino acid metabolism. Transl. Sci. Rare Dis. 2016, 1, 91–110. [Google Scholar] [CrossRef] [Green Version]
- She, P.; Reid, T.M.; Bronson, S.; Vary, T.C.; Hajnal, A.; Lynch, C.J.; Hutson, S.M. Disruption of BCATm in Mice Leads to Increased Energy Expenditure Associated with the Activation of a Futile Protein Turnover Cycle. Cell Metab. 2007, 6, 181–194. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pietiläinen, K.H.; Naukkarinen, J.; Rissanen, A.; Saharinen, J.; Ellonen, P.; Keränen, H.; Suomalainen-Wartiovaara, A.; Götz, A.; Suortti, T.; Yki-Jarvinen, H.; et al. Global Transcript Profiles of Fat in Monozygotic Twins Discordant for BMI: Pathways behind Acquired Obesity. PLoS Med. 2008, 5, e51. [Google Scholar] [CrossRef] [PubMed]
- Stančáková, A.; Civelek, M.; Saleem, N.K.; Soininen, P.; Kangas, A.J.; Cederberg, H.; Paananen, J.; Pihlajamäki, J.; Bonnycastle, L.L.; Morken, M.A.; et al. Hyperglycemia and a Common Variant of GCKR Are Associated with the Levels of Eight Amino Acids in 9,369 Finnish Men. Diabetes 2012, 61, 1895–1902. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lerin, C.; Goldfine, A.B.; Boes, T.; Liu, M.; Kasif, S.; Dreyfuss, J.M.; De Sousa-Coelho, A.L.; Daher, G.; Manoli, I.; Sysol, J.R.; et al. Defects in muscle branched-chain amino acid oxidation contribute to impaired lipid metabolism. Mol. Metab. 2016, 5, 926–936. [Google Scholar] [CrossRef]
- Lotta, L.A.; Scott, R.A.; Sharp, S.J.; Burgess, S.; Luan, J.; Tillin, T.; Schmidt, A.F.; Imamura, F.; Stewart, I.D.; Perry, J.R.B.; et al. Genetic Predisposition to an Impaired Metabolism of the Branched-Chain Amino Acids and Risk of Type 2 Diabetes: A Mendelian Randomisation Analysis. PLoS Med. 2016, 13, e1002179. [Google Scholar] [CrossRef]
- Yoon, M.-S. The Emerging Role of Branched-Chain Amino Acids in Insulin Resistance and Metabolism. Nutrients 2016, 8, 405. [Google Scholar] [CrossRef] [Green Version]
- Newgard, C.B. Interplay between Lipids and Branched-Chain Amino Acids in Development of Insulin Resistance. Cell Metab. 2012, 15, 606–614. [Google Scholar] [CrossRef] [Green Version]
- Gran, P.; Cameron-Smith, D. The actions of exogenous leucine on mTOR signalling and amino acid transporters in human myotubes. BMC Physiol. 2011, 11, 10. [Google Scholar] [CrossRef] [Green Version]
- Zheng, L.; Zhang, W.; Zhou, Y.; Li, F.; Wei, H.; Peng, J. Recent Advances in Understanding Amino Acid Sensing Mechanisms that Regulate mTORC1. Int. J. Mol. Sci. 2016, 17, 1636. [Google Scholar] [CrossRef] [Green Version]
- Pezze, P.D.; Ruf, S.; Sonntag, A.G.; Langelaar-Makkinje, M.; Hall, P.; Heberle, A.M.; Navas, P.R.; Van Eunen, K.; Tölle, R.C.; Schwarz, J.J.; et al. A systems study reveals concurrent activation of AMPK and mTOR by amino acids. Nat. Commun. 2016, 7, 13254. [Google Scholar] [CrossRef]
- Wolfson, R.L.; Chantranupong, L.; Saxton, R.A.; Shen, K.; Scaria, S.M.; Cantor, J.R.; Sabatini, D.M. Sestrin2 is a leucine sensor for the mTORC1 pathway. Science 2015, 351, 43–48. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Maida, A.; Chan, J.S.; Sjøberg, K.A.; Zota, A.; Schmoll, D.; Kiens, B.; Herzig, S.; Rose, A.J. Repletion of branched chain amino acids reverses mTORC1 signaling but not improved metabolism during dietary protein dilution. Mol. Metab. 2017, 6, 873–881. [Google Scholar] [CrossRef] [PubMed]
- Kido, K.; Sase, K.; Yokokawa, T.; Fujita, S. Enhanced skeletal muscle insulin sensitivity after acute resistance-type exercise is upregulated by rapamycin-sensitive mTOR complex 1 inhibition. Sci. Rep. 2020, 10, 1–12. [Google Scholar] [CrossRef]
- Hodson, N.; McGlory, C.; Oikawa, S.Y.; Jeromson, S.; Song, Z.; Rüegg, M.A.; Hamilton, D.L.; Phillips, S.; Philp, A. Differential localization and anabolic responsiveness of mTOR complexes in human skeletal muscle in response to feeding and exercise. Am. J. Physiol. Physiol. 2017, 313, C604–C611. [Google Scholar] [CrossRef] [PubMed]
- Bae, J.Y.; Shin, K.O.; Woo, J.; Woo, S.H.; Jang, K.S.; Lee, Y.H.; Kang, S. Exercise and dietary change ameliorate high fat diet induced obesity and insulin resistance via mTOR signaling pathway. J. Exerc. Nutr. Biochem. 2016, 20, 28–33. [Google Scholar] [CrossRef]
- Zhang, Y.; Ding, S.; Sun, Y. PO-063 Exercise alleviates insulin resistance by regulating MG53 and IR/IRS/AKT/mTOR signaling in db/db mice skeletal muscle. Exerc. Biochem. Rev. 2018, 1. [Google Scholar] [CrossRef]
- Huang, S.; Tang, N.; Zhao, H.; Tang, C. Effect of electrical stimulation combined with diet therapy on insulin resistance via mTOR signaling. Mol. Med. Rep. 2019, 20, 5152–5162. [Google Scholar] [CrossRef] [PubMed]
- Saad, M.J.; Santos, A.; Prada, P.O. Linking Gut Microbiota and Inflammation to Obesity and Insulin Resistance. Physiology 2016, 31, 283–293. [Google Scholar] [CrossRef]
- Heianza, Y.; Sun, D.; Li, X.; Di Donato, A.J.; Bray, G.A.; Sacks, F.M.; Qi, L. Gut microbiota metabolites, amino acid metabolites and improvements in insulin sensitivity and glucose metabolism: The POUNDS Lost trial. Gut 2019, 68, 263–270. [Google Scholar] [CrossRef]
- Bifari, F.; Ruocco, C.; Decimo, I.; Fumagalli, G.; Valerio, A.; Nisoli, E. Amino acid supplements and metabolic health: A potential interplay between intestinal microbiota and systems control. Genes Nutr. 2017, 12, 1–12. [Google Scholar] [CrossRef]
- Lin, R.; Liu, W.; Piao, M.; Zhu, H. A review of the relationship between the gut microbiota and amino acid metabolism. Amino Acids 2017, 49, 2083–2090. [Google Scholar] [CrossRef] [PubMed]
- Ley, R.E.; Turnbaugh, P.J.; Klein, S.; Gordon, J.I. Human gut microbes associated with obesity. Nature 2006, 444, 1022–1023. [Google Scholar] [CrossRef]
- Hullar, M.A.; Fu, B.C. Diet, the Gut Microbiome, and Epigenetics Meredith. Cancer J. 2015, 20, 170–175. [Google Scholar] [CrossRef] [Green Version]
- Collins, S.M.; Surette, M.G.; Bercik, P. The interplay between the intestinal microbiota and the brain. Nat. Rev. Genet. 2012, 10, 735–742. [Google Scholar] [CrossRef] [PubMed]
- Heianza, Y.; Sun, D.; Smith, S.R.; Bray, G.A.; Sacks, F.M.; Qi, L. Changes in Gut Microbiota–Related Metabolites and Long-term Successful Weight Loss in Response to Weight-Loss Diets: The POUNDS Lost Trial. Diabetes Care 2018, 41, 413–419. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pedersen, H.K.; Gudmundsdottir, V.; Nielsen, H.B.; Hyotylainen, T.; Nielsen, T.; Jensen, B.A.H.; Forslund, K.; Hildebrand, F.; Prifti, E.; Falony, G.; et al. Human gut microbes impact host serum metabolome and insulin sensitivity. Nat. Cell Biol. 2016, 535, 376–381. [Google Scholar] [CrossRef]
- Blaut, M. Gut microbiota and energy balance: Role in obesity. Proc. Nutr. Soc. 2015, 74, 227–234. [Google Scholar] [CrossRef] [Green Version]
- Sun, L.; Yu, Z.; Ye, X.; Zou, S.; Li, H.; Yu, D.; Wu, H.; Chen, Y.; Dore, J.; Clément, K.; et al. A Marker of Endotoxemia Is Associated with Obesity and Related Metabolic Disorders in Apparently Healthy Chinese. Diabetes Care 2010, 33, 1925–1932. [Google Scholar] [CrossRef] [Green Version]
- Yang, Z.; Huang, S.; Zou, D.; Dong, D.; He, X.; Liu, N.; Liu, W.; Huang, L. Metabolic shifts and structural changes in the gut microbiota upon branched-chain amino acid supplementation in middle-aged mice. Amino Acids 2016, 48, 2731–2745. [Google Scholar] [CrossRef]
- D’Antona, G.; Ragni, M.; Cardile, A.; Tedesco, L.; Dossena, M.; Bruttini, F.; Caliaro, F.; Corsetti, G.; Bottinelli, R.; Carruba, M.O.; et al. Branched-Chain Amino Acid Supplementation Promotes Survival and Supports Cardiac and Skeletal Muscle Mitochondrial Biogenesis in Middle-Aged Mice. Cell Metab. 2010, 12, 362–372. [Google Scholar] [CrossRef] [Green Version]
- Harris, L.-A.L.; Smith, G.; Patterson, B.W.; Ramaswamy, R.S.; Okunade, A.L.; Kelly, S.C.; Porter, L.C.; Klein, S.; Yoshino, J.; Mittendorfer, B. Alterations in 3-Hydroxyisobutyrate and FGF21 Metabolism Are Associated With Protein Ingestion–Induced Insulin Resistance. Diabetes 2017, 66, 1871–1878. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mardinoglu, A.; Gogg, S.; Lotta, L.A.; Stančáková, A.; Nerstedt, A.; Boren, J.; Blüher, M.; Ferrannini, E.; Langenberg, C.; Wareham, N.J.; et al. Elevated Plasma Levels of 3-Hydroxyisobutyric Acid Are Associated With Incident Type 2 Diabetes. EBioMedicine 2018, 27, 151–155. [Google Scholar] [CrossRef] [Green Version]
- Sánchez-Pintos, P.; De Castro, M.-J.; Roca, I.; Rite, S.; López, M.; Couce, M.-L. Similarities between acylcarnitine profiles in large for gestational age newborns and obesity. Sci. Rep. 2017, 7, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Aguer, C.; McCoin, C.; Knotts, T.A.; Thrush, A.B.; Ono-Moore, K.; McPherson, R.; Dent, R.; Hwang, D.H.; Adams, S.H.; Harper, M. Acylcarnitines: Potential implications for skeletal muscle insulin resistance. FASEB J. 2015, 29, 336–345. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kirchberg, F.F.; Harder, U.; Weber, M.; Grote, V.; Demmelmair, H.; Peissner, W.; Rzehak, P.; Xhonneux, A.; Carlier, C.; Ferré, N.; et al. Dietary Protein Intake Affects Amino Acid and Acylcarnitine Metabolism in Infants Aged 6 Months. J. Clin. Endocrinol. Metab. 2015, 100, 149–158. [Google Scholar] [CrossRef] [Green Version]
- Bergman, H.-M.; Lindfors, L.; Palm, F.; Kihlberg, J.; Lanekoff, I. Metabolite aberrations in early diabetes detected in rat kidney using mass spectrometry imaging. Anal. Bioanal. Chem. 2019, 411, 2809–2816. [Google Scholar] [CrossRef] [Green Version]
- Guasch-Ferré, M.; Ruiz-Canela, M.; Li, J.; Zheng, Y.; Bullo, M.; Wang, D.D.; Toledo, E.; Clish, C.; Corella, D.; Estruch, R.; et al. Plasma Acylcarnitines and Risk of Type 2 Diabetes in a Mediterranean Population at High Cardiovascular Risk. J. Clin. Endocrinol. Metab. 2019, 104, 1508–1519. [Google Scholar] [CrossRef]
- Weiser, A.; Giesbertz, P.; Daniel, H.; Spanier, B. Acylcarnitine Profiles in Plasma and Tissues of Hyperglycemic NZO Mice Correlate with Metabolite Changes of Human Diabetes. J. Diabetes Res. 2018, 2018, 1–9. [Google Scholar] [CrossRef] [PubMed]
- He, F.; Wu, C.; Li, P.; Li, N.; Zhang, D.; Zhu, Q.; Ren, W.; Peng, Y. Functions and Signaling Pathways of Amino Acids in Intestinal Inflammation. BioMed Res. Int. 2018, 2018, 1–13. [Google Scholar] [CrossRef]
- Reddy, P.; Leong, J.; Jialal, I. Amino acid levels in nascent metabolic syndrome: A contributor to the pro-inflammatory burden. J. Diabetes Its Complicat. 2018, 32, 465–469. [Google Scholar] [CrossRef]
- Zhang, F.; Zhao, S.; Yan, W.; Xia, Y.; Chen, X.; Wang, W.; Zhang, J.; Gao, C.; Peng, C.; Yan, F.; et al. Branched Chain Amino Acids Cause Liver Injury in Obese/Diabetic Mice by Promoting Adipocyte Lipolysis and Inhibiting Hepatic Autophagy. EBioMedicine 2016, 13, 157–167. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mu, W.-C.; Vanhoosier, E.; Elks, C.M.; Grant, R.W. Long-Term Effects of Dietary Protein and Branched-Chain Amino Acids on Metabolism and Inflammation in Mice. Nutrients 2018, 10, 918. [Google Scholar] [CrossRef] [Green Version]
- Chen, S.; Akter, S.; Kuwahara, K.; Matsushita, Y.; Nakagawa, T.; Konishi, M.; Honda, T.; Yamamoto, S.; Hayashi, T.; Noda, M.; et al. Serum amino acid profiles and risk of type 2 diabetes among Japanese adults in the Hitachi Health Study. Sci. Rep. 2019, 9, 1–9. [Google Scholar] [CrossRef] [Green Version]
- Korner, J.; Cline, G.W.; Slifstein, M.; Barba, P.; Rayat, G.R.; Febres, G.; Leibel, R.L.; Maffei, A.; Harris, P.E. A role for foregut tyrosine metabolism in glucose tolerance. Mol. Metab. 2019, 23, 37–50. [Google Scholar] [CrossRef] [PubMed]
- Dickinson, J.M.; Rasmussen, B.B. Amino Acid Transporters in the Regulation of Human Skeletal Muscle Protein Metabolism Jared. Curr. Opin. Clin. Nutr. Metab. Care 2014, 16, 638–644. [Google Scholar] [CrossRef] [Green Version]
- Liu, R.; Li, H.; Fan, W.; Jin, Q.; Chao, T.; Wu, Y.; Huang, J.; Hao, L.; Yang, X. Leucine Supplementation Differently Modulates Branched-Chain Amino Acid Catabolism, Mitochondrial Function and Metabolic Profiles at the Different Stage of Insulin Resistance in Rats on High-Fat Diet. Nutrients 2017, 9, 565. [Google Scholar] [CrossRef] [Green Version]
- Rohini, A.; Agrawal, N.; Kumar, H.; Kumar, V. Emerging role of branched chain amino acids in metabolic disorders: A mechanistic review. Pharma Nutr. 2018, 6, 47–54. [Google Scholar] [CrossRef]
- Bekhbat, M.; Treadway, M.T.; Goldsmith, D.R.; Woolwine, B.J.; Haroon, E.; Miller, A.H.; Felger, J.C. Gene signatures in peripheral blood immune cells related to insulin resistance and low tyrosine metabolism define a sub-type of depression with high CRP and anhedonia. Brain Behav. Immun. 2020, 88, 161–165. [Google Scholar] [CrossRef]
- Zhang, S.; Li, X.; Luo, H.; Fang, Z.-Z.; Ai, H. Role of aromatic amino acids in pathogeneses of diabetic nephropathy in Chinese patients with type 2 diabetes. J. Diabetes Its Complicat. 2020, 34, 107667. [Google Scholar] [CrossRef]
- Gong, X.; Yang, C.; Hong, Y.; Chung, A.C.; Cai, Z. PFOA and PFOS promote diabetic renal injury in vitro by impairing the metabolisms of amino acids and purines. Sci. Total Environ. 2019, 676, 72–86. [Google Scholar] [CrossRef]
- Liu, Y.; Chen, X.; Liu, Y.; Chen, T.; Zhang, Q.; Zhang, H.; Zhu, Z.; Chai, Y.; Zhang, J. Metabolomic study of the protective effect of Gandi capsule for diabetic nephropathy. Chem. Interact. 2019, 314, 108815. [Google Scholar] [CrossRef] [PubMed]
- Kormanovski, A.; Lara-Padilla, E.; Gutiérrez-Camacho, L.R.; Castillo-Hernández, M.D.C.; Guevara-Balcázar, G. Alterations in glutathione, nitric oxide and 3-nitrotyrosine levels following exercise and/or hyperbaric oxygen treatment in mice with diet-induced diabetes. Biomed. Rep. 2020, 12, 222–232. [Google Scholar] [CrossRef]
- Bala, C.; Rusu, A.; Ciobanu, D.M.; Craciun, A.E.; Roman, G. The association study of high-sensitivity C-reactive protein, pentraxin 3, nitrotyrosine, and insulin dose in patients with insulin-treated type 2 diabetes mellitus. Ther. Clin. Risk Manag. 2018, 14, 955–963. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, J.; Cao, Y.-F.; Sun, X.-Y.; Han, L.; Li, S.-N.; Gu, W.-Q.; Song, M.; Jiang, C.; Yang, X.; Fang, Z.-Z. Plasma tyrosine and its interaction with low high-density lipoprotein cholesterol and the risk of type 2 diabetes mellitus in Chinese. J. Diabetes Investig. 2019, 10, 491–498. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Galkina, S.I.; Fedorova, N.V.; Ksenofontov, A.L.; Stadnichuk, V.I.; Baratova, L.A.; Sud’Ina, G.F. Neutrophils as a source of branched-chain, aromatic and positively charged free amino acids. Cell Adhes. Migr. 2019, 13, 98–105. [Google Scholar] [CrossRef] [Green Version]
- Vangipurapu, J.; Stancáková, A.; Smith, U.; Kuusisto, J.; Laakso, M. Nine Amino Acids Are Associated With Decreased Insulin Secretion and Elevated Glucose Levels in a 7.4-Year Follow-up Study of 5181 Finnish Men. Diabetes 2019, 68, 1353–1358. [Google Scholar] [CrossRef]
- Oxenkrug, G. Insulin Resistance and Dysregulation of Tryptophan–Kynurenine and Kynurenine–Nicotinamide Adenine Dinucleotide Metabolic Pathways. Mol. Neurobiol. 2013, 48, 294–301. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sas, K.; Szabó, E.; Vécsei, L. Mitochondria, Oxidative Stress and the Kynurenine System, with a Focus on Ageing and Neuroprotection. Molecules 2018, 23, 191. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Komatsu, H.; Fukuchi, M.; Habata, Y. Potential Utility of Biased GPCR Signaling for Treatment of Psychiatric Disorders. Int. J. Mol. Sci. 2019, 20, 3207. [Google Scholar] [CrossRef] [Green Version]
- Sebastiani, G.; Ceccarelli, E.; Castagna, M.G.; Dotta, F. G-protein-coupled receptors (GPCRs) in the treatment of diabetes: Current view and future perspectives. Best Pr. Res. Clin. Endocrinol. Metab. 2018, 32, 201–213. [Google Scholar] [CrossRef]
- Wauson, E.M.; Lorente-Rodríguez, A.; Cobb, M.H. Minireview: Nutrient Sensing by G Protein-Coupled Receptors. Mol. Endocrinol. 2013, 27, 1188–1197. [Google Scholar] [CrossRef] [Green Version]
- Husted, A.S.; Trauelsen, M.; Rudenko, O.; Hjorth, S.A.; Schwartz, T.W. GPCR-Mediated Signaling of Metabolites. Cell Metab. 2017, 25, 777–796. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Law, N.C.; White, M.F.; Hunzicker-Dunn, M.E. G protein-coupled receptors (GPCRs) That Signal via Protein Kinase A (PKA) Cross-talk at Insulin Receptor Substrate 1 (IRS1) to Activate the phosphatidylinositol 3-kinase (PI3K)/AKT Pathway. J. Biol. Chem. 2016, 291, 27160–27169. [Google Scholar] [CrossRef] [Green Version]
- Haxho, F.; Haq, S.; Szewczuk, M.R. Biased G protein-coupled receptor agonism mediates Neu1 sialidase and matrix metalloproteinase-9 crosstalk to induce transactivation of insulin receptor signaling. Cell. Signal. 2018, 43, 71–84. [Google Scholar] [CrossRef] [PubMed]
- Fu, Q.; Shi, Q.; West, T.M.; Xiang, Y.K. Cross-talk between insulin signaling and GPCRs. Physiol. Behav. 2018, 176, 139–148. [Google Scholar] [CrossRef]
- Chun, L.; Zhang, W.-H.; Liu, J.-F. Structure and ligand recognition of class C GPCRs. Acta Pharmacol. Sin. 2012, 33, 312–323. [Google Scholar] [CrossRef] [Green Version]
- Murakoshi, M.; Kuwabara, H.; Nagasaki, M.; Xiong, Y.M.; Reagan, J.D.; Maeda, H.; Nara, F. Discovery and pharmacological effects of a novel GPR142 antagonist. J. Recept. Signal Transduct. 2016, 37, 290–296. [Google Scholar] [CrossRef]
- Wang, J.; Carrillo, J.J.; Lin, H.V. GPR142 Agonists Stimulate Glucose-Dependent Insulin Secretion via Gq-Dependent Signaling. PLoS ONE 2016, 11, e0154452. [Google Scholar] [CrossRef]
- Riddy, D.; Delerive, P.; Summers, R.; Sexton, P.; Langmead, C.J. G Protein–Coupled Receptors Targeting Insulin Resistance, Obesity, and Type 2 Diabetes Mellitus. Pharmacol. Rev. 2017, 70, 39–67. [Google Scholar] [CrossRef] [Green Version]
- Scow, J.S.; Tavakkolizadeh, A.; Zheng, Y.; Sarr, M.G. Acute “adaptation” by the small intestinal enterocyte: A posttranscriptional mechanism involving apical translocation of nutrient transporters. Surgery 2011, 149, 601–605. [Google Scholar] [CrossRef] [Green Version]
- Sierra, H.; Cordova, M.; Chen, C.-S.J. Rajadhyaksha, Milind Confocal Imaging–Guided Laser Ablation of Basal Cell Carcinomas: An Ex Vivo Study. J. Investig. Dermatol. 2015, 135, 612–615. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Foster, S.; Porrello, E.R.; Purdue, B.; Chan, H.-W.; Voigt, A.; Frenzel, S.; Hannan, R.D.; Moritz, K.M.; Simmons, D.G.; Molenaar, P.; et al. Expression, Regulation and Putative Nutrient-Sensing Function of Taste GPCRs in the Heart. PLoS ONE 2013, 8, e64579. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Takai, S.; Watanabe, Y.; Sanematsu, K.; Yoshida, R.; Margolskee, R.F.; Jiang, P.; Atsuta, I.; Koyano, K.; Ninomiya, Y.; Shigemura, N. Effects of insulin signaling on mouse taste cell proliferation. PLoS ONE 2019, 14, e0225190. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Young, R.L.; Sutherland, K.; Pezos, N.; Brierley, S.M.; Horowitz, M.; Rayner, C.K.; Blackshaw, L.A. Expression of taste molecules in the upper gastrointestinal tract in humans with and without type 2 diabetes. Gut 2008, 5, 1–21. [Google Scholar] [CrossRef]
- Wauson, E.M.; Zaganjor, E.; Lee, A.-Y.; Guerra, M.L.; Ghosh, A.B.; Bookout, A.L.; Chambers, C.P.; Jivan, A.; McGlynn, K.; Hutchison, M.R.; et al. The G Protein-Coupled Taste Receptor T1R1/T1R3 Regulates mTORC1 and Autophagy. Mol. Cell 2012, 47, 851–862. [Google Scholar] [CrossRef] [Green Version]
- Yarova, P.L.; Stewart, A.L.; Sathish, V.; Britt, R.D., Jr.; Thompson, M.A.; Lowe, A.; Freeman, M.; Aravamudan, B.; Kita, H.; Brennan, S.; et al. Calcium-sensing receptor antagonists abrogate airway hyperresponsiveness and inflammation in allergic asthma. Sci. Transl. Med. 2015, 7, 284ra60. [Google Scholar] [CrossRef] [Green Version]
- Conigrave, A.D.; Franks, A.H.; Brown, E.M.; Quinn, S.J. L-Amino acid sensing by the calcium-sensing receptor: A general mechanism for coupling protein and calcium metabolism? Eur. J. Clin. Nutr. 2002, 56, 1072–1080. [Google Scholar] [CrossRef]
- Jones, P.M.; Kitsou-Mylona, I.; Gray, E.; Squires, P.E.; Persaud, S.J. Expression and function of the extracellular calcium-sensing receptor in pancreatic β-cells. Arch. Physiol. Biochem. 2007, 113, 98–103. [Google Scholar] [CrossRef] [PubMed]
- Hu, B.; Tong, F.; Xu, L.; Shen, Z.; Yan, L.; Xu, G.; Shen, R. Role of Calcium Sensing Receptor in Streptozotocin-Induced Diabetic Rats Exposed to Renal Ischemia Reperfusion Injury. Kidney Blood Press. Res. 2018, 43, 276–286. [Google Scholar] [CrossRef]
- Xie, J.; Jiang, Y.; Kan, Y.; Zhao, J.; Kuang, H.; Ge, P. Calcium-sensing receptor is involved in the pathogenesis of fat emulsion-induced insulin resistance in rats. Mol. Med. Rep. 2015, 12, 2043–2048. [Google Scholar] [CrossRef] [Green Version]
- Abdulla, H.; Bass, J.J.; Stokes, T.; Gorissen, S.H.M.; McGlory, C.; Phillips, B.E.; Phillips, S.M.; Smith, K.; Idris, I.; Atherton, P.J. The effect of oral essential amino acids on incretin hormone production in youth and ageing. Endocrinol. Diabetes Metab. 2019, 2, e00085. [Google Scholar] [CrossRef] [Green Version]
- Rigamonti, A.E.; Leoncini, R.; De Col, A.; Tamini, S.; Cicolini, S.; Abbruzzese, L.; Cella, S.G.; Sartorio, A. The Appetite−Suppressant and GLP-1-Stimulating Effects of Whey Proteins in Obese Subjects are Associated with Increased Circulating Levels of Specific Amino Acids. Nutrients 2020, 12, 775. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gojda, J.; Straková, R.; Plíhalová, A.; Tuma, P.; Potočková, J.; Polák, J.; Anděl, M. Increased Incretin But Not Insulin Response after Oral versus Intravenous Branched Chain Amino Acids. Ann. Nutr. Metab. 2017, 70, 293–302. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Liu, H.; Chen, J.; Li, Y.; Qu, S. Multiple Factors Related to the Secretion of Glucagon-Like Peptide-1. Int. J. Endocrinol. 2015, 2015, 1–11. [Google Scholar] [CrossRef] [Green Version]
- Modvig, I.M.; Kuhre, R.E.; Jepsen, S.L.; Xu, S.F.S.; Engelstoft, M.S.; Egerod, K.L.; Schwartz, T.W.; Ørskov, C.; Rosenkilde, M.M.; Holst, J.J. Amino acids differ in their capacity to stimulate GLP-1 release from the perfused rat small intestine and stimulate secretion by different sensing mechanisms. Am. J. Physiol. Metab. 2021, 320, E874–E885. [Google Scholar] [CrossRef]

Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zakaria, N.F.; Hamid, M.; Khayat, M.E. Amino Acid-Induced Impairment of Insulin Signaling and Involvement of G-Protein Coupling Receptor. Nutrients 2021, 13, 2229. https://doi.org/10.3390/nu13072229
Zakaria NF, Hamid M, Khayat ME. Amino Acid-Induced Impairment of Insulin Signaling and Involvement of G-Protein Coupling Receptor. Nutrients. 2021; 13(7):2229. https://doi.org/10.3390/nu13072229
Chicago/Turabian StyleZakaria, Nur Fatini, Muhajir Hamid, and Mohd Ezuan Khayat. 2021. "Amino Acid-Induced Impairment of Insulin Signaling and Involvement of G-Protein Coupling Receptor" Nutrients 13, no. 7: 2229. https://doi.org/10.3390/nu13072229
APA StyleZakaria, N. F., Hamid, M., & Khayat, M. E. (2021). Amino Acid-Induced Impairment of Insulin Signaling and Involvement of G-Protein Coupling Receptor. Nutrients, 13(7), 2229. https://doi.org/10.3390/nu13072229