
Nutritional, Gastrointestinal and Endo-Metabolic Challenges in the Management of Children with Spinal Muscular Atrophy Type 1

 ,
, 
 , ,
, ,  ,
, 
 ,
,  and
and 
Abstract
:1. Introduction
2. Methods
3. Nutritional Aspects: Critical Issues and Possible Solutions
3.1. Undernutrition and Overnutrition: A Common Problem
3.2. Ventilated Patients and Non-Ventilated Patients: What Are the Differences?
3.3. Predictive Energy and Fat Mass Equations
3.4. Enteral Nutrition
3.4.1. Nasogastric and Nasojejunal Tube vs. Percutaneous Endoscopic Gastrostomy (PEG) and Percutaneous Endoscopic Transgastric Jejunostomy (PEG-j)
3.4.2. Administration Schedules
3.4.3. Enteral Formulas
4. Gastrointestinal Aspects and Clinical Challenges
4.1. Symptoms, Mechanisms and Pathophysiology
4.1.1. Upper GI
4.1.2. Lower GI
5. Metabolic and Endocrine Aspects
5.1. Dysregulation of Lipid Metabolism
5.2. Dysregulation of Glucose Metabolism
5.3. Liver Disorders
5.4. Vitamin Deficiencies and Bone Health
5.5. Endocrine Disorders
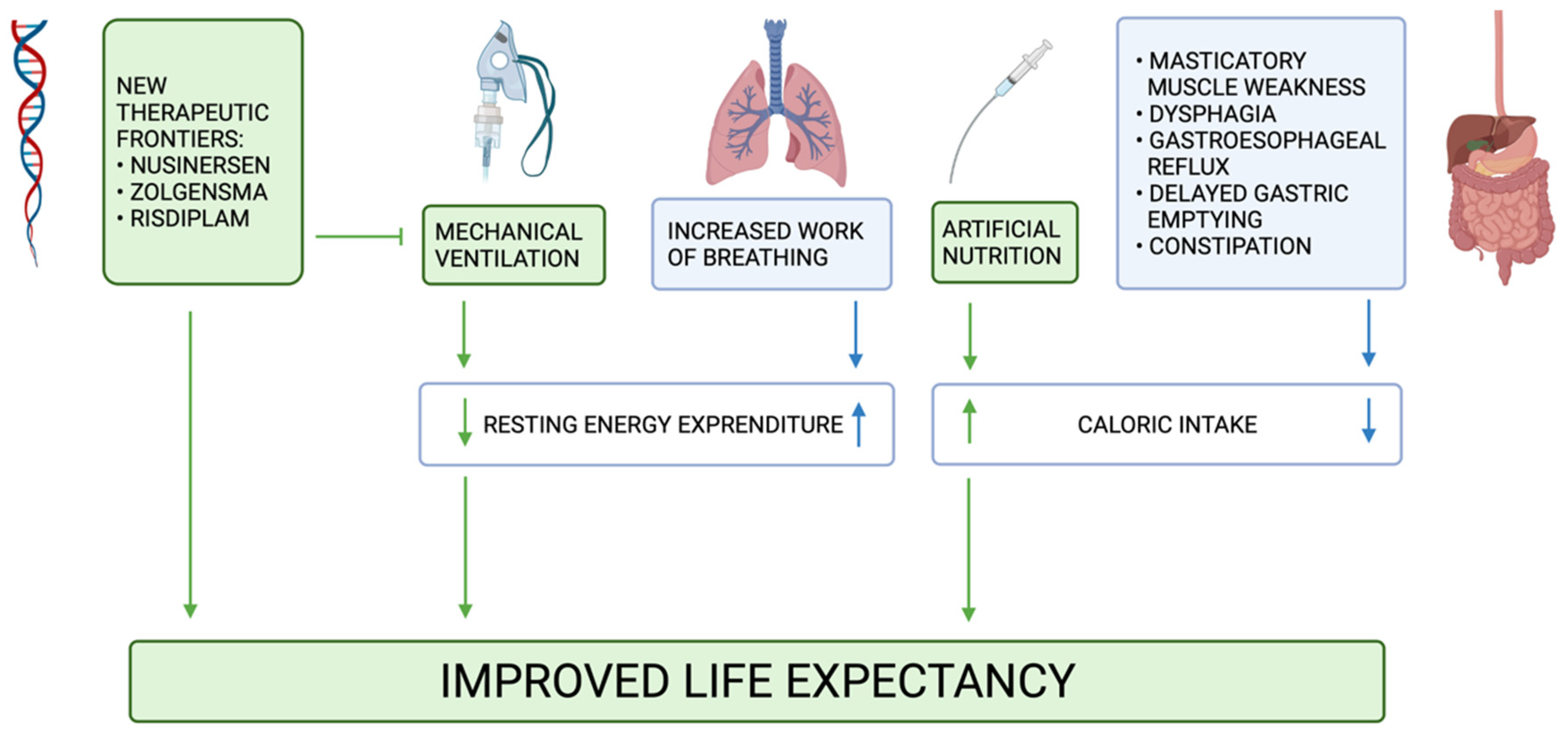
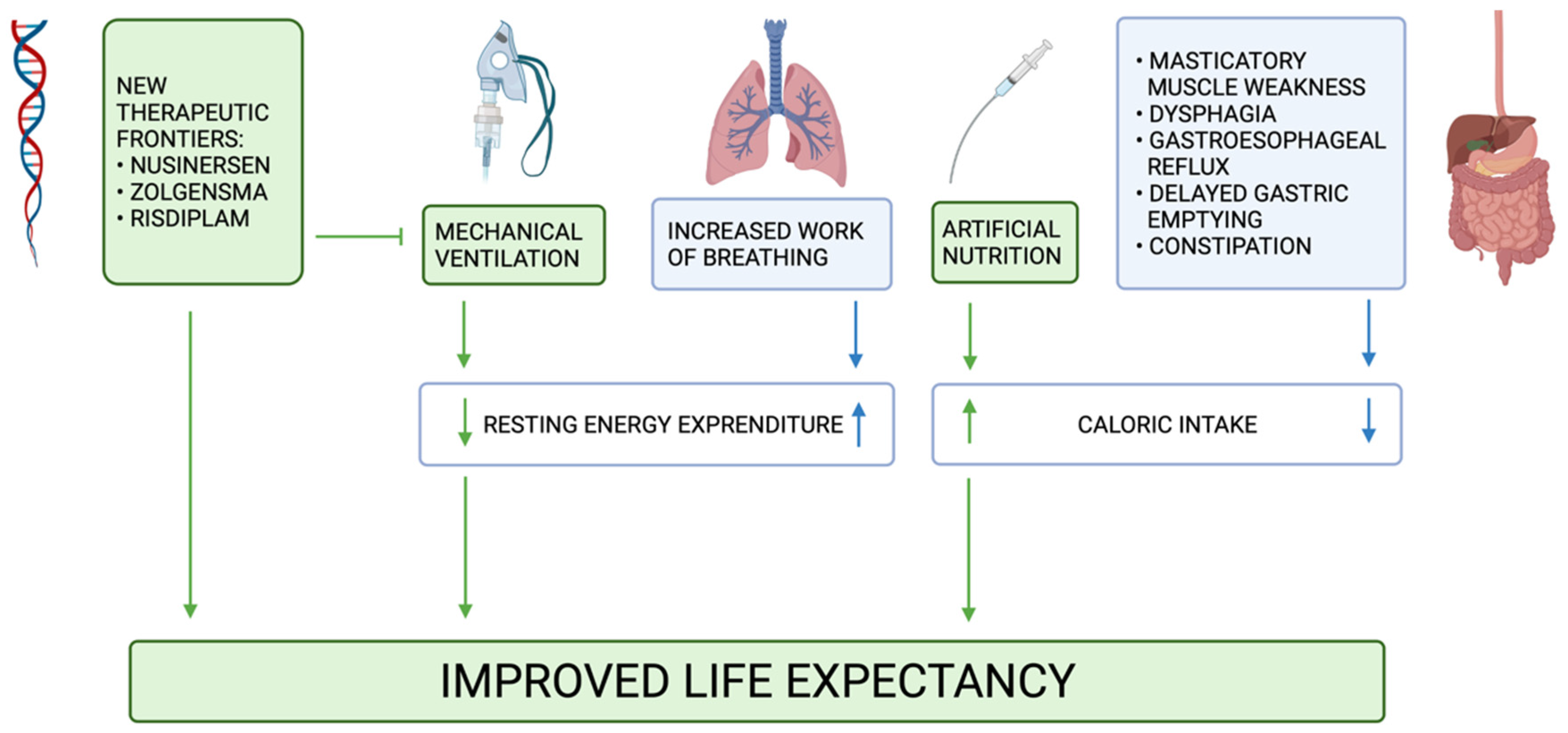
6. New Therapeutic Frontiers
7. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Yeo, C.J.J.; Darras, B.T. Overturning the Paradigm of Spinal Muscular Atrophy as Just a Motor Neuron Disease. Pediatr. Neurol. 2020, 109, 12–19. [Google Scholar] [CrossRef]
- Mercuri, E.; Finkel, R.S.; Muntoni, F.; Wirth, B.; Montes, J.; Main, M.; Mazzone, E.S.; Vitale, M.; Snyder, B.; Quijano-Roy, S.; et al. Diagnosis and management of spinal muscular atrophy: Part 1: Recommendations for diagnosis, rehabilitation, orthopedic and nutritional care. Neuromuscul. Disord. 2018, 28, 103–115. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, X.; Sanchis-Juan, A.; French, C.E.; Connell, A.J.; Delon, I.; Kingsbury, Z.; Chawla, A.; Halpern, A.L.; Taft, R.J.; Bentley, D.R.; et al. Spinal muscular atrophy diagnosis and carrier screening from genome sequencing data. Genet. Med. 2020, 22, 945–953. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lunn, M.R.; Wang, C.H. Spinal muscular atrophy. Lancet 2008, 371, 2120–2133. [Google Scholar] [CrossRef]
- Pera, M.C.; Coratti, G.; Berti, B.; D’Amico, A.; Sframeli, M.; Albamonte, E.; de Sanctis, R.; Messina, S.; Catteruccia, M.; Brigati, G.; et al. Diagnostic journey in Spinal Muscular Atrophy: Is it still an odyssey? PLoS ONE 2020, 15, e0230677. [Google Scholar] [CrossRef]
- Bach, J.R.; Baird, J.S.; Plosky, D.; Navado, J.; Weaver, B. Spinal muscular atrophy type 1: Management and outcomes. Pediatr. Pulmonol. 2002, 34, 16–22. [Google Scholar] [CrossRef]
- Gregoretti, C.; Ottonello, G.; Chiarini Testa, M.B.; Mastella, C.; Ravà, L.; Bignamini, E.; Veljkovic, A.; Cutrera, R. Survival of patients with spinal muscular atrophy type 1. Pediatrics 2013, 131, e1509–e1514. [Google Scholar] [CrossRef] [Green Version]
- Park, H.B.; Lee, S.M.; Lee, J.S.; Park, M.S.; Park, K.I.; Namgung, R.; Lee, C. Survival analysis of spinal muscular atrophy type I. Korean J. Pediatr. 2010, 53, 965–970. [Google Scholar] [CrossRef]
- Ioos, C.; Leclair-Richard, D.; Mrad, S.; Barois, A.; Estournet-Mathiaud, B. Respiratory capacity course in patients with infantile spinal muscular atrophy. Chest 2004, 126, 831–837. [Google Scholar] [CrossRef]
- Gidaro, T.; Servais, L. Nusinersen treatment of spinal muscular atrophy: Current knowledge and existing gaps. Dev. Med. Child. Neurol. 2019, 61, 19–24. [Google Scholar] [CrossRef] [Green Version]
- Wadman, R.I.; Bosboom, W.M.; van den Berg, L.H.; Wokke, J.H.; Iannaccone, S.T.; Vrancken, A.F. Drug treatment for spinal muscular atrophy type I. Cochrane Database Syst. Rev. 2011, CD006281. [Google Scholar] [CrossRef] [Green Version]
- Neil, E.E.; Bisaccia, E.K. Nusinersen: A Novel Antisense Oligonucleotide for the Treatment of Spinal Muscular Atrophy. J. Pediatr. Pharmacol. Ther. 2019, 24, 194–203. [Google Scholar] [CrossRef]
- Kirschner, J.; Butoianu, N.; Goemans, N.; Haberlova, J.; Kostera-Pruszczyk, A.; Mercuri, E.; van der Pol, W.L.; Quijano-Roy, S.; Sejersen, T.; Tizzano, E.F.; et al. European ad-hoc consensus statement on gene replacement therapy for spinal muscular atrophy. Eur. J. Paediatr. Neurol. 2020, 28, 38–43. [Google Scholar] [CrossRef] [PubMed]
- Mendell, J.R.; Al-Zaidy, S.A.; Lehman, K.J.; McColly, M.; Lowes, L.P.; Alfano, L.N.; Reash, N.F.; Iammarino, M.A.; Church, K.R.; Kleyn, A.; et al. Five-Year Extension Results of the Phase 1 START Trial of Onasemnogene Abeparvovec in Spinal Muscular Atrophy. JAMA Neurol. 2021. [Google Scholar] [CrossRef] [PubMed]
- Lowes, L.P.; Alfano, L.N.; Arnold, W.D.; Shell, R.; Prior, T.W.; McColly, M.; Lehman, K.J.; Church, K.; Sproule, D.M.; Nagendran, S.; et al. Impact of Age and Motor Function in a Phase 1/2A Study of Infants with SMA Type 1 Receiving Single-Dose Gene Replacement Therapy. Pediatr. Neurol. 2019, 98, 39–45. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mendell, J.R.; Lehman, K.J.; McColly, M.; Lowes, L.P.; Alfano, L.N.; Miller, N.F.; Iammarino, M.A.; Church, K.; Ogrinc, F.G.; L’Italien, J.; et al. AVXS-101 Gene-Replacement Therapy (GRT) in Spinal Muscular Atrophy Type 1 (SMA1): Long-Term Follow-Up From the Phase 1 Clinical Trial (S25.006). Neurology 2019, 92, S25.006. [Google Scholar] [CrossRef] [Green Version]
- Baranello, G.; Darras, B.T.; Day, J.W.; Deconinck, N.; Klein, A.; Masson, R.; Mercuri, E.; Rose, K.; El-Khairi, M.; Gerber, M.; et al. Risdiplam in Type 1 Spinal Muscular Atrophy. N. Engl. J. Med. 2021, 384, 915–923. [Google Scholar] [CrossRef]
- Messina, S.; Sframeli, M. New Treatments in Spinal Muscular Atrophy: Positive Results and New Challenges. J. Clin. Med. 2020, 9, 2222. [Google Scholar] [CrossRef]
- Pierzchlewicz, K.; Kępa, I.; Podogrodzki, J.; Kotulska, K. Spinal Muscular Atrophy: The Use of Functional Motor Scales in the Era of Disease-Modifying Treatment. Child. Neurol. Open 2021, 8, 2329048X211008725. [Google Scholar] [CrossRef]
- Singh, R.N.; Ottesen, E.W.; Singh, N.N. The First Orally Deliverable Small Molecule for the Treatment of Spinal Muscular Atrophy. Neurosci. Insights 2020, 15, 2633105520973985. [Google Scholar] [CrossRef]
- Jędrzejowska, M. Advances in Newborn Screening and Presymptomatic Diagnosis of Spinal Muscular Atrophy. Degener. Neurol. Neuromuscul. Dis. 2020, 10, 39–47. [Google Scholar] [CrossRef]
- Kariyawasam, D.S.T.; Russell, J.S.; Wiley, V.; Alexander, I.E.; Farrar, M.A. The implementation of newborn screening for spinal muscular atrophy: The Australian experience. Genet. Med. 2020, 22, 557–565. [Google Scholar] [CrossRef]
- Vill, K.; Schwartz, O.; Blaschek, A.; Gläser, D.; Nennstiel, U.; Wirth, B.; Burggraf, S.; Röschinger, W.; Becker, M.; Czibere, L.; et al. Newborn screening for spinal muscular atrophy in Germany: Clinical results after 2 years. Orphanet J. Rare Dis. 2021, 16, 153. [Google Scholar] [CrossRef]
- De Vivo, D.C.; Bertini, E.; Swoboda, K.J.; Hwu, W.-L.; Crawford, T.O.; Finkel, R.S.; Kirschner, J.; Kuntz, N.L.; Parsons, J.A.; Ryan, M.M.; et al. Nusinersen initiated in infants during the presymptomatic stage of spinal muscular atrophy: Interim efficacy and safety results from the Phase 2 NURTURE study. Neuromuscul. Disord. 2019, 29, 842–856. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bertoli, S.; De Amicis, R.; Bedogni, G.; Foppiani, A.; Leone, A.; Ravella, S.; Mastella, C.; Baranello, G.; Masson, R.; Bertini, E.; et al. Predictive energy equations for spinal muscular atrophy type I children. Am. J. Clin. Nutr. 2020, 111, 983–996. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.-J.; Chen, T.-H.; Wu, Y.-Z.; Tseng, Y.-H. Metabolic and Nutritional Issues Associated with Spinal Muscular Atrophy. Nutrients 2020, 12, 3842. [Google Scholar] [CrossRef]
- Poruk, K.E.; Davis, R.H.; Smart, A.L.; Chisum, B.S.; Lasalle, B.A.; Chan, G.M.; Gill, G.; Reyna, S.P.; Swoboda, K.J. Observational study of caloric and nutrient intake, bone density, and body composition in infants and children with spinal muscular atrophy type I. Neuromuscul. Disord. 2012, 22, 966–973. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Klanjsek, P.; Pajnkihar, M.; Marcun Varda, N.; Povalej Brzan, P. Screening and assessment tools for early detection of malnutrition in hospitalised children: A systematic review of validation studies. BMJ Open 2019, 9. [Google Scholar] [CrossRef] [PubMed]
- Baranello, G.; De Amicis, R.; Arnoldi, M.T.; Zanin, R.; Mastella, C.; Masson, R.; Leone, A.; Alberti, K.; Foppiani, A.; Battezzati, A.; et al. Evaluation of body composition as a potential biomarker in spinal muscular atrophy. Muscle Nerve 2020, 61, 530–534. [Google Scholar] [CrossRef]
- Sproule, D.M.; Montes, J.; Dunaway, S.; Montgomery, M.; Battista, V.; Koenigsberger, D.; Martens, B.; Shen, W.; Punyanitya, M.; Benton, M.; et al. Adiposity is increased among high-functioning, non-ambulatory patients with spinal muscular atrophy. Neuromuscul. Disord. 2010, 20, 448–452. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Moore, G.E.; Lindenmayer, A.W.; McConchie, G.A.; Ryan, M.M.; Davidson, Z.E. Describing nutrition in spinal muscular atrophy: A systematic review. Neuromuscul. Disord. 2016, 26, 395–404. [Google Scholar] [CrossRef]
- Bach, J.R. The use of mechanical ventilation is appropriate in children with genetically proven spinal muscular atrophy type 1: The motion for. Paediatr. Respir. Rev. 2008, 9, 45–46. [Google Scholar] [CrossRef] [PubMed]
- Bach, J.R. POINT: Is Noninvasive Ventilation Always the Most Appropriate Manner of Long-term Ventilation for Infants With Spinal Muscular Atrophy Type 1? Yes, Almost Always. Chest 2017, 151, 962–965. [Google Scholar] [CrossRef] [Green Version]
- Bach, J.R. Noninvasive Respiratory Management of Patients with Neuromuscular Disease. Ann. Rehabil. Med. 2017, 41, 519–538. [Google Scholar] [CrossRef] [Green Version]
- Tobin, M.J.; Laghi, F.; Jubran, A. Narrative review: Ventilator-induced respiratory muscle weakness. Ann. Intern. Med. 2010, 153, 240–245. [Google Scholar] [CrossRef]
- LoMauro, A.; Aliverti, A.; Mastella, C.; Arnoldi, M.T.; Banfi, P.; Baranello, G. Spontaneous Breathing Pattern as Respiratory Functional Outcome in Children with Spinal Muscular Atrophy (SMA). PLoS ONE 2016, 11, e0165818. [Google Scholar] [CrossRef] [PubMed]
- Delsoglio, M.; Achamrah, N.; Berger, M.M.; Pichard, C. Indirect Calorimetry in Clinical Practice. J. Clin. Med. 2019, 8, 1387. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Savarino, G.; Corsello, A.; Corsello, G. Macronutrient balance and micronutrient amounts through growth and development. Ital. J. Pediatr. 2021, 47, 109. [Google Scholar] [CrossRef]
- Krick, J.; Murphy, P.E.; Markham, J.F.; Shapiro, B.K. A proposed formula for calculating energy needs of children with cerebral palsy. Dev. Med. Child. Neurol. 1992, 34, 481–487. [Google Scholar] [CrossRef]
- Foppiani, A.; De Amicis, R.; Leone, A.; Ravella, S.; Bedogni, G.; Battezzati, A.; D’Amico, A.; Bertini, E.; Pedemonte, M.; Bruno, C.; et al. Predictive fat mass equations for spinal muscular atrophy type I children: Development and internal validation. Clin. Nutr. 2021, 40, 1578–1587. [Google Scholar] [CrossRef]
- Finkel, R.S.; Mercuri, E.; Meyer, O.H.; Simonds, A.K.; Schroth, M.K.; Graham, R.J.; Kirschner, J.; Iannaccone, S.T.; Crawford, T.O.; Woods, S.; et al. Diagnosis and management of spinal muscular atrophy: Part 2: Pulmonary and acute care; medications, supplements and immunizations; other organ systems; and ethics. Neuromuscul. Disord. 2018, 28, 197–207. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ørngreen, M.C.; Zacho, M.; Hebert, A.; Laub, M.; Vissing, J. Patients with severe muscle wasting are prone to develop hypoglycemia during fasting. Neurology 2003, 61, 997–1000. [Google Scholar] [CrossRef] [PubMed]
- Mehta, N.M.; Newman, H.; Tarrant, S.; Graham, R.J. Nutritional Status and Nutrient Intake Challenges in Children With Spinal Muscular Atrophy. Pediatr. Neurol. 2016, 57, 80–83. [Google Scholar] [CrossRef] [PubMed]
- Wadman, R.I.; van Bruggen, H.W.; Witkamp, T.D.; Sparreboom-Kalaykova, S.I.; Stam, M.; van den Berg, L.H.; Steenks, M.H.; van der Pol, W.L. Bulbar muscle MRI changes in patients with SMA with reduced mouth opening and dysphagia. Neurology 2014, 83, 1060–1066. [Google Scholar] [CrossRef]
- Wales, P.W.; Diamond, I.R.; Dutta, S.; Muraca, S.; Chait, P.; Connolly, B.; Langer, J.C. Fundoplication and gastrostomy versus image-guided gastrojejunal tube for enteral feeding in neurologically impaired children with gastroesophageal reflux. J. Pediatr. Surg. 2002, 37, 407–412. [Google Scholar] [CrossRef]
- Durkin, E.T.; Schroth, M.K.; Helin, M.; Shaaban, A.F. Early laparoscopic fundoplication and gastrostomy in infants with spinal muscular atrophy type I. J. Pediatr. Surg. 2008, 43, 2031–2037. [Google Scholar] [CrossRef] [PubMed]
- Birnkrant, D.J.; Pope, J.F.; Martin, J.E.; Repucci, A.H.; Eiben, R.M. Treatment of type I spinal muscular atrophy with noninvasive ventilation and gastrostomy feeding. Pediatr. Neurol. 1998, 18, 407–410. [Google Scholar] [CrossRef]
- Oskoui, M.; Levy, G.; Garland, C.J.; Gray, J.M.; O’Hagen, J.; De Vivo, D.C.; Kaufmann, P. The changing natural history of spinal muscular atrophy type 1. Neurology 2007, 69, 1931–1936. [Google Scholar] [CrossRef]
- Yerushalmy-Feler, A.; Levy, D.; Sagi, L.; Fattal-Valevski, A.; Shiff, Y.E.; Brener, A.; Cohen, S. Nutritional Therapy in Children with Spinal Muscular Atrophy in the Era of Nusinersen. J. Pediatr. Gastroenterol. Nutr. 2021, 72, e154–e160. [Google Scholar] [CrossRef]
- Davis, R.H.; Godshall, B.J.; Seffrood, E.; Marcus, M.; LaSalle, B.A.; Wong, B.; Schroth, M.K.; Swoboda, K.J. Nutritional practices at a glance: Spinal muscular atrophy type I nutrition survey findings. J. Child. Neurol. 2014, 29, 1467–1472. [Google Scholar] [CrossRef] [Green Version]
- Bach, J.R. Medical considerations of long-term survival of Werdnig-Hoffmann disease. Am. J. Phys. Med. Rehabil. 2007, 86, 349–355. [Google Scholar] [CrossRef] [PubMed]
- Dipasquale, V.; Serra, G.; Corsello, G.; Romano, C. Standard and Specialized Infant Formulas in Europe: Making, Marketing, and Health Outcomes. Nutr. Clin. Pr. 2020, 35, 273–281. [Google Scholar] [CrossRef]
- Dipasquale, V.; Catena, M.A.; Cardile, S.; Romano, C. Standard Polymeric Formula Tube Feeding in Neurologically Impaired Children: A Five-Year Retrospective Study. Nutrients 2018, 10, 684. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ibrahim, H.; Mansour, M.; El Gendy, Y.G. Peptide-based formula versus standard-based polymeric formula for critically ill children: Is it superior for patients’ tolerance? Arch. Med. Sci. 2020, 16, 592–596. [Google Scholar] [CrossRef] [PubMed]
- Chatwin, M.; Bush, A.; Macrae, D.J.; Clarke, S.A.; Simonds, A.K. Risk management protocol for gastrostomy and jejunostomy insertion in ventilator dependent infants. Neuromuscul. Disord. 2013, 23, 289–297. [Google Scholar] [CrossRef]
- Sy, K.; Mahant, S.; Taback, N.; Vajsar, J.; Chait, P.G.; Friedman, J.N. Enterostomy tube placement in children with spinal muscular atrophy type 1. J. Pediatr. 2006, 149, 837–839. [Google Scholar] [CrossRef]
- Yuan, N.; Wang, C.H.; Trela, A.; Albanese, C.T. Laparoscopic Nissen fundoplication during gastrostomy tube placement and noninvasive ventilation may improve survival in type I and severe type II spinal muscular atrophy. J. Child. Neurol. 2007, 22, 727–731. [Google Scholar] [CrossRef]
- Corsello, A.; Pugliese, D.; Gasbarrini, A.; Armuzzi, A. Diet and Nutrients in Gastrointestinal Chronic Diseases. Nutrients 2020, 12, 2693. [Google Scholar] [CrossRef]
- Arnold, W.D.; Kassar, D.; Kissel, J.T. Spinal muscular atrophy: Diagnosis and management in a new therapeutic era. Muscle Nerve 2015, 51, 157–167. [Google Scholar] [CrossRef]
- Gombash, S.E.; Cowley, C.J.; Fitzgerald, J.A.; Iyer, C.C.; Fried, D.; McGovern, V.L.; Williams, K.C.; Burghes, A.H.M.; Christofi, F.L.; Gulbransen, B.D.; et al. SMN deficiency disrupts gastrointestinal and enteric nervous system function in mice. Hum. Mol. Genet. 2015, 24, 3847–3860. [Google Scholar] [CrossRef] [Green Version]
- Hsieh-Li, H.M.; Chang, J.G.; Jong, Y.J.; Wu, M.H.; Wang, N.M.; Tsai, C.H.; Li, H. A mouse model for spinal muscular atrophy. Nat. Genet. 2000, 24, 66–70. [Google Scholar] [CrossRef]
- Sintusek, P.; Catapano, F.; Angkathunkayul, N.; Marrosu, E.; Parson, S.H.; Morgan, J.E.; Muntoni, F.; Zhou, H. Histopathological Defects in Intestine in Severe Spinal Muscular Atrophy Mice Are Improved by Systemic Antisense Oligonucleotide Treatment. PLoS ONE 2016, 11, e0155032. [Google Scholar] [CrossRef] [PubMed]
- Wan, B.; Feng, P.; Guan, Z.; Sheng, L.; Liu, Z.; Hua, Y. A severe mouse model of spinal muscular atrophy develops early systemic inflammation. Hum. Mol. Genet. 2018, 27, 4061–4076. [Google Scholar] [CrossRef] [PubMed]
- Shababi, M.; Lorson, C.L.; Rudnik-Schöneborn, S.S. Spinal muscular atrophy: A motor neuron disorder or a multi-organ disease? J. Anat. 2014, 224, 15–28. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wijngaarde, C.A.; Blank, A.C.; Stam, M.; Wadman, R.I.; van den Berg, L.H.; van der Pol, W.L. Cardiac pathology in spinal muscular atrophy: A systematic review. Orphanet J. Rare Dis. 2017, 12, 67. [Google Scholar] [CrossRef]
- Florie, M.G.M.H.; Pilz, W.; Dijkman, R.H.; Kremer, B.; Wiersma, A.; Winkens, B.; Baijens, L.W.J. The Effect of Cranial Nerve Stimulation on Swallowing: A Systematic Review. Dysphagia 2021, 36, 216–230. [Google Scholar] [CrossRef]
- van den Engel-Hoek, L.; de Groot, I.J.M.; de Swart, B.J.M.; Erasmus, C.E. Feeding and Swallowing Disorders in Pediatric Neuromuscular Diseases: An Overview. J. Neuromuscul. Dis. 2015, 2, 357–369. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Audag, N.; Goubau, C.; Toussaint, M.; Reychler, G. Screening and evaluation tools of dysphagia in adults with neuromuscular diseases: A systematic review. Ther. Adv. Chronic Dis. 2019, 10, 2040622318821622. [Google Scholar] [CrossRef] [PubMed]
- Wada, A.; Kawakami, M.; Liu, M.; Otaka, E.; Nishimura, A.; Liu, F.; Otsuka, T. Development of a new scale for dysphagia in patients with progressive neuromuscular diseases: The Neuromuscular Disease Swallowing Status Scale (NdSSS). J. Neurol. 2015, 262, 2225–2231. [Google Scholar] [CrossRef]
- Tilton, A.H.; Miller, M.D.; Khoshoo, V. Nutrition and swallowing in pediatric neuromuscular patients. Semin. Pediatr. Neurol. 1998, 5, 106–115. [Google Scholar] [CrossRef]
- Marques, V.D.; Barreira, A.A.; Davis, M.B.; Abou-Sleiman, P.M.; Silva, W.A.J.; Zago, M.A.; Sobreira, C.; Fazan, V.; Marques, W.J. Expanding the phenotypes of the Pro56Ser VAPB mutation: Proximal SMA with dysautonomia. Muscle Nerve 2006, 34, 731–739. [Google Scholar] [CrossRef]
- Romano, C.; van Wynckel, M.; Hulst, J.; Broekaert, I.; Bronsky, J.; Dall’Oglio, L.; Mis, N.F.; Hojsak, I.; Orel, R.; Papadopoulou, A.; et al. European Society for Paediatric Gastroenterology, Hepatology and Nutrition Guidelines for the Evaluation and Treatment of Gastrointestinal and Nutritional Complications in Children With Neurological Impairment. J. Pediatr. Gastroenterol. Nutr. 2017, 65, 242–264. [Google Scholar] [CrossRef] [Green Version]
- Krishna, S.G.; Bryant, J.F.; Tobias, J.D. Management of the Difficult Airway in the Pediatric Patient. J. Pediatr. Intensive Care 2018, 7, 115–125. [Google Scholar] [CrossRef]
- Choi, Y.-A.; Suh, D.I.; Chae, J.-H.; Shin, H.-I. Trajectory of change in the swallowing status in spinal muscular atrophy type I. Int. J. Pediatr. Otorhinolaryngol. 2020, 130, 109818. [Google Scholar] [CrossRef]
- Messina, S. New Directions for SMA Therapy. J. Clin. Med. 2018, 7, 251. [Google Scholar] [CrossRef] [Green Version]
- Schorling, D.C.; Pechmann, A.; Kirschner, J. Advances in Treatment of Spinal Muscular Atrophy—New Phenotypes, New Challenges, New Implications for Care. J. Neuromuscul. Dis. 2020, 7, 1–13. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Day, J.W.; Finkel, R.S.; Chiriboga, C.A.; Connolly, A.M.; Crawford, T.O.; Darras, B.T.; Iannaccone, S.T.; Kuntz, N.L.; Peña, L.D.M.; Shieh, P.B.; et al. Onasemnogene abeparvovec gene therapy for symptomatic infantile-onset spinal muscular atrophy in patients with two copies of SMN2 (STR1VE): An open-label, single-arm, multicentre, phase 3 trial. Lancet. Neurol. 2021, 20, 284–293. [Google Scholar] [CrossRef]
- Wang, C.H.; Finkel, R.S.; Bertini, E.S.; Schroth, M.; Simonds, A.; Wong, B.; Aloysius, A.; Morrison, L.; Main, M.; Crawford, T.O.; et al. Consensus statement for standard of care in spinal muscular atrophy. J. Child. Neurol. 2007, 22, 1027–1049. [Google Scholar] [CrossRef] [PubMed]
- Yang, Y.; Vassilakos, G.; Hammers, D.W.; Yang, Z.; Barton, E.R.; Sweeney, H.L. Smooth muscle atrophy and colon pathology in SMN deficient mice. Am. J. Transl. Res. 2019, 11, 1789–1799. [Google Scholar] [PubMed]
- Deguise, M.-O.; Baranello, G.; Mastella, C.; Beauvais, A.; Michaud, J.; Leone, A.; De Amicis, R.; Battezzati, A.; Dunham, C.; Selby, K.; et al. Abnormal fatty acid metabolism is a core component of spinal muscular atrophy. Ann. Clin. Transl. Neurol. 2019, 6, 1519–1532. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Watson, K.S.; Boukhloufi, I.; Bowerman, M.; Parson, S.H. The Relationship between Body Composition, Fatty Acid Metabolism and Diet in Spinal Muscular Atrophy. Brain Sci. 2021, 11, 131. [Google Scholar] [CrossRef]
- Tein, I.; Sloane, A.E.; Donner, E.J.; Lehotay, D.C.; Millington, D.S.; Kelley, R.I. Fatty acid oxidation abnormalities in childhood-onset spinal muscular atrophy: Primary or secondary defect(s)? Pediatr. Neurol. 1995, 12, 21–30. [Google Scholar] [CrossRef]
- Tein, I. Disorders of fatty acid oxidation. Handb. Clin. Neurol. 2013, 113, 1675–1688. [Google Scholar] [CrossRef] [PubMed]
- Crawford, T.O.; Sladky, J.T.; Hurko, O.; Besner-Johnston, A.; Kelley, R.I. Abnormal fatty acid metabolism in childhood spinal muscular atrophy. Ann. Neurol. 1999, 45, 337–343. [Google Scholar] [CrossRef]
- Harpey, J.P.; Charpentier, C.; Paturneau-Jouas, M.; Renault, F.; Romero, N.; Fardeau, M. Secondary metabolic defects in spinal muscular atrophy type II. Lancet 1990, 336, 629–630. [Google Scholar] [CrossRef]
- Berti, B.; Onesimo, R.; Leone, D.; Palermo, C.; Giorgio, V.; Buonsenso, D.; Pane, M.; Mercuri, E. Hypoglycaemia in patients with type 1 SMA: An underdiagnosed problem? Arch. Dis. Child. 2020, 105, 707. [Google Scholar] [CrossRef] [PubMed]
- Djordjevic, S.A.; Milic-Rasic, V.; Brankovic, V.; Kosac, A.; Dejanovic-Djordjevic, I.; Markovic-Denic, L.; Djuricic, G.; Milcanovic, N.; Kovacevic, S.; Petrovic, H.; et al. Glucose and lipid metabolism disorders in children and adolescents with spinal muscular atrophy types 2 and 3. Neuromuscul. Disord. 2021, 31, 291–299. [Google Scholar] [CrossRef] [PubMed]
- Bowerman, M.; Michalski, J.-P.; Beauvais, A.; Murray, L.M.; DeRepentigny, Y.; Kothary, R. Defects in pancreatic development and glucose metabolism in SMN-depleted mice independent of canonical spinal muscular atrophy neuromuscular pathology. Hum. Mol. Genet. 2014, 23, 3432–3444. [Google Scholar] [CrossRef] [Green Version]
- Bowerman, M.; Swoboda, K.J.; Michalski, J.-P.; Wang, G.-S.; Reeks, C.; Beauvais, A.; Murphy, K.; Woulfe, J.; Screaton, R.A.; Scott, F.W.; et al. Glucose metabolism and pancreatic defects in spinal muscular atrophy. Ann. Neurol. 2012, 72, 256–268. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Di Cola, G.; Cool, M.H.; Accili, D. Hypoglycemic effect of insulin-like growth factor-1 in mice lacking insulin receptors. J. Clin. Investig. 1997, 99, 2538–2544. [Google Scholar] [CrossRef] [Green Version]
- Sun, C.; Zhou, J. Trichostatin A improves insulin stimulated glucose utilization and insulin signaling transduction through the repression of HDAC2. Biochem. Pharmacol. 2008, 76, 120–127. [Google Scholar] [CrossRef]
- Brener, A.; Lebenthal, Y.; Shtamler, A.; Levy, S.; Stein, R.; Fattal-Valevski, A.; Sagi, L. The endocrine manifestations of spinal muscular atrophy, a real-life observational study. Neuromuscul. Disord. 2020, 30, 270–276. [Google Scholar] [CrossRef] [PubMed]
- Hong, S.-H.; Choi, K.M. Sarcopenic Obesity, Insulin Resistance, and Their Implications in Cardiovascular and Metabolic Consequences. Int. J. Mol. Sci. 2020, 21, 494. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brener, A.; Sagi, L.; Shtamler, A.; Levy, S.; Fattal-Valevski, A.; Lebenthal, Y. Insulin-like growth factor-1 status is associated with insulin resistance in young patients with spinal muscular atrophy. Neuromuscul. Disord. 2020, 30, 888–896. [Google Scholar] [CrossRef]
- Feldman, A.G.; Parsons, J.A.; Dutmer, C.M.; Veerapandiyan, A.; Hafberg, E.; Maloney, N.; Mack, C.L. Subacute Liver Failure Following Gene Replacement Therapy for Spinal Muscular Atrophy Type 1. J. Pediatr. 2020, 225, 252–258.e1. [Google Scholar] [CrossRef]
- Friesen, W.J.; Massenet, S.; Paushkin, S.; Wyce, A.; Dreyfuss, G. SMN, the product of the spinal muscular atrophy gene, binds preferentially to dimethylarginine-containing protein targets. Mol. Cell 2001, 7, 1111–1117. [Google Scholar] [CrossRef]
- Aton, J.; Davis, R.H.; Jordan, K.C.; Scott, C.B.; Swoboda, K.J. Vitamin D intake is inadequate in spinal muscular atrophy type I cohort: Correlations with bone health. J. Child. Neurol. 2014, 29, 374–380. [Google Scholar] [CrossRef] [Green Version]
- Nair, R.; Maseeh, A. Vitamin D: The “sunshine” vitamin. J. Pharmacol. Pharmacother. 2012, 3, 118–126. [Google Scholar] [CrossRef]
- Tai, V.; Leung, W.; Grey, A.; Reid, I.R.; Bolland, M.J. Calcium intake and bone mineral density: Systematic review and meta-analysis. BMJ 2015, 351, h4183. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wasserman, H.M.; Hornung, L.N.; Stenger, P.J.; Rutter, M.M.; Wong, B.L.; Rybalsky, I.; Khoury, J.C.; Kalkwarf, H.J. Low bone mineral density and fractures are highly prevalent in pediatric patients with spinal muscular atrophy regardless of disease severity. Neuromuscul. Disord. 2017, 27, 331–337. [Google Scholar] [CrossRef]
- Felderhoff-Mueser, U.; Grohmann, K.; Harder, A.; Stadelmann, C.; Zerres, K.; Bührer, C.; Obladen, M. Severe spinal muscular atrophy variant associated with congenital bone fractures. J. Child. Neurol. 2002, 17, 718–721. [Google Scholar] [CrossRef]
- Vai, S.; Bianchi, M.L.; Moroni, I.; Mastella, C.; Broggi, F.; Morandi, L.; Arnoldi, M.T.; Bussolino, C.; Baranello, G. Bone and Spinal Muscular Atrophy. Bone 2015, 79, 116–120. [Google Scholar] [CrossRef] [PubMed]
- Shanmugarajan, S.; Tsuruga, E.; Swoboda, K.J.; Maria, B.L.; Ries, W.L.; Reddy, S. V Bone loss in survival motor neuron (Smn(-/-) SMN2) genetic mouse model of spinal muscular atrophy. J. Pathol. 2009, 219, 52–60. [Google Scholar] [CrossRef] [Green Version]
- Shanmugarajan, S.; Swoboda, K.J.; Iannaccone, S.T.; Ries, W.L.; Maria, B.L.; Reddy, S. V Congenital bone fractures in spinal muscular atrophy: Functional role for SMN protein in bone remodeling. J. Child. Neurol. 2007, 22, 967–973. [Google Scholar] [CrossRef] [PubMed]
- Hensel, N.; Brickwedde, H.; Tsaknakis, K.; Grages, A.; Braunschweig, L.; Lüders, K.A.; Lorenz, H.M.; Lippross, S.; Walter, L.M.; Tavassol, F.; et al. Altered bone development with impaired cartilage formation precedes neuromuscular symptoms in spinal muscular atrophy. Hum. Mol. Genet. 2020, 29, 2662–2673. [Google Scholar] [CrossRef]
- Cebeci, A.N.; Taş, A. Higher body fat and lower fat-free mass in girls with premature adrenarche. J. Clin. Res. Pediatr. Endocrinol. 2015, 7, 45–48. [Google Scholar] [CrossRef]
- Ibáñez, L.; Díaz, R.; López-Bermejo, A.; Marcos, M.V. Clinical spectrum of premature pubarche: Links to metabolic syndrome and ovarian hyperandrogenism. Rev. Endocr. Metab. Disord. 2009, 10, 63–76. [Google Scholar] [CrossRef]
- Dorn, L.D.; Hostinar, C.E.; Susman, E.J.; Pervanidou, P. Conceptualizing Puberty as a Window of Opportunity for Impacting Health and Well-Being Across the Life Span. J. Res. Adolesc. 2019, 29, 155–176. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sunyach, C.; Michaud, M.; Arnoux, T.; Bernard-Marissal, N.; Aebischer, J.; Latyszenok, V.; Gouarné, C.; Raoul, C.; Pruss, R.M.; Bordet, T.; et al. Olesoxime delays muscle denervation, astrogliosis, microglial activation and motoneuron death in an ALS mouse model. Neuropharmacology 2012, 62, 2346–2352. [Google Scholar] [CrossRef]
- Bertini, E.; Dessaud, E.; Mercuri, E.; Muntoni, F.; Kirschner, J.; Reid, C.; Lusakowska, A.; Comi, G.P.; Cuisset, J.-M.; Abitbol, J.-L.; et al. Safety and efficacy of olesoxime in patients with type 2 or non-ambulatory type 3 spinal muscular atrophy: A randomised, double-blind, placebo-controlled phase 2 trial. Lancet. Neurol. 2017, 16, 513–522. [Google Scholar] [CrossRef]
- Kim, H.-Y. Neuroprotection by docosahexaenoic acid in brain injury. Mil. Med. 2014, 179, 106–111. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gasior, M.; Rogawski, M.A.; Hartman, A.L. Neuroprotective and disease-modifying effects of the ketogenic diet. Behav. Pharmacol. 2006, 17, 431–439. [Google Scholar] [CrossRef] [Green Version]
- Hwee, D.T.; Kennedy, A.R.; Hartman, J.J.; Ryans, J.; Durham, N.; Malik, F.I.; Jasper, J.R. The small-molecule fast skeletal troponin activator, CK-2127107, improves exercise tolerance in a rat model of heart failure. J. Pharmacol. Exp. Ther. 2015, 353, 159–168. [Google Scholar] [CrossRef] [Green Version]
- Bowerman, M.; Becker, C.G.; Yáñez-Muñoz, R.J.; Ning, K.; Wood, M.J.A.; Gillingwater, T.; Talbot, K.; The UK SMA Research Consortium. Therapeutic strategies for spinal muscular atrophy: SMN and beyond. Dis. Model. Mech. 2017, 10, 943–954. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mercuri, E.; Pera, M.C.; Scoto, M.; Finkel, R.; Muntoni, F. Spinal muscular atrophy—Insights and challenges in the treatment era. Nat. Rev. Neurol. 2020, 16, 706–715. [Google Scholar] [CrossRef] [PubMed]


{kind=link}
{kind=link}
| Gastroenterological management |
|
| Nutritional management |
|
| Metabolic and endocrine management |
|
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Corsello, A.; Scatigno, L.; Pascuzzi, M.C.; Calcaterra, V.; Dilillo, D.; Vizzuso, S.; Pelizzo, G.; Zoia, E.; Mandelli, A.; Govoni, A.; et al. Nutritional, Gastrointestinal and Endo-Metabolic Challenges in the Management of Children with Spinal Muscular Atrophy Type 1. Nutrients 2021, 13, 2400. https://doi.org/10.3390/nu13072400
Corsello A, Scatigno L, Pascuzzi MC, Calcaterra V, Dilillo D, Vizzuso S, Pelizzo G, Zoia E, Mandelli A, Govoni A, et al. Nutritional, Gastrointestinal and Endo-Metabolic Challenges in the Management of Children with Spinal Muscular Atrophy Type 1. Nutrients. 2021; 13(7):2400. https://doi.org/10.3390/nu13072400
Chicago/Turabian StyleCorsello, Antonio, Lorenzo Scatigno, Martina Chiara Pascuzzi, Valeria Calcaterra, Dario Dilillo, Sara Vizzuso, Gloria Pelizzo, Elena Zoia, Anna Mandelli, Annalisa Govoni, and et al. 2021. "Nutritional, Gastrointestinal and Endo-Metabolic Challenges in the Management of Children with Spinal Muscular Atrophy Type 1" Nutrients 13, no. 7: 2400. https://doi.org/10.3390/nu13072400
APA StyleCorsello, A., Scatigno, L., Pascuzzi, M. C., Calcaterra, V., Dilillo, D., Vizzuso, S., Pelizzo, G., Zoia, E., Mandelli, A., Govoni, A., Bosetti, A., Francavilla, R., Indrio, F., Fabiano, V., Zuccotti, G. V., & Verduci, E. (2021). Nutritional, Gastrointestinal and Endo-Metabolic Challenges in the Management of Children with Spinal Muscular Atrophy Type 1. Nutrients, 13(7), 2400. https://doi.org/10.3390/nu13072400