
Faecal Microbiota Composition Varies between Patients with Breast Cancer and Healthy Women: A Comparative Case-Control Study

 , and
, and
Abstract
:1. Introduction
2. Materials and Methods
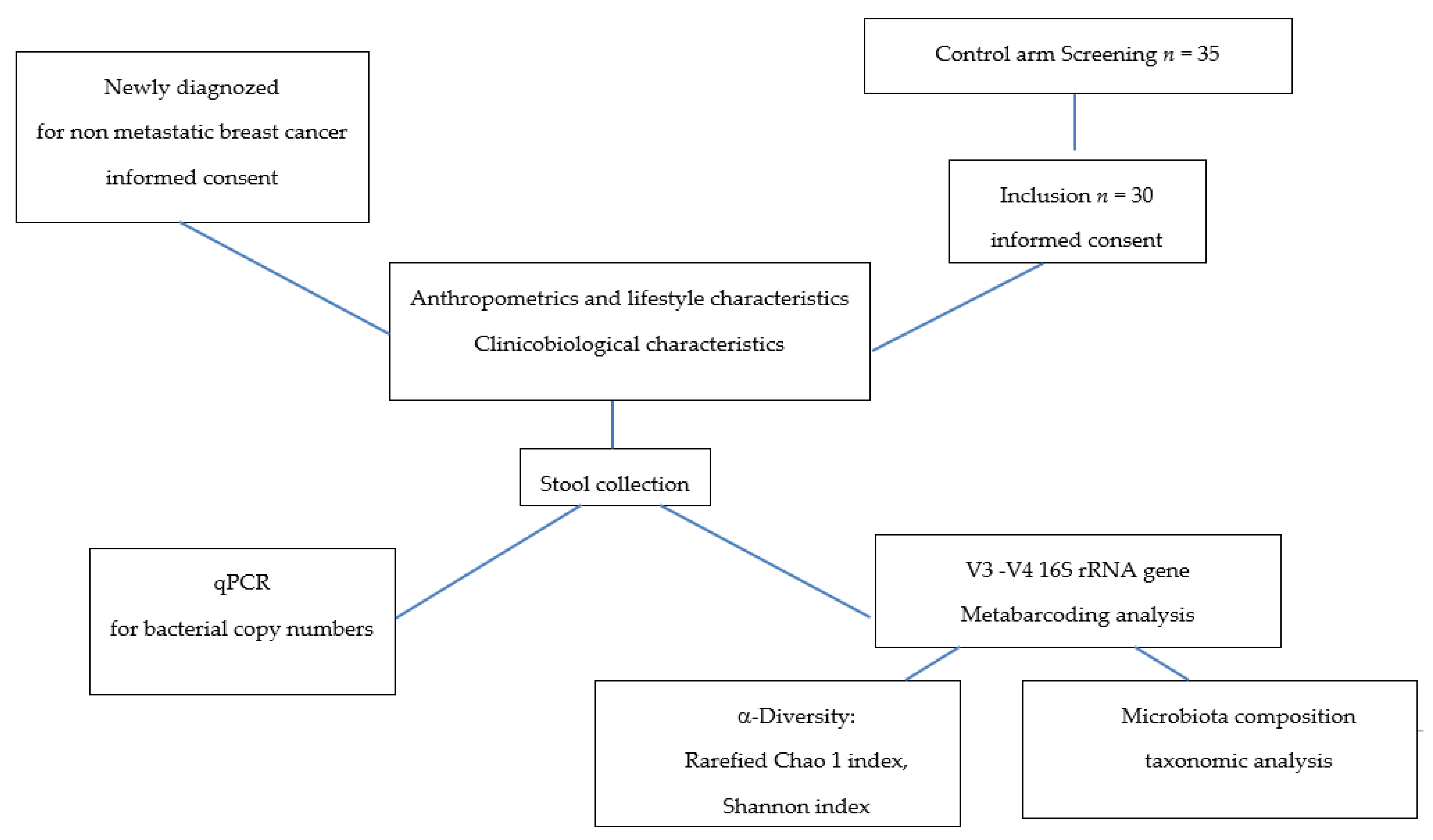
2.1. Recruitment and Sample Collection
2.2. V3–V4 16S rRNA Gene Sequencing
2.3. Bioinformatic Analysis
2.4. Quantitative PCR (qPCR) for Bacterial Copy Numbers
2.5. Statistical Analysis
3. Results
3.1. Clinical Characteristics of the Studied Population
3.2. Faecal Microbiota Analysis
3.2.1. qPCR for Specific Bacterial Copy Numbers
3.2.2. V3–V4 16S r RNA Gene Sequencing
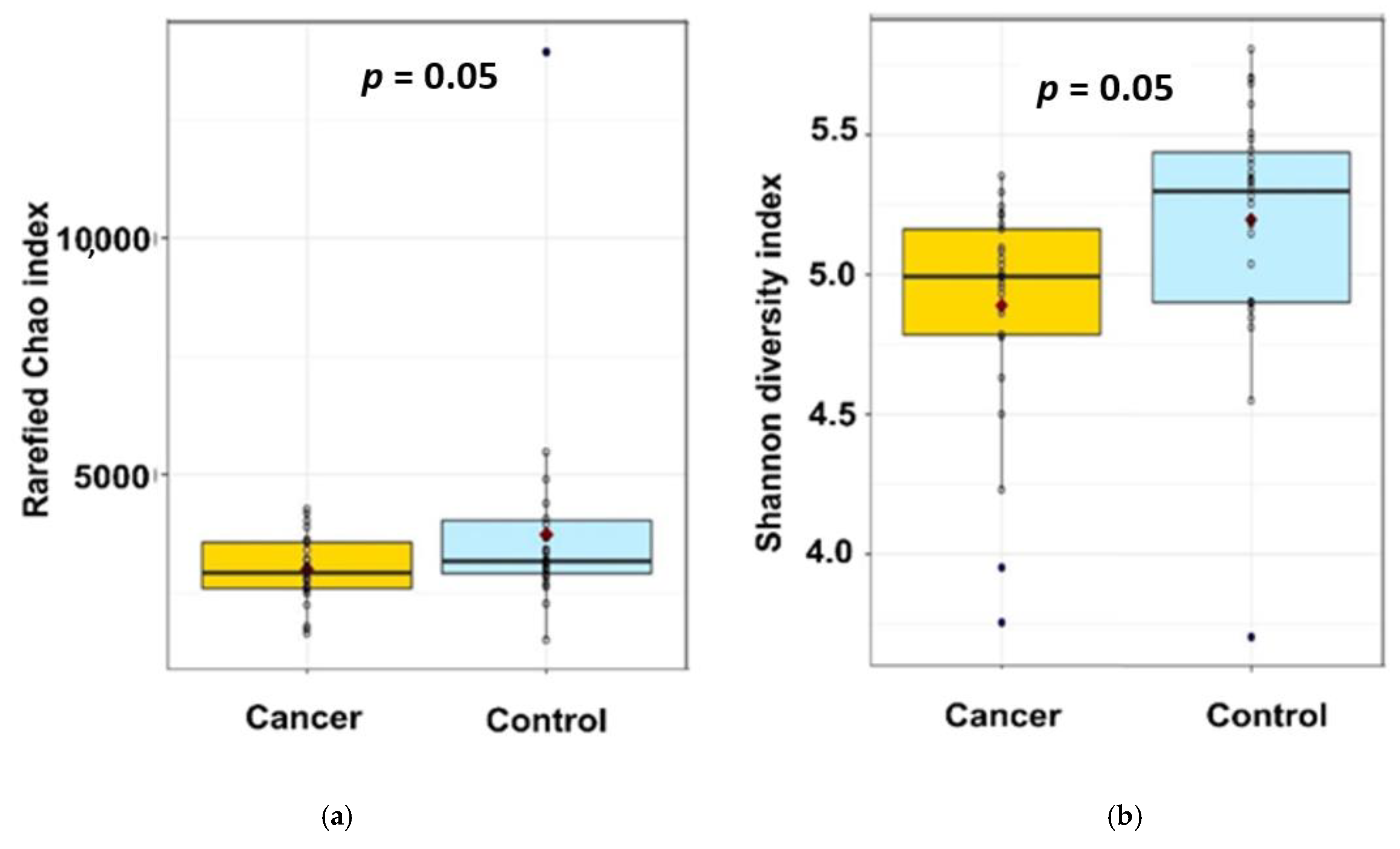
- α-diversity analysis
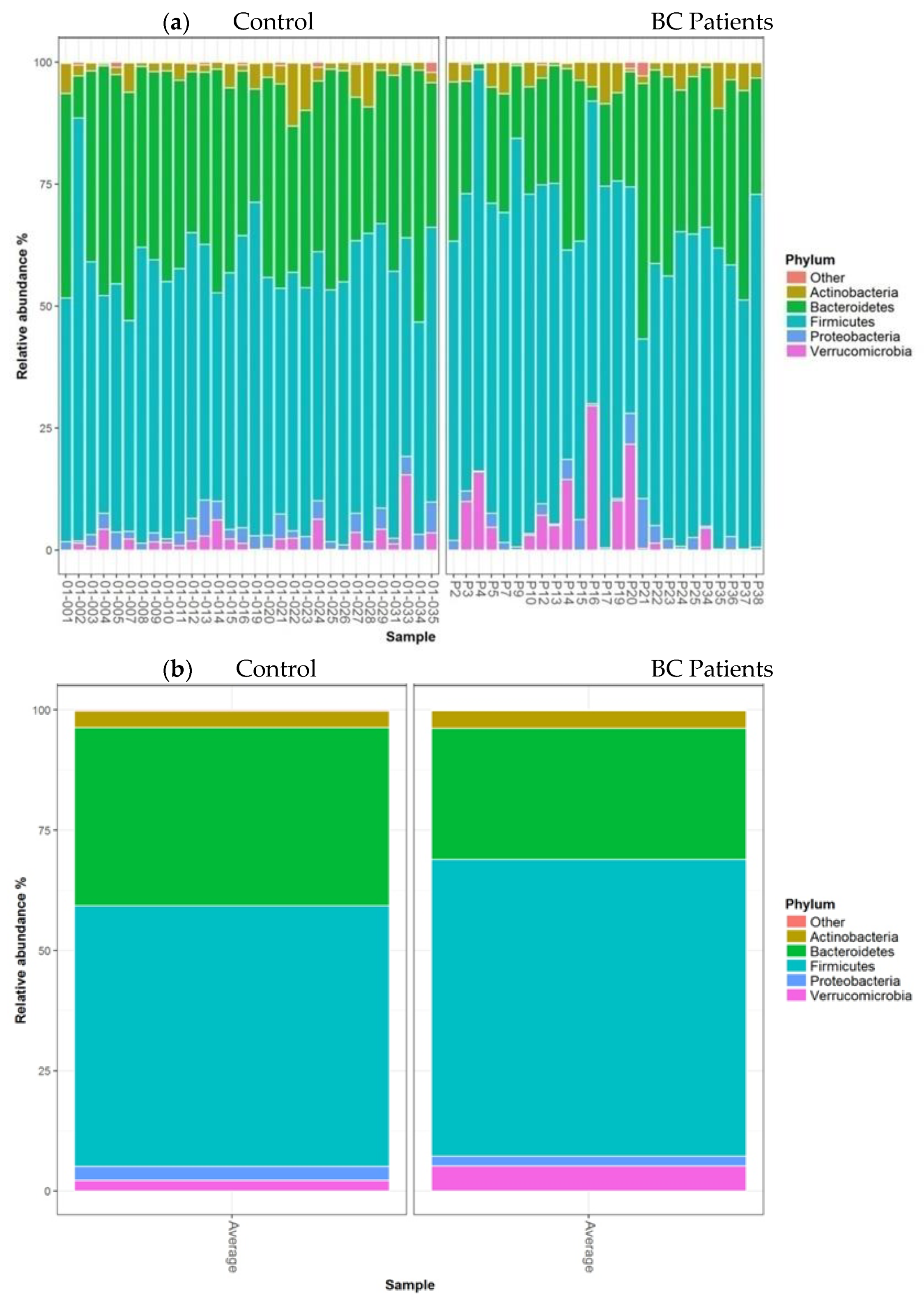
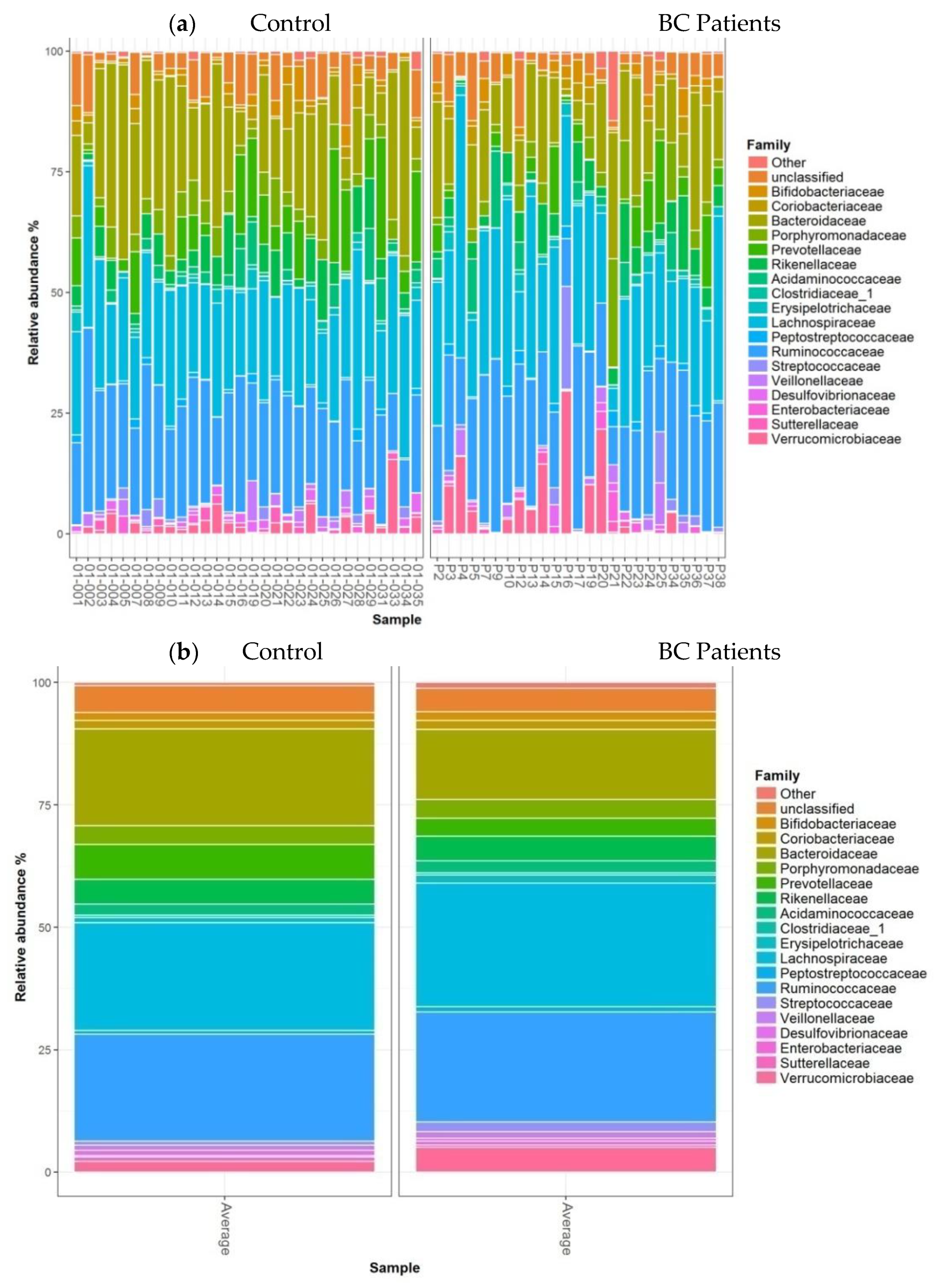
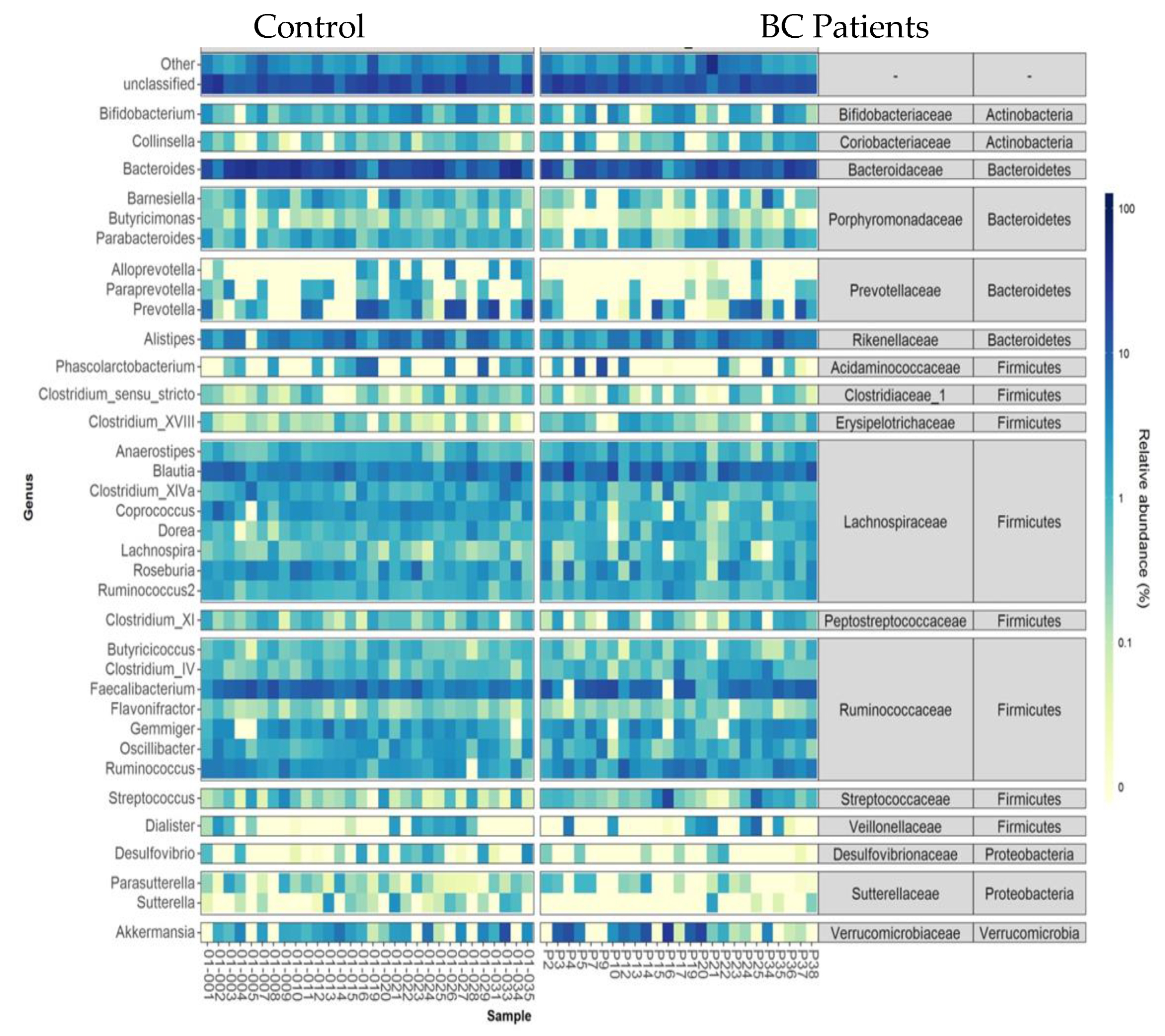
- Taxonomic analysis of faecal microbiota composition at the phylum, family, and genus levels
4. Discussion
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| BC | Breast Cancer |
| BMI | Body Mass Index |
| BRCA1 | Breast Cancer 1 |
| BRCA2 | Breast Cancer 2 |
| ER/PgR | Oestrogen Receptor/Progesterone Receptor |
| HER2 | Human Epidermal Growth Factor Receptor-2 |
| HR | Hormonal Receptor |
| RT-qPCR | Reverse Transcription Quantitative Polymerase |
References
- Global Burden of Disease Cancer Collaboration; Fitzmaurice, C.; Allen, C.; Barber, R.M.; Barregard, L.; Bhutta, Z.A.; Brenner, H.; Dicker, D.J.; Chimed-Orchir, O.; Dandona, R.; et al. Global, Regional, and National Cancer Incidence, Mortality, Years of Life Lost, Years Lived with Disability, and Disability-Adjusted Life-years for 32 Cancer Groups, 1990 to 2015: A Systematic Analysis for the Global Burden of Disease Study. JAMA Oncol. 2017, 3, 524–548. [Google Scholar]
- Wiseman, M.J. Nutrition and cancer: Prevention and survival. Br. J. Nutr. 2018, 122, 481–487. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huybrechts, I.; Zouiouich, S.; Loobuyck, A.; Vandenbulcke, Z.; Vogtmann, E.; Pisanu, S.; Iguacel, I.; Scalbert, A.; Indave, I.; Smelov, V.; et al. The Human Microbiome in Relation to Cancer Risk: A Systematic Review of Epidemiologic Studies. Cancer Epidemiol. Biomark. Prev. 2020, 29, 1856–1868. [Google Scholar] [CrossRef]
- Zitvogel, L.; Galluzzi, L.; Viaud, S.; Vétizou, M.; Daillère, R.; Merad, M.; Kroemer, G. Cancer and the gut microbiota: An unexpected link. Sci. Transl. Med. 2015, 7, 271ps1. [Google Scholar] [CrossRef] [Green Version]
- Bhatt, A.P.; Redinbo, M.R.; Bultman, S.J. The role of the microbiome in cancer development and therapy. CA Cancer J. Clin. 2017, 67, 326–344. [Google Scholar] [CrossRef] [Green Version]
- Sheflin, A.M.; Whitney, A.K.; Weir, T.L. Cancer-promoting effects of microbial dysbiosis. Curr. Oncol. Rep. 2014, 16, 406. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rajagopala, S.V.; Vashee, S.; Oldfield, L.M.; Suzuki, Y.; Venter, J.C.; Telenti, A.; Nelson, K.E. The Human Microbiome and Cancer. Cancer Prev Res. Am. Assoc. Cancer Res. 2017, 10, 226–234. [Google Scholar]
- Lakritz, J.; Poutahidis, T.; Mirabal, S.; Varian, B.J.; Levkovich, T.; Ibrahim, Y.M.; Ward, J.M.; Teng, E.C.; Fisher, B.; Parry, N.; et al. Gut bacteria require neutrophils to promote mammary tumorigenesis. Oncotarget 2015, 6, 9387–9396. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wirtz, H.S.; Buist, D.S.; Gralow, J.R.; Barlow, W.E.; Gray, S.; Chubak, J.; Yu, O.; Bowles, E.J.; Fujii, M.; Boudreau, D.M. Frequent antibiotic use and second breast cancer events. Cancer Epidemiol. Biomark. Prev. 2013, 22, 1588–1599. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Luu, T.H.; Michel, C.; Bard, J.-M.; Dravet, F.; Nazih, H.; Bobin-Dubigeon, C. Intestinal Proportion of Blautia sp. is Associated with Clinical Stage and Histoprognostic Grade in Patients with Early-Stage Breast Cancer. Nutr. Cancer 2017, 69, 267–275. [Google Scholar] [CrossRef] [PubMed]
- Heymann, D.; Kerdraon, O.; Verriele, V.; Verhille, E.; Veron, V.; Vitre, M.; Delmas, F.; Henry, C.; Gouy, Y.; Amiand, M.; et al. Centre de Ressources Biologiques-Tumorothèque: Bioresources and Associated Clinical Data Dedicated to Translational Research in Oncology at the Institut de Cancérologie de l’Ouest, France. Open J. Bioresour. 2020, 7, 5. [Google Scholar] [CrossRef] [Green Version]
- Klindworth, A.; Pruesse, E.; Schweer, T.; Peplies, J.; Quast, C.; Horn, M.; Glöckner, F.O. Evaluation of general 16S ribosomal RNA gene PCR primers for classical and next-generation sequencing-based diversity studies. Nucleic Acids Res. 2012, 41, e1. [Google Scholar] [CrossRef]
- Schloss, P.D.; Westcott, S.L.; Ryabin, T.; Hall, J.R.; Hartmann, M.; Hollister, E.B.; Lesniewski, R.A.; Oakley, B.B.; Parks, D.H.; Robinson, C.J.; et al. Introducing mothur: Open-Source, Platform-Independent, Community-Supported Software for Describing and Comparing Microbial Communities. Appl. Environ. Microbiol. 2009, 75, 7537–7541. [Google Scholar] [CrossRef] [Green Version]
- Golombos, D.M.; Ayangbesan, A.; O’Malley, P.; Lewicki, P.; Barlow, L.; Barbieri, C.E.; Chan, C.; DuLong, C.; Abu-Ali, G.; Huttenhower, C.; et al. The Role of Gut Microbiome in the Pathogenesis of Prostate Cancer: A Prospective, Pilot Study. Urology 2018, 111, 122–128. [Google Scholar] [CrossRef] [PubMed]
- Gao, Z.; Guo, B.; Gao, R.; Zhu, Q.; Qin, H. Microbiota disbiosis is associated with colorectal cancer. Front. Microbiol. 2015, 6, 20. [Google Scholar] [CrossRef]
- Weiss, G.A.; Hennet, T. Mechanisms and consequences of intestinal dysbiosis. Cell. Mol. Life Sci. 2017, 74, 2959–2977. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pellegrini, M.; Ippolito, M.; Monge, T.; Violi, R.; Cappello, P.; Ferrocino, I.; Cocolin, L.S.; De Francesco, A.; Bo, S.; Finocchiaro, C. Gut microbiota composition after diet and probiotics in overweight breast cancer survivors: A randomized open-label pilot intervention trial. Nutrition 2020, 74, 110749. [Google Scholar] [CrossRef] [PubMed]
- Goedert, J.J.; Jones, G.; Hua, X.; Xu, X.; Yu, G.; Flores, R.; Falk, R.T.; Gail, M.H.; Shi, J.; Ravel, J.; et al. Investigation of the association between the fecal microbiota and breast cancer in postmenopausal women: A population-based case-control pilot study. J. Natl. Cancer Inst. 2015, 107. [Google Scholar] [CrossRef] [PubMed]
- Arslan, A.; Koenig, K.L.; Lenner, P.; Afanasyeva, Y.; Shore, R.; Chen, Y.; Lundin, E.; Toniolo, P.; Hallmans, G.; Zeleniuch-Jacquotte, A. Circulating estrogen metabolites and risk of breast cancer in postmenopausal women. Cancer Epidemiol. Biomarkers Prev. 2014, 23, 1290–1297. [Google Scholar] [CrossRef] [Green Version]
- Fernández, M.F.; Reina-Pérez, I.; Astorga, J.M.; Rodríguez-Carrillo, A.; Plaza-Díaz, J.; Fontana, L. Breast Cancer and Its Relationship with the Microbiota. Int. J. Environ. Res. Public Health 2018, 15, 1747. [Google Scholar] [CrossRef] [Green Version]
- Odamaki, T.; Kato, K.; Sugahara, H.; Hashikura, N.; Takahashi, S.; Xiao, J.-Z.; Abe, F.; Osawa, R. Age-related changes in gut microbiota composition from newborn to centenarian: A cross-sectional study. BMC Microbiol. 2016, 16, 90. [Google Scholar] [CrossRef] [Green Version]
- Claesson, M.J.; Cusack, S.; O’Sullivan, O.; Greene-Diniz, R.; De Weerd, H.; Flannery, E.; Marchesi, J.R.; Falush, D.; Dinan, T.G.; Fitzgerald, G.F.; et al. Composition, variability, and temporal stability of the intestinal microbiota of the elderly. Proc. Natl. Acad. Sci. USA 2011, 108 (Suppl. 1), 4586–4591. [Google Scholar] [CrossRef] [Green Version]
- Lahti, L.; Salojärvi, J.; Salonen, A.; Scheffer, M.; De Vos, W.M. Tipping elements in the human intestinal ecosystem. Nat. Commun. 2014, 5, 4344. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- An, R.; Wilms, E.; Masclee, A.A.M.; Smidt, H.; Zoetendal, E.G.; Jonkers, D. Age-Dependent Changes in GI Physiology and Microbiota: Time to Reconsider? Gut 2018, 67, 2213–2222. [Google Scholar] [CrossRef]
- Mueller, S.; Saunier, K.; Hanisch, C.; Norin, E.; Alm, L.; Midtvedt, T.; Cresci, A.; Silvi, S.; Orpianesi, C.; Verdenelli, M.C.; et al. Differences in Fecal Microbiota in Different European Study Populations in Relation to Age, Gender, and Country: A Cross-Sectional Study. Appl. Environ. Microbiol. 2006, 72, 1027–1033. [Google Scholar] [CrossRef] [Green Version]
- Hamilton, A.J. Species diversity or biodiversity? J. Environ. Manag. 2005, 75, 89–92. [Google Scholar] [CrossRef] [PubMed]
- Ahn, J.; Sinha, R.; Pei, Z.; Dominianni, C.; Wu, J.; Shi, J.; Goedert, J.J.; Hayes, R.; Yang, L. Human gut microbiome and risk for colorectal cancer. J. Natl. Cancer Inst. 2013, 105, 1907–1911. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kang, D.-W.; Park, J.G.; Ilhan, Z.E.; Wallstrom, G.; LaBaer, J.; Adams, J.B.; Krajmalnik-Brown, R. Reduced incidence of Prevotella and other fermenters in intestinal microflora of autistic children. PLoS ONE 2013, 8, e68322. [Google Scholar]
- Sha, S.; Xu, B.; Wang, X.; Zhang, Y.; Wang, H.; Kong, X.; Zhu, H.; Wu, K. The biodiversity and composition of the dominant fecal microbiota in patients with inflammatory bowel disease. Diagn. Microbiol. Infect. Dis. 2013, 75, 245–251. [Google Scholar] [CrossRef]
- Labus, J.S.; Hollister, E.B.; Jacobs, J.; Kirbach, K.; Oezguen, N.; Gupta, A.; Acosta, J.; Luna, R.A.; Aagaard, K.; Versalovic, J.; et al. Differences in gut microbial composition correlate with regional brain volumes in irritable bowel syndrome. Microbiome 2017, 5, 49. [Google Scholar] [CrossRef] [PubMed]
- Ling, Z.; Jin, C.; Xie, T.; Cheng, Y.; Li, L.; Wu, N. Alterations in the Fecal Microbiota of Patients with HIV-1 Infection: An Observational Study in A Chinese Population. Sci. Rep. 2016, 6, 30673. [Google Scholar] [CrossRef] [Green Version]
- Vaiserman, A.M.; Koliada, A.; Marotta, F. Gut Microbiota: A Player in Aging and a Target for Anti-Aging Intervention. Ageing Res. Rev. 2017, 35, 36–45. [Google Scholar] [CrossRef] [PubMed]
- Plottel, C.S.; Blaser, M.J. Microbiome and malignancy. Cell Host Microbe. 2011, 10, 324–335. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Parida, S.; Sharma, D. The Microbiome-Estrogen Connection and Breast Cancer Risk. Cells 2019, 8, 1642. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dabek, M.; McCrae, S.I.; Stevens, V.J.; Duncan, S.H.; Louis, P. Distribution of beta-glucosidase and beta-glucuronidase activity and of beta-glucuronidase gene gus in human colonic bacteria. FEMS Microbiol. Ecol. 2008, 66, 487–495. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Butel, M.-J. Probiotics, gut microbiota and health. Médecine Mal. Infectieuses. 2014, 44, 1–8. [Google Scholar] [CrossRef]
- Sivan, A.; Corrales, L.; Hubert, N.; Williams, J.B.; Aquino-Michaels, K.; Earley, Z.M.; Benyamin, F.W.; Lei, Y.M.; Jabri, B.; Alegre, M.L.; et al. Commensal Bifidobacterium promotes antitumor immunity and facilitates anti-PD-L1 efficacy. Science 2015, 350, 1084–1089. [Google Scholar] [CrossRef] [Green Version]
- Shen, X.J.; Rawls, J.; Randall, T.; Burcall, L.; Mpande, C.N.; Jenkins, N.; Jovov, B.; Abdo, Z.; Sandler, R.S.; Keku, T.O. Molecular characterization of mucosal adherent bacteria and associations with colorectal adenomas. Gut Microbes. 2010, 1, 138–347. [Google Scholar] [CrossRef] [Green Version]
- Bailey, M.T.; Dowd, S.E.; Galley, J.D.; Hufnagle, A.R.; Allen, R.G.; Lyte, M. Exposure to a social stressor alters the structure of the intestinal microbiota: Implications for stressor-induced immunomodulation. Brain Behav. Immun. 2011, 25, 397–407. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bhargava, P.; Mowry, E.M. Gut Microbiome and Multiple Sclerosis. Curr. Neurol. Neurosci. Rep. 2014, 14, 492. [Google Scholar] [CrossRef]
- Montassier, E.; Al-Ghalith, G.A.; Ward, T.; Corvec, S.; Gastinne, T.; Potel, G.; Moreau, P.; De La Cochetiere, M.F.; Batard, E.; Knights, D. Pretreatment gut microbiome predicts chemotherapy-related bloodstream infection. Genome Med. 2016, 8, 49. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Thomas, A.M.; Jesus, E.C.; Lopes, A.; Aguiar, S.; Begnami, M.D.; Rocha, R.M.; Carpinetti, P.A.; Camargo, A.A.; Hoffmann, C.; Freitas, H.C.; et al. Tissue-Associated Bacterial Alterations in Rectal Carcinoma Patients Revealed by 16S rRNA Community Profiling. Front. Cell. Infect. Microbiol. 2016, 6, 179. [Google Scholar] [CrossRef] [Green Version]
- Mikó, E.; Kovács, T.; Sebő, É.; Tóth, J.; Csonka, T.; Ujlaki, G.; Sipos, A.; Szabó, J.; Méhes, G.; Bai, P. Microbiome—Microbial Metabolome—Cancer Cell Interactions in Breast Cancer—Familiar, but Unexplored. Cells 2019, 8, 293. [Google Scholar] [CrossRef] [Green Version]
- Saad, M.J.A.; Santos, A.; Prada, P.O. Linking Gut Microbiota and Inflammation to Obesity and Insulin Resistance. Physiology 2016, 31, 283–293. [Google Scholar] [CrossRef] [PubMed]
- Morris, P.G.; Hudis, C.A.; Giri, D.; Morrow, M.; Falcone, D.J.; Zhou, X.K.; Du, B.; Brogi, E.; Crawford, C.B.; Kopelovich, L.; et al. Inflammation and increased aromatase expression occur in the breast tissue of obese women with breast cancer. Cancer Prev. Res. 2011, 4, 1021–1029. [Google Scholar] [CrossRef] [Green Version]
- Wu, A.H.; Tseng, C.; Vigen, C.; Yu, Y.; Cozen, W.; Garcia, A.A.; Spicer, D. Gut Microbiome Associations with Breast Cancer Risk Factors and Tumor Characteristics: A Pilot Study. Breast Cancer Res. Treat. 2020, 182, 451–463. [Google Scholar] [CrossRef] [PubMed]
- Nejman, D.; Livyatan, I.; Fuks, G.; Gavert, N.; Zwang, Y.; Geller, L.T.; Rotter-Maskowitz, A.; Weiser, R.; Mallel, G.; Gigi, E.; et al. The human tumor microbiome is composed of tumor type-specific intracellular bacteria. Science 2020, 368, 973–980. [Google Scholar] [CrossRef]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Characteristics | Healthy Controls (n = 30) | BC Patients (n = 25) | p |
|---|---|---|---|
| Age (year) * | 53.5 (7.0–62.0) | 63.0 (53.8–71.0) | 0.01 |
| BMI (kg·m−2) * | 23.8 (22.4–25.6) | 24.0 (21.5–26.4) | 0.85 |
| Menopausal status ** | 20 (66.6) | 22 (88.0) | 0.10 |
| Alcohol consumption | 1 | ||
| <7 glasses/w | 29 (96.6) | 24 (96.0) | |
| >10 to 14/w | 1 (3.3) | 1 (4.0) | |
| smoking | 1 | ||
| 0 | 28 (93.3) | 25 (100) | |
| 1–5/d | 1 (3.3) | 0 | |
| 6–10/d | 1 (3.3) | 0 | |
| >20/d | 0 | 0 | |
| Medication treatment | |||
| Hormone therapy | 4 (13.3) | 7 (28.0) | 0.20 |
| Type of Cancer | |
|---|---|
| Invasive Carcinoma of No Special Type (Ductal) | 19 (76.0%) |
| Invasive Lobular Carcinoma | 6 (24.0%) |
| Histoprognostic Grade | |
| Grade I | 5 (20.0%) |
| Grade II | 16 (64.0%) |
| Grade III | 4 (16.0%) |
| Clinical Stage | |
| I | 21 (84.0%) |
| IIa | 3 (12.0%) |
| IIIb | 1 (4.0%) |
| Tumoral Overexpression Receptors | |
| ER/Pg+ | 25 (100%) |
| HER2+ | 0 |
| Tumour Localization | |
| unifocal | 23 (92.0%) |
| multifocal | 2 (8.0%) |
| size (mm) * | 12.5 (9.0–14.7) |
| Gene BRCA1/BRCA2 | 0 |
| Bacterial Population | Healthy Controls (n = 29) | BC Patients (n = 25) | p |
|---|---|---|---|
| Bacteroidetes phylum | 19.14 (16.27–22.75) | 14.24 (12.52–19.21) | <0.001 |
| Firmicutes phylum | 15.46 (10.75–33.57] | 36.10 (24.91–43.46) | 0.003 |
| Lactobacillales sp. | 0.006 (0.004–0.015) | 0.006 (0.004–0.02) | 0.91 |
| Clostridium cluster IV | 6.1 (3.63–11.91) | 12.02 (9.33–21.56) | 0.003 |
| Faecalibacterium prausnitzii | 4.88 (2.93–7.48) | 7.63 (4.25–10.77) | 0.25 |
| Clostridium cluster XIVa | 5.97 (4.02–15.59) | 18.64 (10.94–22.43) | 0.012 |
| R. intestinalis | 1.16 (0.86–1.96) | 1.21 (0.64–2.11) | 0.73 |
| Blautia sp. | 1.19 (0.99–1.79) | 1.90 (1.28–2.37) | 0.045 |
| Lactonifactor longoviformis | 0.002 (0.001–0.003) | 0.002 (0.001–0.004) | 0.45 |
| Actinobacteria phylum | |||
| Bifidobacterium sp. | 0.126 (0.061–0.293) | 0.070 (0.016–0.162) | 0.11 |
| Coriobacteriaceae | 0.328 (0.169–0.619) | 0.23 (0.14–0.60) | 0.65 |
| E. lenta | 0.011 (0.007–0.020) | 0.009 (0.005–0.017) | 0.55 |
| Proteobacteria phylum | |||
| Escherichia + Shigella | 0.005 (0.000–0.013) | 0.006 (0.001–0.018) | 0.50 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bobin-Dubigeon, C.; Luu, H.T.; Leuillet, S.; Lavergne, S.N.; Carton, T.; Le Vacon, F.; Michel, C.; Nazih, H.; Bard, J.-M. Faecal Microbiota Composition Varies between Patients with Breast Cancer and Healthy Women: A Comparative Case-Control Study. Nutrients 2021, 13, 2705. https://doi.org/10.3390/nu13082705
Bobin-Dubigeon C, Luu HT, Leuillet S, Lavergne SN, Carton T, Le Vacon F, Michel C, Nazih H, Bard J-M. Faecal Microbiota Composition Varies between Patients with Breast Cancer and Healthy Women: A Comparative Case-Control Study. Nutrients. 2021; 13(8):2705. https://doi.org/10.3390/nu13082705
Chicago/Turabian StyleBobin-Dubigeon, Christine, Huyen Trang Luu, Sébastien Leuillet, Sidonie N. Lavergne, Thomas Carton, Françoise Le Vacon, Catherine Michel, Hassane Nazih, and Jean-Marie Bard. 2021. "Faecal Microbiota Composition Varies between Patients with Breast Cancer and Healthy Women: A Comparative Case-Control Study" Nutrients 13, no. 8: 2705. https://doi.org/10.3390/nu13082705
APA StyleBobin-Dubigeon, C., Luu, H. T., Leuillet, S., Lavergne, S. N., Carton, T., Le Vacon, F., Michel, C., Nazih, H., & Bard, J.-M. (2021). Faecal Microbiota Composition Varies between Patients with Breast Cancer and Healthy Women: A Comparative Case-Control Study. Nutrients, 13(8), 2705. https://doi.org/10.3390/nu13082705