
Malnutrition in Pediatric Chronic Cholestatic Disease: An Up-to-Date Overview

 and
and
Abstract
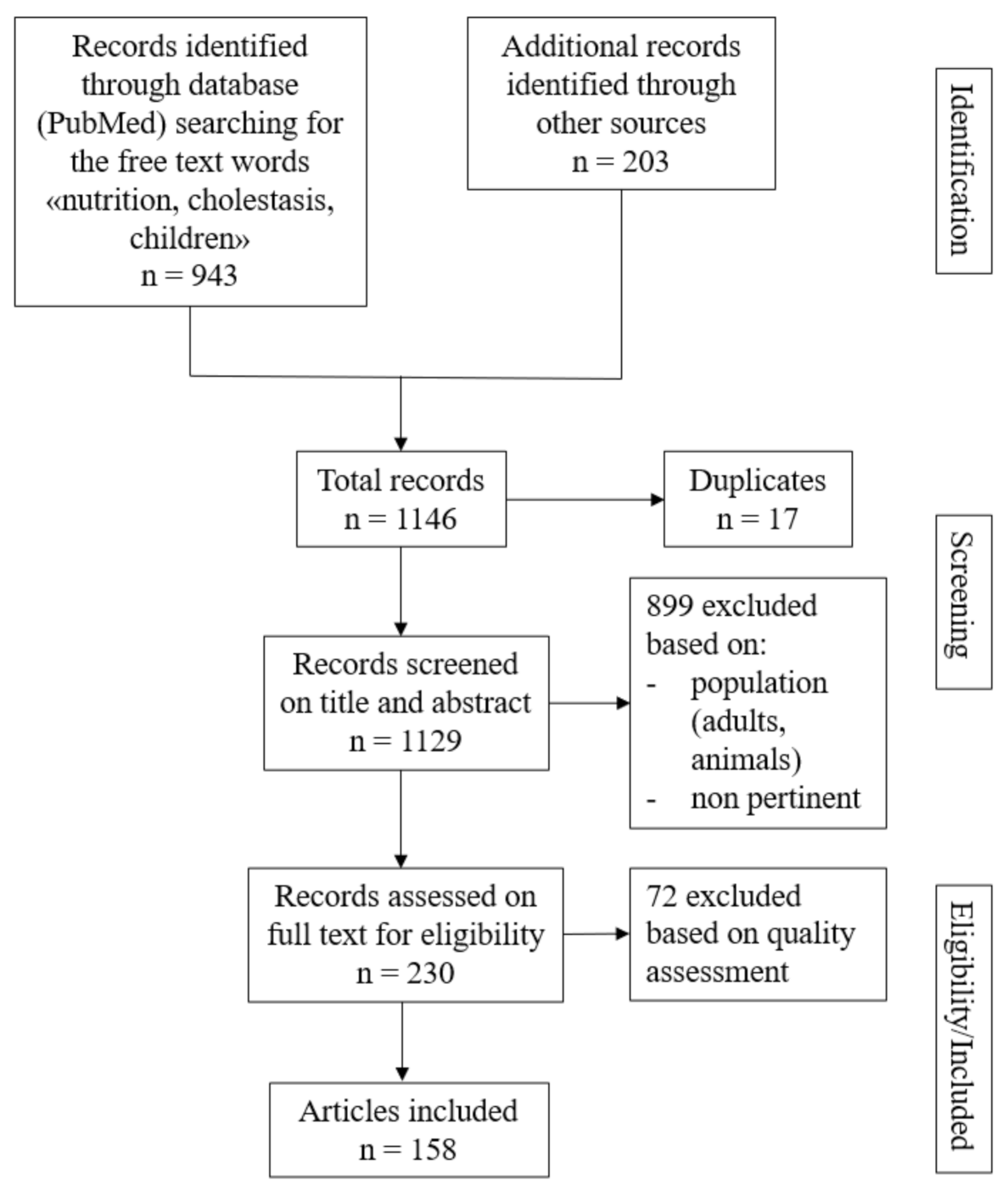
:1. Introduction
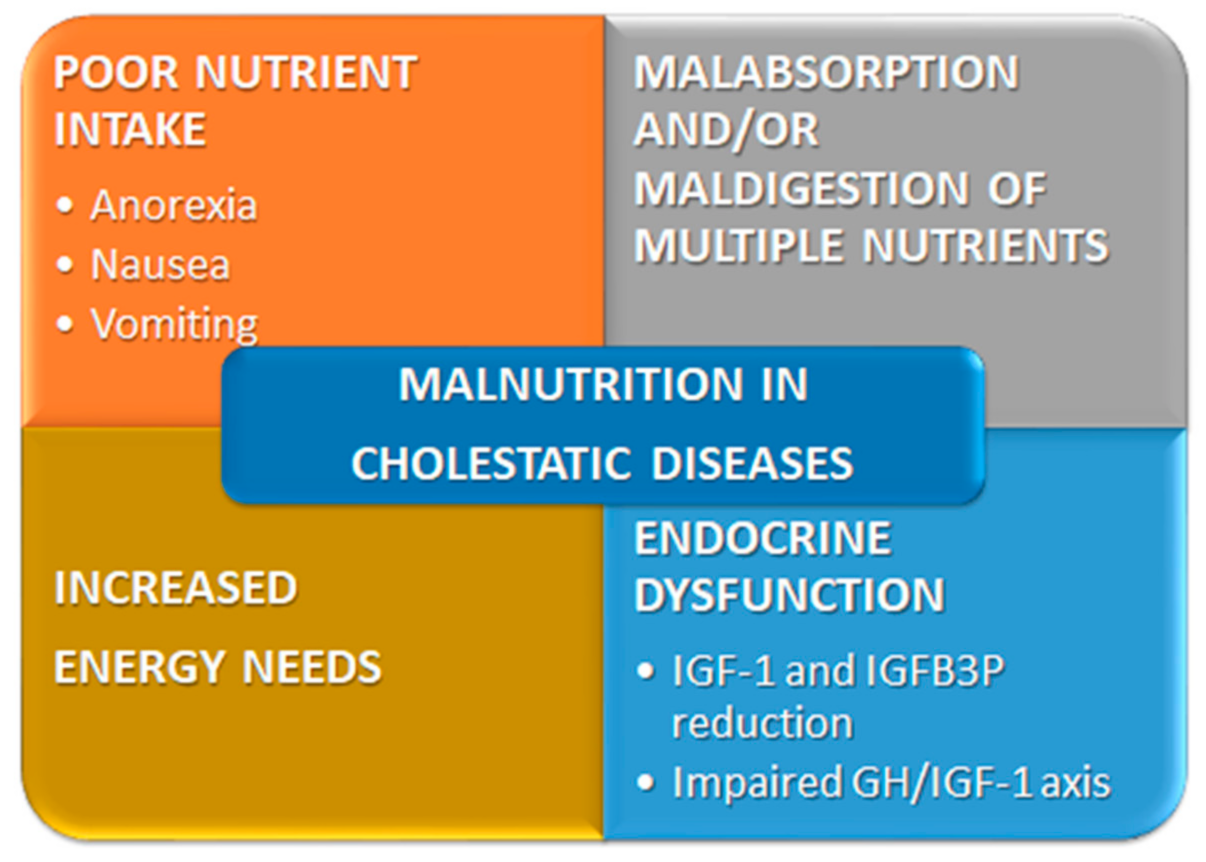
2. Causes of Malnutrition in Cholestatic Children
2.1. Metabolic Changes
2.2. Poor Nutrient Intake
2.3. Increased Requirements or Malabsorption/Maldigestion of Multiple Nutrients
2.3.1. Increased Energy Needs
2.3.2. Water and Electrolytes
2.3.3. Carbohydrates
2.3.4. Proteins
2.3.5. Lipids and Bile–Acid-Dependent Absorption of Fats and Fat-Soluble Nutrients
2.3.6. Medium Chain Triglycerides and Long-Chain Triglycerides
2.3.7. Essential Fatty Acids
2.3.8. Fat-Soluble Vitamins
2.3.9. Water-Soluble Vitamins and Minerals
2.4. Endocrine Dysfunction
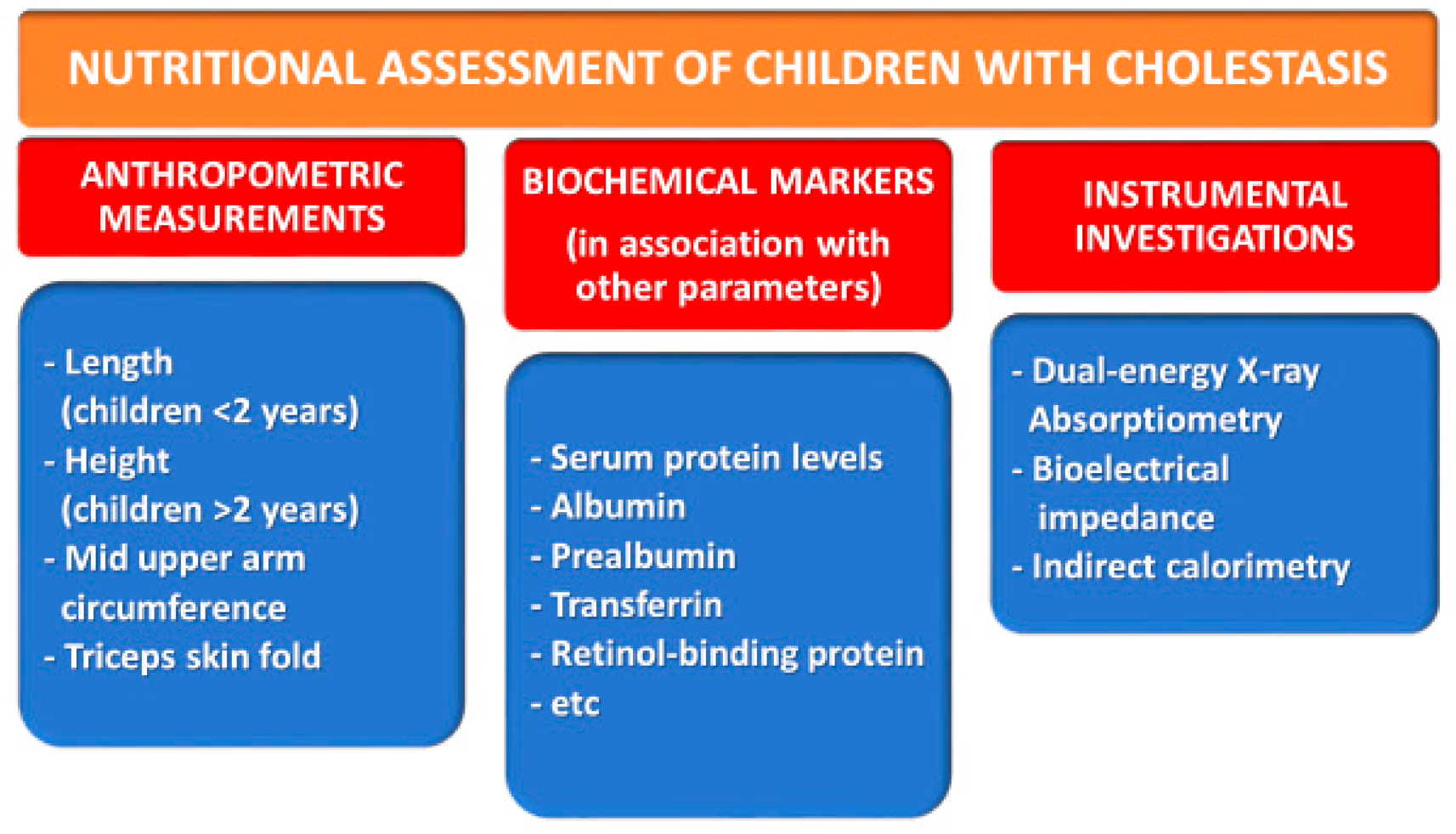
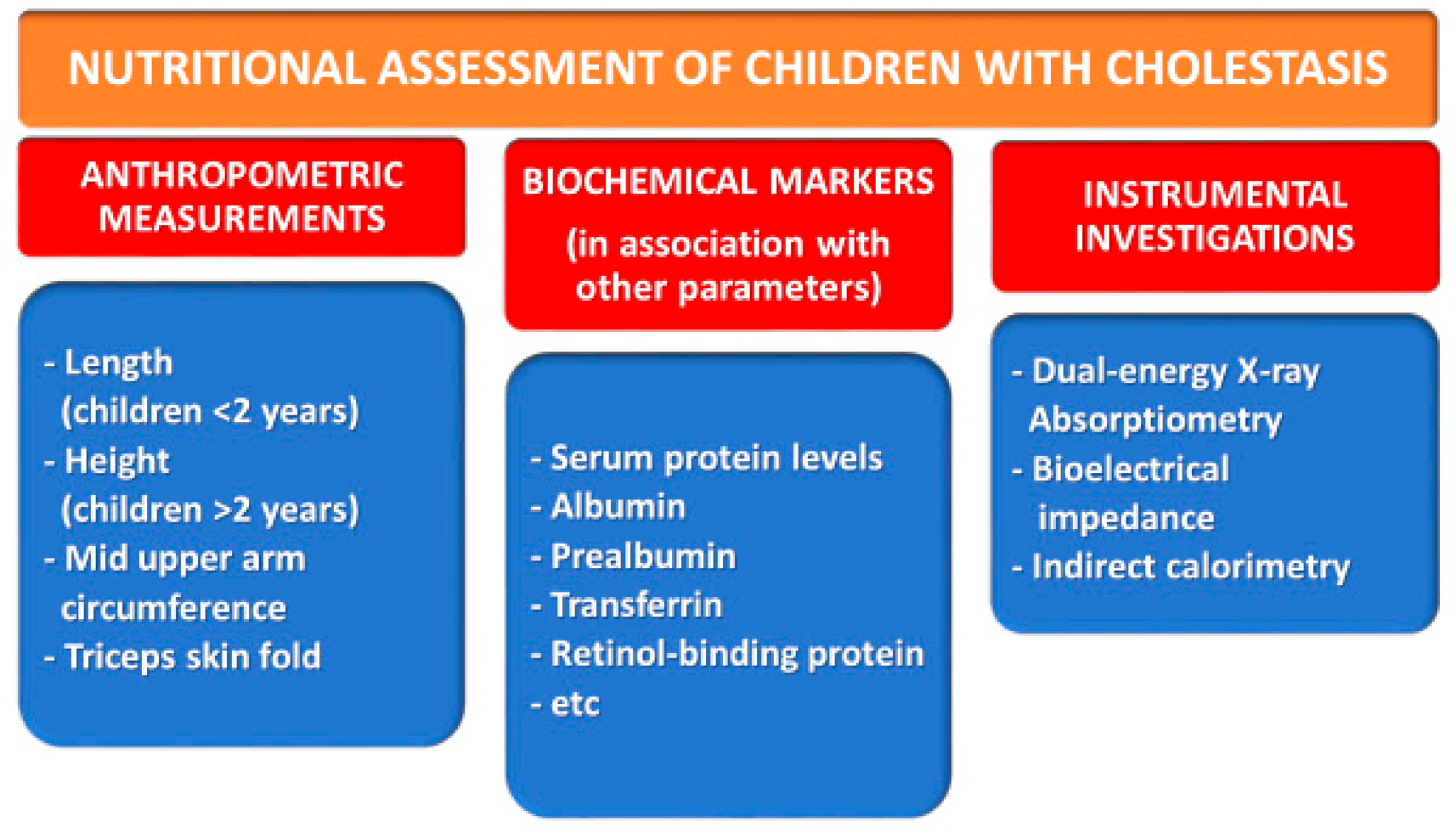
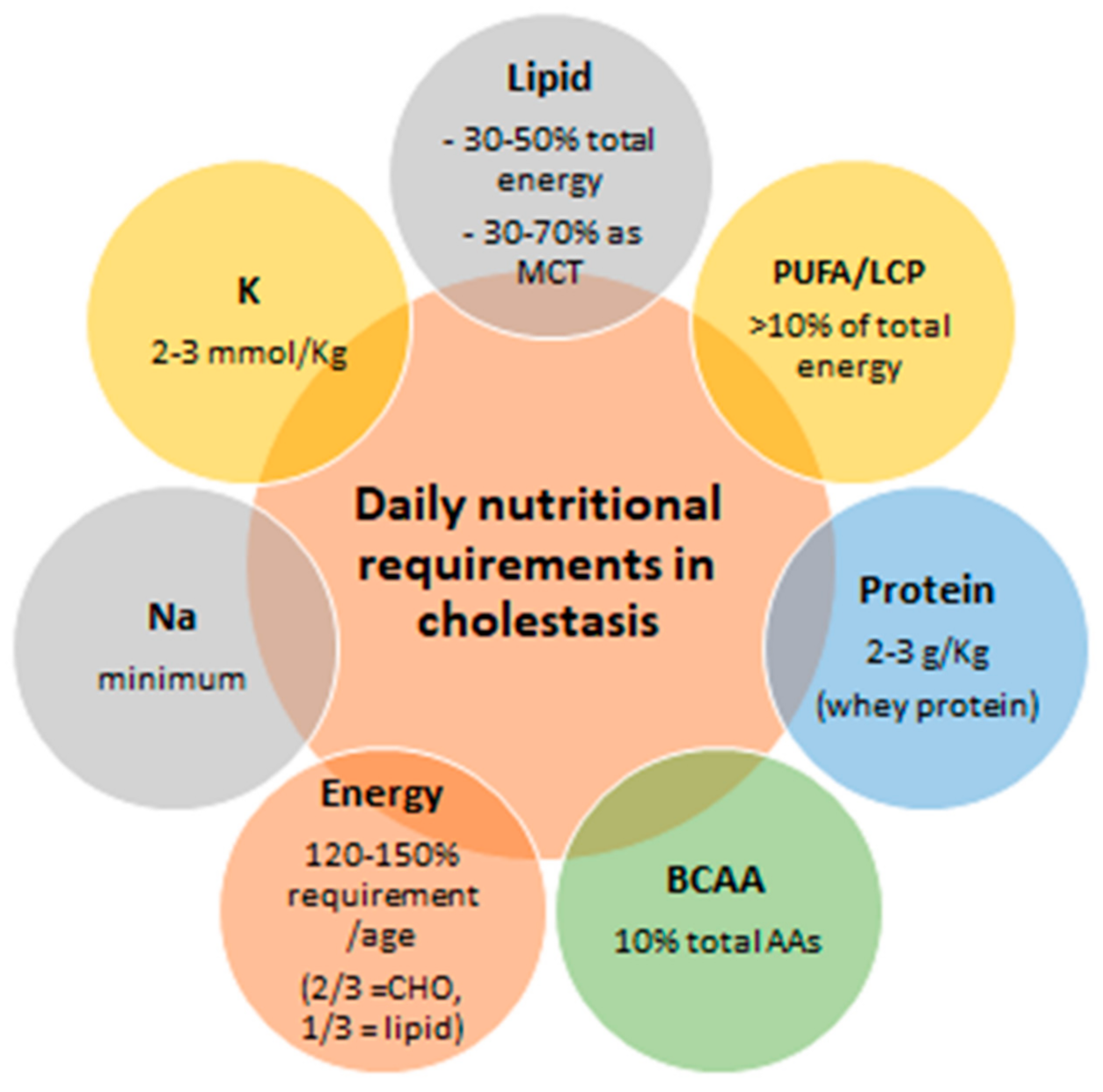
3. Issues in the Nutritional Management of Children with Cholestasis
3.1. Anthropometric Measurements
3.2. Biochemical Markers
3.3. Other Investigations
4. Special Diets in Some Common or Special IEM Causing Cholestasis
4.1. Tyrosinemia Type 1
4.2. Galactosemia
4.3. Hereditary Fructose Intolerance
4.4. Citrin Deficiency
5. Pre- and Post-Transplant Nutritional Status of Children with End-Stage Cholestatic Liver Disease
6. Post-Transplant Obesity with Fatty Liver and MetS Risk: The Malnutrition in Excess paradox
Sarcopenia
7. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Conflicts of Interest
References
- Catzola, A.; Vajro, P. Management options for cholestatic liver disease in children. Expert Rev. Gastroenterol. Hepatol. 2017, 11, 1019–1030. [Google Scholar] [CrossRef] [PubMed]
- Venigalla, S.; Gourley, G.R. Neonatal cholestasis. Semin Perinatol. 2004, 28, 348–355. [Google Scholar] [CrossRef] [PubMed]
- Fawaz, R.; Baumann, U.; Ekong, U.; Fischler, B.; Hadzic, N.; Mack, C.L.; McLin, V.A.; Molleston, J.P.; Neimark, E.; Ng, V.L.; et al. Guideline for the Evaluation of Cholestatic Jaundice in Infants: Joint Recommendations of the North American Society for Pediatric Gastroenterology, Hepatology, and Nutrition and the European Society for Pediatric Gastroenterology, Hepatology, and Nutrition. J. Pediatr. Gastroenterol. Nutr. 2017, 64, 154–168. [Google Scholar] [CrossRef] [PubMed]
- Mandato, C.; Siano, M.A.; Nazzaro, L.; Gelzo, M.; Francalanci, P.; Rizzo, F.; D’Agostino, Y.; Morleo, M.; Brillante, S.; Weisz, A.; et al. A ZFYVE19 gene mutation associated with neonatal cholestasis and cilia dysfunction: Case report with a novel pathogenic variant. Orphanet J. Rare Dis. 2021, 16, 1–9. [Google Scholar] [CrossRef]
- Mandato, C.; Zollo, G.; Vajro, P. Cholestatic jaundice in infancy: Struggling with many old and new phenotypes. Ital. J. Pediatr. 2019, 45, 1–5. [Google Scholar] [CrossRef] [Green Version]
- Mouzaki, M.; Bronsky, J.; Gupte, G.; Hojsak, I.; Jahnel, J.; Pai, N.; Quiros-Tejeira, R.E.; Wieman, R.; Sundaram, S. Nutrition Support of Children with Chronic Liver Diseases: A Joint Position Paper of the North American Society for Pediatric Gastroenterology, Hepatology, and Nutrition and the European Society for Pediatric Gastroenterology, Hepatology, and Nutrition. J. Pediatr. Gastroenterol. Nutr. 2019, 69, 498–511. [Google Scholar] [CrossRef] [Green Version]
- Mandato, C.; Di Nuzzi, A.; Vajro, P. Nutrition and Liver Disease. Nutrients 2017, 10, 9. [Google Scholar] [CrossRef] [Green Version]
- Reuter, B.; Shaw, J.; Hanson, J.; Tate, V.; Acharya, C.; Bajaj, J.S. Nutritional Assessment in Inpatients with Cirrhosis Can Be Improved After Training and Is Associated with Lower Readmissions. Liver Transplant. 2019, 25, 1790–1799. [Google Scholar] [CrossRef] [PubMed]
- Chen, X.; Wang, J.; Lu, Y.; Xie, X.; Gu, Y.; Latour, J.M.; Zhang, Y. Feeding practices in 6–24-month-old children with chronic cholestatic liver diseases: A mixed-method study. BMC Pediatr. 2020, 20, 395. [Google Scholar] [CrossRef]
- Nightingale, S.; Ng, V.L. Optimizing Nutritional Management in Children with Chronic Liver Disease. Pediatr. Clin. North Am. 2009, 56, 1161–1183. [Google Scholar] [CrossRef]
- Laviano, A.; Cangiano, C.; Preziosa, I. Plasma tryptophan levels and anorexia in liver cirrhosis. Int. J. Eat. Disord. 1997, 21, 181–186. [Google Scholar] [CrossRef]
- Aranda-Michel, J. Nutrition in hepatic failure and liver transplantation. Curr. Gastroenterol. Rep. 2001, 3, 362–370. [Google Scholar] [CrossRef] [PubMed]
- Aqel, B.A.; Scolapio, J.; Dickson, R.C.; Burton, D.D.; Bouras, E.P. Contribution of Ascites to Impaired Gastric Function and Nutritional Intake in Patients with Cirrhosis and Ascites. Clin. Gastroenterol. Hepatol. 2005, 3, 1095–1100. [Google Scholar] [CrossRef]
- Portincasa, P.; Di Ciaula, A.; Garruti, G.; Vacca, M.; De Angelis, M.; Wang, D.Q.-H. Bile Acids and GPBAR-1: Dynamic Interaction Involving Genes, Environment and Gut Microbiome. Nutrients 2020, 12, 3709. [Google Scholar] [CrossRef] [PubMed]
- Kriegermeier, A.; Green, R. Pediatric Cholestatic Liver Disease: Review of Bile Acid Metabolism and Discussion of Current and Emerging Therapies. Front. Med. 2020, 7. [Google Scholar] [CrossRef]
- Shneider, B.L. Intestinal Bile Acid Transport: Biology, Physiology, and Pathophysiology. J. Pediatr. Gastroenterol. Nutr. 2001, 32, 407–417. [Google Scholar] [CrossRef]
- Lemoine, C.; Superina, R. Surgical diversion of enterohepatic circulation in pediatric cholestasis. Semin. Pediatr. Surg. 2020, 29, 150946. [Google Scholar] [CrossRef]
- Utterson, E.C.; Shepherd, R.W.; Sokol, R.J.; Bucuvalas, J.; Magee, J.C.; McDiarmid, S.V.; Anand, R.; the SPLIT Research Group. Biliary Atresia: Clinical Profiles, Risk Factors, and Outcomes of 755 Patients Listed for Liver Transplantation. J. Pediatr. 2005, 147, 180–185. [Google Scholar] [CrossRef]
- Kyrana, E.; Williams, J.E.; Wells, J.C.; Dhawan, A. Resting Energy Expenditure of Children with End-stage Chronic Liver Disease Before and After Liver Transplantation. J. Pediatr. Gastroenterol. Nutr. 2019, 69, 102–107. [Google Scholar] [CrossRef] [PubMed]
- Watanabe, M.; Houten, S.; Mataki, C.; Christoffolete, M.; Kim, B.W.; Sato, H.; Messaddeq, N.; Harney, J.W.; Ezaki, O.; Kodama, T.; et al. Bile acids induce energy expenditure by promoting intracellular thyroid hormone activation. Nat. Cell Biol. 2006, 439, 484–489. [Google Scholar] [CrossRef]
- DeRusso, P.A.; Ye, W.; Shepherd, R.; Haber, B.A.; Shneider, B.L.; Whitington, P.F.; Schwarz, K.B.; Bezerra, J.A.; Rosenthal, P.; Karpen, S.; et al. Growth failure and outcomes in infants with biliary atresia: A report from the Biliary Atresia Research Consortium. Hepatology 2007, 46, 1632–1638. [Google Scholar] [CrossRef]
- Kelly, D.A.; Proteroe, S.; Clarke, S. Acute and chronic liver disease. In Nutrition in Pediatris, 5th ed.; Duggan, C., Watkins, J.B., Koletzko, B., Walker, W.A., Eds.; People’s Medical Publishing House-USA: Shelton, CT, USA, 2016; pp. 851–863. [Google Scholar] [CrossRef] [Green Version]
- Schofield, W.N. Predicting basal metabolic rate, new standards and review of previous work. Hum. Nutr. Clin. Nutr. 1985, 39 (Suppl. 1), 5–41. [Google Scholar]
- Carpenter, A.; Ng, V.L.; Chapman, K.; Ling, S.C.; Mouzaki, M. Predictive equations are inaccurate in the estimation of the resting energy expenditure of children with end-stage liver disease. J. Parenter. Enter Nutr. 2017, 41, 507–511. [Google Scholar] [CrossRef]
- Socha, P. Nutritional Management of Cholestatic Syndromes in Childhood. Ann. Nestlé 2008, 66, 137–147. [Google Scholar] [CrossRef]
- Tavill, A.S. The synthesis and degradation of liver-produced proteins. Gut 1972, 13, 225–241. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Feranchak, A.P.; Suchy, F.J.; Sokol, R.J. Medical management of cholestasis in infants and children. In Liver Disease in Children, 4th ed.; Suchy, F.J., Sokol, R.J., Balistreri, W., Eds.; Published by Cambridge University Press: Cambridge, UK, 2014; pp. 111–139. [Google Scholar] [CrossRef]
- Tomkins, A.M.; Garlick, P.J.; Schofield, W.N.; Waterlow, J.C. The Combined Effects of Infection and Malnutrition on Protein Metabolism in Children. Clin. Sci. 1983, 65, 313–324. [Google Scholar] [CrossRef] [Green Version]
- Charlton, C.P.; Buchanan, E.; Holden, C.E.; Preece, M.A.; Green, A.; Booth, I.W.; Tarlow, M.J. Intensive enteral feeding in advanced cirrhosis: Reversal of malnutrition without precipitation of hepatic encephalopathy. Arch. Dis. Child. 1992, 67, 603–607. [Google Scholar] [CrossRef] [Green Version]
- Weisdorf, S.A.; Freese, D.K.; Fath, J.J.; Tsai, M.Y.; Cerra, F.B. Amino Acid Abnormalities in Infants with Extrahepatic Biliary Atresia and Cirrhosis. J. Pediatr. Gastroenterol. Nutr. 1987, 6, 860–864. [Google Scholar] [CrossRef]
- Sokal, E.M.; Baudoux, M.C.; Collette, E.; Hausleithner, V.; Lambotte, L.; Buts, J.P. Branched Chain Amino Acids Improve Body Composition and Nitrogen Balance in a Rat Model of Extra Hepatic Biliary Atresia. Pediatr. Res. 1996, 40, 66–71. [Google Scholar] [CrossRef] [Green Version]
- Chin, S.E.; Shepherd, R.; Thomas, B.J.; Cleghorn, G.; Patrick, M.K.; Wilcox, J.A.; Ong, T.H.; Lynch, S.V.; Strong, R. Nutritional support in children with end-stage liver disease: A randomized crossover trial of a branched-chain amino acid supplement. Am. J. Clin. Nutr. 1992, 56, 158–163. [Google Scholar] [CrossRef] [PubMed]
- Baker, A.; Stevenson, R.; Dhawan, A.; Goncalves, I.; Socha, P.; Sokal, E. Guidelines for nutritional care for infants with cholestatic liver disease before liver transplantation. Pediatr. Transplant. 2007, 11, 825–834. [Google Scholar] [CrossRef] [PubMed]
- Mager, D.R.; Wykes, L.J.; Roberts, E.A.; Ball, R.O.; Pencharz, P.B. Branched-Chain Amino Acid Needs in Children with Mild-to-Moderate Chronic Cholestatic Liver Disease. J. Nutr. 2006, 136, 133–139. [Google Scholar] [CrossRef] [Green Version]
- Plauth, M.; Bernal, W.; Dasarathy, S.; Merli, M.; Plank, L.D.; Schütz, T.; Bischoff, S.C. ESPEN guideline on clinical nutrition in liver disease. Clin. Nutr. 2019, 38, 485–521. [Google Scholar] [CrossRef] [Green Version]
- Young, S.; Kwarta, E.; Azzam, R.; Sentongo, T. Nutrition Assessment and Support in Children with End-Stage Liver Disease. Nutr. Clin. Pract. 2013, 28, 317–329. [Google Scholar] [CrossRef] [PubMed]
- Koletzko, B.; Demmelmair, H.; Socha, P. 3 Nutritional support of infants and children: Supply and metabolism of lipids. Baillieres Clin Gastroenterol. 1998, 12, 671–696. [Google Scholar] [CrossRef]
- Kalivianakis, M.; Minich, D.M.; Havinga, R.; Kuipers, F.; Stellaard, F.; Vonk, R.J.; Verkade, H.J. Detection of impaired intestinal absorption of long-chain fatty acids: Validation studies of a novel test in a rat model of fat malabsorption. Am. J. Clin. Nutr. 2000, 72, 174–180. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Carey, M.C.; Small, D.M.; Bliss, C.M. Lipid Digestion and Absorption. Annu. Rev. Physiol. 1983, 45, 651–677. [Google Scholar] [CrossRef]
- Shah, N.; Limketkai, B. The use of medium chain triglycerides in gastrointestinal disorders. Pract. Gastroenterol. 2017, XLI, 20–28. Available online: https://med.virginia.edu/ginutrition/wp-content/uploads/sites/199/2014/06/Parrish-February-17.pdf (accessed on 12 August 2021).
- Beath, S.V.; Booth, I.W.; Kelly, D.A. Nutritional support in liver disease. Arch. Dis. Child. 1993, 69, 545–547. [Google Scholar] [CrossRef] [Green Version]
- Medium-Chain Triglycerides. Available online: https://www.drugbank.ca/drugs/DB13959 (accessed on 12 August 2021).
- Kelly, D.A.; Davenport, M. Current management of biliary atresia. Arch. Dis. Child. 2007, 92, 1132–1135. [Google Scholar] [CrossRef] [Green Version]
- Koletzko, B.; Agostoni, C.; Carlson, S.; Clandinin, T.; Hornstra, G.; Neuringer, M.; Uauy, R.; Yamashiro, Y.; Willatts, P. Long chain polyunsaturated fatty acids (LC-PUFA) and perinatal development. Acta Paediatr. 2007, 90, 460–464. [Google Scholar] [CrossRef]
- Kaufman, S.S.; Scrivner, D.J.; Murray, N.D.; Vanderhoof, J.A.; Hart, M.H.; Antonson, D.L. Influence of portagen and pregestimil on essential fatty acid status in infantile liver disease. Pediatrics 1992, 89, 151–154. [Google Scholar]
- Macías-Rosales, R.; Larrosa-Haro, A.; Ortíz-Gabriel, G.; Trujillo-Hernandez, B. Effectiveness of Enteral Versus Oral Nutrition with a Medium-Chain Triglyceride Formula to Prevent Malnutrition and Growth Impairment in Infants with Biliary Atresia. J. Pediatr. Gastroenterol. Nutr. 2016, 62, 101–109. [Google Scholar] [CrossRef]
- Kumar, N.G.; Contaifer, D.; Madurantakam, P.; Carbone, S.; Price, E.T.; Van Tassell, B.; Brophy, D.F.; Wijesinghe, D.S. Dietary Bioactive Fatty Acids as Modulators of Immune Function: Implications on Human Health. Nutrients 2019, 11, 2974. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pettei, M.J.; Daftary, S.; Levine, J.J. Essential fatty acid deficiency associated with the use of a medium-chain-triglyceride infant formula in pediatric hepatobiliary disease. Am. J. Clin. Nutr. 1991, 53, 1217–1221. [Google Scholar] [CrossRef] [PubMed]
- Holman, R.T. The Slow Discovery of the Importance of ω3 Essential Fatty Acids in Human Health. J. Nutr. 1998, 128, 427S–433S. [Google Scholar] [CrossRef] [Green Version]
- Pavlovski, C.J. Screening for essential fatty acid deficiency in at risk infants. Med. Hypotheses 2009, 73, 910–916. [Google Scholar] [CrossRef] [PubMed]
- Koletzko, B. Fats for brains. Eur. J. Clin. Nutr. 1992, 46, S51–S62. [Google Scholar]
- Yang, C.H.; Perumpail, B.J.; Yoo, E.R.; Ahmed, A.; Kerner, J.A., Jr. Nutritional Needs and Support for Children with Chronic Liver Disease. Nutrients 2017, 9, 1127. [Google Scholar] [CrossRef]
- Sundaram, S.S.; Mack, C.L.; Feldman, A.G.; Sokol, R.J. Biliary atresia: Indications and timing of liver transplantation and optimization of pretransplant care. Liver Transplant. 2016, 23, 96–109. [Google Scholar] [CrossRef] [Green Version]
- Smart, K.M.; Alex, G.; Hardikar, W. Feeding the child with liver disease: A review and practical clinical guide. J. Gastroenterol. Hepatol. 2011, 26, 810–815. [Google Scholar] [CrossRef]
- Shneider, B.L.; Magee, J.C.; Karpen, S.J.; Rand, E.B.; Narkewicz, M.R.; Bass, L.M.; Schwarz, K.; Whitington, P.F.; Bezerra, J.A.; Kerkar, N.; et al. Total Serum Bilirubin within 3 Months of Hepatoportoenterostomy Predicts Short-Term Outcomes in Biliary Atresia. J. Pediatr. 2015, 170, 211–217. [Google Scholar] [CrossRef] [Green Version]
- Venkat, V.L.; Shneider, B.L.; Magee, J.C.; Turmelle, Y.; Arnon, R.; Bezerra, J.A.; Hertel, P.M.; Karpen, S.J.; Kerkar, N.; Loomes, K.M.; et al. Total Serum Bilirubin Predicts Fat-Soluble Vitamin Deficiency Better Than Serum Bile Acids in Infants with Biliary Atresia. J. Pediatr. Gastroenterol. Nutr. 2014, 59, 702–707. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Samra, N.M.; El Abrak, S.E.; El Dash, H.H.; Raziky, M.E.S.E.; El Sheikh, M.A. Evaluation of vitamin D status bone mineral density and dental health in children with cholestasis. Clin. Res. Hepatol. Gastroenterol. 2018, 42, 368–377. [Google Scholar] [CrossRef] [PubMed]
- Sokol, R.J. Frequency and Clinical Progression of the Vitamin E Deficiency Neurologic Disorder in Children with Prolonged Neonatal Cholestasis. Arch. Pediatr. Adolesc. Med. 1985, 139, 1211–1215. [Google Scholar] [CrossRef] [PubMed]
- Ramaccioni, V.; Soriano, H.E.; Arumugam, R.; Klish, W.J. Nutritional Aspects of Chronic Liver Disease and Liver Transplantation in Children. J. Pediatr. Gastroenterol. Nutr. 2000, 30, 361–367. [Google Scholar] [CrossRef] [PubMed]
- Himoto, T.; Masaki, T. Current Trends of Essential Trace Elements in Patients with Chronic Liver Diseases. Nutrients 2020, 12, 2084. [Google Scholar] [CrossRef]
- Karakochuk, C.D.; Barr, S.I.; Boy, E.; Bahizire, E.; Tugirimana, P.L.; Akilimali, P.Z.; Houghton, L.A.; Green, T.J. The effect of inflammation on serum zinc concentrations and the prevalence estimates of population-level zinc status among Congolese children aged 6–59 months. Eur. J. Clin. Nutr. 2017, 71, 1467–1470. [Google Scholar] [CrossRef]
- Gibson, R.S.; Hess, S.Y.; Hotz, C.; Brown, K.H. Indicators of zinc status at the population level: A review of the evidence. Br. J. Nutr. 2008, 99 (Suppl. 3), S14–S23. [Google Scholar] [CrossRef] [Green Version]
- Göksu, N.; Özsoylu, S. Hepatic and Serum Levels of Zinc, Copper, and Magnesium in Childhood Cirrhosis. J. Pediatr. Gastroenterol. Nutr. 1986, 5, 459–462. [Google Scholar] [CrossRef]
- Argao, E.A.; Balistreri, W.F.; Hollis, B.W.; Ryckman, F.C.; Heubi, J.E. Effect of Orthotopic Liver Transplantation on Bone Mineral Content and Serum Vitamin D Metabolites in Infants and Children with Chronic Cholestasis. Hepatology 1994, 20, 598–603. [Google Scholar] [CrossRef]
- Bonkovsky, H.L.; Banner, B.F.; Lambrecht, R.W.; Rubin, R.B. Iron in Liver Diseases Other than Hemochromatosis. Semin. Liver Dis. 1996, 16, 65–82. [Google Scholar] [CrossRef]
- Burt, A.; Ferrell, L.; Hubscher, S.; Portmann, B. Mac Sween’s Pathology of the Liver, 6th ed.; Churchill Livingstone: Edinburgh, UK, 2012. [Google Scholar]
- Kohut, T.J.; Gilbert, M.A.; Loomes, K.M. Alagille Syndrome: A Focused Review on Clinical Features, Genetics, and Treatment. Semin. Liver Dis. 2021. [Google Scholar] [CrossRef]
- Los, E.L.; Lukovac, S.; Werner, A.; Dijkstra, T.; Verkade, H.J.; Rings, E.H. Nutrition for Children with Cholestatic Liver Disease. Nestle Nutr. Workshop Ser. Pediatr. Program. 2007, 59, 147–159. [Google Scholar] [CrossRef] [PubMed]
- Nastasio, S.; Maggiore, G. Malattie croniche epatobiliari. In Manuale Sigenp di Nutrizione Pediatrica; Catassi, C., Diamanti, A., Agostoni, C., Eds.; Pensiero Scientifico Editore: Roma, Italy, 2016; pp. 251–258. ISBN 9788849005592. [Google Scholar]
- Barshes, N.R.; Chang, I.-F.; Karpen, S.J.; Carter, B.A.; Goss, J.A. Impact of Pretransplant Growth Retardation in Pediatric Liver Transplantation. J. Pediatr. Gastroenterol. Nutr. 2006, 43, 89–94. [Google Scholar] [CrossRef]
- Da Silva, F.V.; Ferri, P.M.; Queiroz, T.C.N.; Barbosa, P.D.S.H.; De Oliveira, M.C.C.; Pereira, L.J.D.M.; e Silva, A.C.S.; Penna, F.J.; Fagundes, E.D.T.; Ferreira, A.R. Nutritional evaluation of children with chronic cholestatic disease. J. Pediatr. 2016, 92, 197–205. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Diamanti, A.; Capriati, T.; Elia, D. Malnutrizione in difetto. In Manuale Sigenp di Gastroenterologia ed Epatologia Pediatrica; Il Pensiero Scientifico Editore: Rome, Italy, 2014; pp. 3–6. [Google Scholar]
- Goulet, O.J.; de Ville de Goyet, J.; Otte, J.B.; Ricour, C. Preoperative Nutritional Evaluation and Support for Liver Transplantation in Children. Transpl. Proc. 1987, 19, 3249–3255. [Google Scholar]
- Yuksekkaya, H.A.; Cakir, M.; Tumgor, G.; Baran, M.; Arikan, C.; Yagci, R.V.; Aydogdu, S. Nutritional status of infants with neonatal cholestasis. Dig. Dis. Sci. 2007, 53, 803–808. [Google Scholar] [CrossRef]
- Trowbridge, F.L.; Sommer, A. Nutritional anthropometry and mortality risk. Am. J. Clin. Nutr. 1981, 34, 2591–2592. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hall, G.; Chowdhury, S.; Bloem, M. Use of mid-upper-arm circumference Z scores in nutritional assessment. Lancet 1993, 341, 1481. [Google Scholar] [CrossRef]
- Zemel, B.S.; Riley, E.M.; Stallings, V.A. EVALUATION OF METHODOLOGY FOR NUTRITIONAL ASSESSMENT IN CHILDREN: Anthropometry, Body Composition, and Energy Expenditure. Annu. Rev. Nutr. 1997, 17, 211–235. [Google Scholar] [CrossRef]
- Sokol, R.J.; Stall, C. Anthropometric evaluation of children with chronic liver disease. Am. J. Clin. Nutr. 1990, 52, 203–208. [Google Scholar] [CrossRef] [PubMed]
- Wasserman, D.; Zemel, B.S.; Mulberg, A.E.; John, H.A.; Emerick, K.M.; Barden, E.M.; Piccoli, D.A.; Stallings, V.A. Growth, nutritional status, body composition, and energy expenditure in prepubertal children with Alagille syndrome. J. Pediatr. 1999, 134, 172–177. [Google Scholar] [CrossRef]
- Taylor, R.M.; Dhawan, A. Assessing nutritional status in children with chronic liver disease. J. Gastroenterol. Hepatol. 2005, 20, 1817–1824. [Google Scholar] [CrossRef]
- Klein, S. The myth of serum albumin as a measure of nutritional status. Gastroenterology 1990, 99, 1845–1846. [Google Scholar] [CrossRef]
- Beck, F.K.; Rosenthal, T.C. Prealbumin: A marker for nutritional evaluation. Am. Fam. Physician. 2002, 65, 1575–1578. [Google Scholar]
- Mouzaki, M.; Ng, V.; Kamath, B.M.; Selzner, N.; Pencharz, P.; Ling, S. Enteral Energy and Macronutrients in End-Stage Liver Disease. J. Parenter. Enter. Nutr. 2014, 38, 673–681. [Google Scholar] [CrossRef] [PubMed]
- Houchin, M.; Kamat, D.; Leibbrandt, C.; Hind, J.; Dhawan, A. Safety and efficacy of Heparon Junior in infants with cholestatic liver disease. J. Pediatr. Gastroenterol. Nutr. 2010, 50, E53. [Google Scholar] [CrossRef]
- Broekaert, I.J.; Falconer, J.; Bronsky, J.; Gottrand, F.; Dall’Oglio, L.; Goto, E.; Hojsak, I.; Hulst, J.; Kochavi, B.; Papadopoulou, A.; et al. The Use of Jejunal Tube Feeding in Children: A Position Paper by the Gastroenterology and Nutrition Committees of the European Society for Paediatric Gastroenterology, Hepatology, and Nutrition 2019. J. Pediatr. Gastroenterol. Nutr. 2019, 69, 239–258. [Google Scholar] [CrossRef]
- Duché, M.; Habès, D.; Lababidi, A.; Chardot, C.; Wenz, J.; Bernard, O. Percutaneous Endoscopic Gastrostomy for Continuous Feeding in Children with Chronic Cholestasis. J. Pediatr. Gastroenterol. Nutr. 1999, 29, 42–45. [Google Scholar] [CrossRef]
- Heuschkel, R.; Gottrand, F.; Devarajan, K.; Poole, H.; Callan, J.; Dias, J.; Karkelis, S.; Papadopoulou, A.; Husby, S.; Ruemmele, F.; et al. ESPGHAN Position Paper on Management of Percutaneous Endoscopic Gastrostomy in Children and Adolescents. J. Pediatr. Gastroenterol. Nutr. 2015, 60, 131–141. [Google Scholar] [CrossRef]
- Burgmaier, K.; Brandt, J.; Shroff, R.; Witters, P.; Weber, L.T.; Dötsch, J.; Schaefer, F.; Mekahli, D.; Liebau, M.C. Gastrostomy Tube Insertion in Pediatric Patients with Autosomal Recessive Polycystic Kidney Disease (ARPKD): Current Practice. Front. Pediatr. 2018, 6, 164. [Google Scholar] [CrossRef] [PubMed]
- Protheroe, S.M. Feeding the child with chronic liver disease. Nutrition 1998, 14, 796–800. [Google Scholar] [CrossRef]
- Cortez, A.R.; Warren, P.W.; Goddard, G.R.; Jenkins, T.M.; Sauser, J.A.; Gerrein, B.T.; Rymeski, B.A. Primary Placement of a Low-Profile Gastrostomy Button Is Safe and Associated with Improved Outcomes in Children. J. Surg. Res. 2020, 249, 156–162. [Google Scholar] [CrossRef] [PubMed]
- Kelly, D.A.; Tong, C. Neonatal and paediatric infection. In Viral Hepatitis, 4th ed.; Thomas, H.C., Lok, A.S., Locarnini, S.A., Arie JZuckerman, A.J., Eds.; Wiley Blackwell: Hoboken, NJ, USA, 2012; pp. 529–543. [Google Scholar]
- Orso, G.; Mandato, C.; Veropalumbo, C.; Cecchi, N.; Garzi, A.; Vajro, P. Pediatric parenteral nutrition-associated liver disease and cholestasis: Novel advances in pathomechanisms-based prevention and treatment. Dig. Liver Dis. 2015, 48, 215–222. [Google Scholar] [CrossRef] [PubMed]
- Cahova, M.; Bratova, M.; Wohl, P. Parenteral Nutrition-Associated Liver Disease: The Role of the Gut Microbiota. Nutrients 2017, 9, 987. [Google Scholar] [CrossRef] [Green Version]
- Lindor, K.D.; Burnes, J. Ursodeoxycholic acid for the treatment of home parenteral nutrition-associated cholestasis. Gastroenterology 1991, 101, 250–253. [Google Scholar] [CrossRef]
- Spagnuolo, M.I.; Iorio, R.; Vegnente, A.; Guarino, A. Ursodeoxycholic acid for treatment of cholestasis in children on long- term total parenteral nutrition: A pilot study. Gastroenterology 1996, 111, 716–719. [Google Scholar] [CrossRef]
- Sullivan, J.S.; Sundaram, S.S.; Pan, Z.; Sokol, R.J. Parenteral nutrition supplementation in biliary atresia patients listed for liver transplantation. Liver Transplant. 2011, 18, 120–128. [Google Scholar] [CrossRef] [Green Version]
- Rochling, F. Intravenous Lipid Emulsions in the Prevention and Treatment of Liver Disease in Intestinal Failure. Nutrients 2021, 13, 895. [Google Scholar] [CrossRef]
- Wendel, D.; Mortensen, M.; Harmeson, A.; Shaffer, M.L.; Hsu, E.; Horslen, S. Resolving Malnutrition With Parenteral Nutrition Before Liver Transplant in Biliary Atresia. J. Pediatr. Gastroenterol. Nutr. 2018, 66, 212–217. [Google Scholar] [CrossRef] [PubMed]
- Holt, R.I.; Broide, E.; Buchanan, C.R.; Miell, J.P.; Baker, A.J.; Mowat, A.P.; Mieli-Vergani, G. Orthotopic liver transplantation reverses the adverse nutritional changes of end-stage liver disease in children. Am. J. Clin. Nutr. 1997, 65, 534–542. [Google Scholar] [CrossRef] [Green Version]
- Colomb, V.; Goulet, O.; Ricour, C. 12 Home enteral and parenteral nutrition in children. Baillieres Clin. Gastroenterol. 1998, 12, 877–894. [Google Scholar] [CrossRef]
- Diamanti, A.; Calvitti, G.; Martinelli, D.; Santariga, E.; Capriati, T.; Bolasco, G.; Iughetti, L.; Pujia, A.; Knafelz, D.; Maggiore, G. Etiology and Management of Pediatric Intestinal Failure: Focus on the Non-Digestive Causes. Nutrients 2021, 13, 786. [Google Scholar] [CrossRef]
- Van Ginkel, W.G.; Van Reemst, H.E.; Kienstra, N.S.; Daly, A.; Rodenburg, I.L.; Macdonald, A.; Burgerhof, J.G.; De Blaauw, P.; Van De Krogt, J.; Santra, S.; et al. The Effect of Various Doses of Phenylalanine Supplementation on Blood Phenylalanine and Tyrosine Concentrations in Tyrosinemia Type 1 Patients. Nutrients 2019, 11, 2816. [Google Scholar] [CrossRef] [Green Version]
- Van Vliet, K.; Rodenburg, I.L.; Van Ginkel, W.G.; Lubout, C.M.; Wolffenbuttel, B.H.; Van Der Klauw, M.M.; Heiner-Fokkema, M.R.; Van Spronsen, F.J. Biomarkers of Micronutrients in Regular Follow-Up for Tyrosinemia Type 1 and Phenylketonuria Patients. Nutrients 2019, 11, 2011. [Google Scholar] [CrossRef] [Green Version]
- Van Spronsen, F.J.; Van Rijn, M.; Meyer, U.; Das, A.M. Dietary Considerations in Tyrosinemia Type I. Adv. Exp. Med. Biol. 2017, 959, 197–204. [Google Scholar] [CrossRef]
- Berry, G.T.; Walter, J.H. Disorders of galactose metabolism. In Inborn Metabolic Diseases: Diagnosis and Treatment; Saudubray, J.M., van den Berghe, G., Walter, J.H., Eds.; Springer: Berlin/Heidelberg, Germany, 2012. [Google Scholar] [CrossRef]
- Bosch, A.M. Classic galactosemia: Dietary dilemmas. J. Inherit. Metab. Dis. 2010, 34, 257–260. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Welling, L.; Bernstein, L.E.; Berry, G.T.; Burlina, A.B.; Eyskens, F.; Gautschi, M.; Grünewald, S.; Gubbels, C.S.; Knerr, I.; Labrune, P.; et al. International clinical guideline for the management of classical galactosemia: Diagnosis, treatment, and follow-up. J. Inherit. Metab. Dis. 2016, 40, 171–176. [Google Scholar] [CrossRef] [Green Version]
- Coelho, A.I.; Rubio-Gozalbo, M.E.; Vicente, J.; Rivera, I. Sweet and sour: An update on classic galactosemia. J. Inherit. Metab. Dis. 2017, 40, 325–342. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Batey, L.A.; Welt, C.K.; Rohr, F.; Wessel, A.; Anastasoaie, V.; Feldman, H.A.; Guo, C.-Y.; Rubio-Gozalbo, E.; Berry, G.; Gordon, C.M. Skeletal health in adult patients with classic galactosemia. Osteoporos. Int. 2012, 24, 501–509. [Google Scholar] [CrossRef] [PubMed]
- Ali, M.; Rellos, P.; Cox, T.M. Hereditary fructose intolerance. J. Med Genet. 1998, 35, 353–365. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vajro, P.; Veropalumbo, C. Citrin Deficiency: Learn More, and Don’t Forget to Add It to the List of Neonatal Cholestasis and the NASH Trash Bin. J. Pediatr. Gastroenterol. Nutr. 2010, 50, 578–579. [Google Scholar] [CrossRef]
- Pinto, A.; Ashmore, C.; Batzios, S.; Daly, A.; Dawson, C.; Dixon, M.; Evans, S.; Green, D.; Gribben, J.; Hunjan, I.; et al. Dietary Management, Clinical Status and Outcome of Patients with Citrin Deficiency in the UK. Nutrients 2020, 12, 3313. [Google Scholar] [CrossRef] [PubMed]
- Okano, Y.; Ohura, T.; Sakamoto, O.; Inui, A. Current treatment for citrin deficiency during NICCD and adaptation/compensation stages: Strategy to prevent CTLN2. Mol. Genet. Metab. 2019, 127, 175–183. [Google Scholar] [CrossRef]
- Hayasaka, K.; Numakura, C. Adult-onset type II citrullinemia: Current insights and therapy. Appl. Clin. Genet. 2018, 11, 163–170. [Google Scholar] [CrossRef] [Green Version]
- Okano, Y.; Okamoto, M.; Yazaki, M.; Inui, A.; Ohura, T.; Murayama, K.; Watanabe, Y.; Tokuhara, D.; Takeshima, Y. Analysis of daily energy, protein, fat, and carbohydrate intake in citrin-deficient patients: Towards prevention of adult-onset type II citrullinemia. Mol. Genet. Metab. 2021, 133, 63–70. [Google Scholar] [CrossRef]
- Hayasaka, K.; Numakura, C.; Toyota, K.; Kimura, T. Treatment with Lactose (Galactose)-Restricted and Medium-Chain Triglyceride-Supplemented Formula for Neonatal Intrahepatic Cholestasis Caused by Citrin Deficiency. JIMD Rep. 2011, 2, 37–44. [Google Scholar] [CrossRef] [Green Version]
- Inui, A.; Hashimoto, T.; Sogo, T.; Komatsu, H.; Saheki, T.; Fujisawa, T. Chronic hepatitis without hepatic steatosis caused by citrin deficiency in a child. Hepatol. Res. 2015, 46, 357–362. [Google Scholar] [CrossRef]
- Mutoh, K.; Kurokawa, K.; Kobayashi, K.; Saheki, T. Treatment of a citrin-deficient patient at the early stage of adult-onset type II citrullinaemia with arginine and sodium pyruvate. J. Inherit. Metab. Dis. 2008, 31 (Suppl. 2), 343–347. [Google Scholar] [CrossRef]
- Figueiredo, F.; Dickson, E.R.; Pasha, T.; Kasparova, P.; Therneau, T.; Malinchoc, M.; DiCecco, S.; Francisco-Ziller, N.; Charlton, M. Impact of nutritional status on outcomes after liver transplantation1. Transplantation 2000, 70, 1347–1352. [Google Scholar] [CrossRef]
- Hammad, A.; Kaido, T.; Aliyev, V.; Mandato, C.; Uemoto, S. Nutritional Therapy in Liver Transplantation. Nutrients 2017, 9, 1126. [Google Scholar] [CrossRef]
- McDiarmid, S.V.; Anand, R.; Lindblad, A.S. Development of a pediatric end-stage liver disease score to predict poor outcome in children awaiting liver transplantation1. Transplantation 2002, 74, 173–181. [Google Scholar] [CrossRef] [PubMed]
- Mohammad, S.; Grimberg, A.; Rand, E.; Anand, R.; Yin, W.; Alonso, E.M. Long-Term Linear Growth and Puberty in Pediatric Liver Transplant Recipients. J. Pediatr. 2013, 163, 1354–1360. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Almaas, R.; Haflidadottir, S.; Kaldestad, R.H.; Matthews, I.L. Asthma, Eczema, and Food Allergy in Children Following Liver Transplantation. J. Pediatr. 2019, 204, 263–269. [Google Scholar] [CrossRef] [PubMed]
- Valta, H.; Jalanko, H.; Holmberg, C.; Helenius, I.; Mäkitie, O. Impaired Bone Health in Adolescents After Liver Transplantation. Arab. Archaeol. Epigr. 2007, 8, 150–157. [Google Scholar] [CrossRef]
- Ng, V.L.; Alonso, E.M.; Bucuvalas, J.C.; Cohen, G.; Limbers, C.A.; Varni, J.W.; Mazariegos, G.; Magee, J.; McDiarmid, S.V.; Anand, R. Health Status of Children Alive 10 Years after Pediatric Liver Transplantation Performed in the US and Canada: Report of the Studies of Pediatric Liver Transplantation Experience. J. Pediatr. 2011, 160, 820–826. [Google Scholar] [CrossRef] [Green Version]
- Saito, T.; Mizuta, K.; Hishikawa, S.; Kawano, Y.; Sanada, Y.; Fujiwara, T.; Yasuda, Y.; Sugimoto, K.; Sakamoto, K.; Kawarasaki, H. Growth curves of pediatric patients with biliary atresia following living donor liver transplantation: Factors that influence post-transplantation growth. Pediatr. Transplant. 2007, 11, 764–770. [Google Scholar] [CrossRef]
- McDiarmid, S.V.; Gornbein, J.A.; DeSilva, P.J.; Goss, J.A.; Vargas, J.H.; Martín, M.G.; Ament, M.E.; Busuttil, R.W. Factors affecting growth after pediatric liver transplantation. Transplantation 1999, 67, 404–411. [Google Scholar] [CrossRef]
- Swenson, S.M.; Perito, E.R. Weight Gain Trajectory Predicts Long-term Overweight and Obesity After Pediatric Liver Transplant. J. Pediatr. Gastroenterol. Nutr. 2019, 68, 89–95. [Google Scholar] [CrossRef]
- Nair, S.; Verma, S.; Thuluvath, P.J. Obesity and its effect on survival in patients undergoing orthotopic liver transplantation in the United States. Hepatology 2002, 35, 105–109. [Google Scholar] [CrossRef]
- Conzen, K.D.; Vachharajani, N.; Collins, K.M.; Anderson, C.D.; Lin, Y.; Wellen, J.R.; Shenoy, S.; Lowell, J.A.; Doyle, M.B.M.; Chapman, W.C. Morbid obesity in liver transplant recipients adversely affects longterm graft and patient survival in a single-institution analysis. HPB 2015, 17, 251–257. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fouad, Y.; Waked, I.; Bollipo, S.; Gomaa, A.; Ajlouni, Y.; Attia, D. What’s in a name? Renaming ‘NAFLD’ to ‘MAFLD’. Liver Int. 2020, 40, 1254–1261. [Google Scholar] [CrossRef] [Green Version]
- Prasad, G.V.R.; Huang, M.; Silver, S.; Al-Lawati, A.I.; Rapi, L.; Nash, M.M.; Zaltzman, J.S. Metabolic syndrome definitions and components in predicting major adverse cardiovascular events after kidney transplantation. Transpl. Int. 2014, 28, 79–88. [Google Scholar] [CrossRef]
- Thoefner, L.B.; Rostved, A.A.; Pommergaard, H.-C.; Rasmussen, A. Risk factors for metabolic syndrome after liver transplantation: A systematic review and meta-analysis. Transplant. Rev. 2018, 32, 69–77. [Google Scholar] [CrossRef] [PubMed]
- Perito, E.R.; Glidden, D.; Roberts, J.P.; Rosenthal, P. Overweight and obesity in pediatric liver transplant recipients: Prevalence and predictors before and after transplant, United Network for Organ Sharing Data, 1987–2010. Pediatr. Transplant. 2011, 16, 41–49. [Google Scholar] [CrossRef] [Green Version]
- Nobili, V.; Goyet, J.D.V.D. Pediatric post-transplant metabolic syndrome: New clouds on the horizon. Pediatr. Transplant. 2013, 17, 216–223. [Google Scholar] [CrossRef] [PubMed]
- Högler, W.; Baumann, U.; Kelly, D. Endocrine and Bone Metabolic Complications in Chronic Liver Disease and After Liver Transplantation in Children. J. Pediatr. Gastroenterol. Nutr. 2012, 54, 313–321. [Google Scholar] [CrossRef]
- Seo, S.; Maganti, K.; Khehra, M.; Ramsamooj, R.; Tsodikov, A.; Bowlus, C.; McVicar, J.; Zern, M.; Torok, N. De novo nonalcoholic fatty liver disease after liver transplantation. Liver Transplant. 2007, 13, 844–847. [Google Scholar] [CrossRef]
- Heisel, O.; Heisel, R.; Balshaw, R.; Keown, P. New Onset Diabetes Mellitus in Patients Receiving Calcineurin Inhibitors: A Systematic Review and Meta-Analysis. Arab. Archaeol. Epigr. 2004, 4, 583–595. [Google Scholar] [CrossRef]
- Perito, E.R.; Lustig, R.H.; Rosenthal, P. Metabolic Syndrome Components After Pediatric Liver Transplantation: Prevalence and the Impact of Obesity and Immunosuppression. Arab. Archaeol. Epigr. 2016, 16, 1909–1916. [Google Scholar] [CrossRef] [Green Version]
- Di Cosmo, N.; Vajro, P.; Debray, D.; Valerio, G.; Giugliano, M.; Buono, P.; Franzese, A. Normal -Cell Function in Post-Liver Transplantation Diabetes Treated with Tacrolimus. Diabetes Care 2004, 27, 1837–1838. [Google Scholar] [CrossRef] [Green Version]
- Watt, K.D. Metabolic syndrome. Is immunosuppression to blame? Liver Transpl. 2011, 17 (Suppl. 3), S38–S42. [Google Scholar] [CrossRef] [PubMed]
- Vajro, P.; Pecoraro, C.; De Vincenzo, A.; Genovese, E.; Silvestre, C.; Brancato, T.; Migliaro, F. Monitoring of renal function in children before and after liver transplantation. Transplant. Proc. 1998, 30, 1991–1992. [Google Scholar] [CrossRef]
- Munoz, S.J. Hyperlipidemia and other coronary risk factors after orthotopic liver transplantation: Pathogenesis, diagnosis and management. Liver Transpl. Surg. 1995, 1 (Suppl. 1), 29–38. [Google Scholar] [PubMed]
- Galvin, Z.; Rajakumar, R.; Chen, E.; Adeyi, O.; Selzner, M.; Grant, D.; Sapisochin, G.; Greig, P.; Cattral, M.; McGilvray, I.; et al. Predictors of De Novo Nonalcoholic Fatty Liver Disease After Liver Transplantation and Associated Fibrosis. Liver Transplant. 2019, 25, 56–67. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kelly, D.A.; Bucuvalas, J.C.; Alonso, E.M.; Karpen, S.J.; Allen, U.; Green, M.; Farmer, D.; Shemesh, E.; McDonald, R.A. Long-term medical management of the pediatric patient after liver transplantation: 2013 practice guideline by the American Association for the Study of Liver Diseases and the American Society of Transplantation. Liver Transplant. 2013, 19, 798–825. [Google Scholar] [CrossRef]
- Hilk, K.; Zerofsky, M.; Rhee, S.; Rosenthal, P.; Perito, E.R. Center variation in screening for and management of metabolic syndrome in pediatric liver transplant recipients: A survey of SPLIT centers. Pediatr. Transplant. 2019, 23, e13347. [Google Scholar] [CrossRef] [PubMed]
- Mangus, R.; Bush, W.J.; Miller, C.; Kubal, C. Severe Sarcopenia and Increased Fat Stores in Pediatric Patients with Liver, Kidney, or Intestine Failure. J. Pediatr. Gastroenterol. Nutr. 2017, 65, 579–583. [Google Scholar] [CrossRef] [PubMed]
- Lurz, E.; Patel, H.; Frimpong, R.G.; Ricciuto, A.; Kehar, M.; Wales, P.W.; Towbin, A.J.; Chavhan, G.B.; Kamath, B.M.; Ng, V.L. Sarcopenia in patients with end-stage liver disease. J. Pediatr Gastroenterol Nutr. 2018, 66, 222–226. [Google Scholar] [CrossRef]
- Mager, D.R.; Hager, A.; Ooi, P.H.; Siminoski, K.; Gilmour, S.M.; Yap, J.Y. Persistence of Sarcopenia After Pediatric Liver Transplantation Is Associated with Poorer Growth and Recurrent Hospital Admissions. J. Parenter. Enter. Nutr. 2018, 43, 271–280. [Google Scholar] [CrossRef] [PubMed]
- Rezende, I.F.B.; Conceição-Machado, M.E.P.; Souza, V.S.; Dos Santos, E.M.; Silva, L.R. Sarcopenia in children and adolescents with chronic liver disease. J. Pediatr. 2019, 96, 439–446. [Google Scholar] [CrossRef] [PubMed]
- Merli, M. Pediatric sarcopenia: Exploring a new concept in children with chronic liver disease. J. Pediatr. 2019, 96, 406–408. [Google Scholar] [CrossRef] [PubMed]
- Meyer, F.; Bannert, K.; Wiese, M.; Esau, S.; Sautter, L.F.; Ehlers, L.; Aghdassi, A.A.; Metges, C.C.; Garbe, L.-A.; Jaster, R.; et al. Molecular Mechanism Contributing to Malnutrition and Sarcopenia in Patients with Liver Cirrhosis. Int. J. Mol. Sci. 2020, 21, 5357. [Google Scholar] [CrossRef]
- Verhagen, M.V.; Levolger, S.; Hulshoff, J.B.; Werner, M.J.; van der Doef, H.P.; Viddeleer, A.R.; de Kleine, R.H.; de Haas, R.J. Utility of preoperative CT-based body metrics in relation to postoperative complications in pediatric liver transplant recipients. Liver Transplant. 2021. [Google Scholar] [CrossRef]
- Jitwongwai, S.; Lertudomphonwanit, C.; Junhasavasdikul, T.; Fuangfa, P.; Tanpowpong, P.; Gesprasert, G.; Treepongkaruna, S. Low psoas muscle index as an unfavorable factor in children with end-stage liver disease undergoing liver transplantation. Pediatr. Transplant. 2021, 25, e13996. [Google Scholar] [CrossRef]
- Boster, J.M.; Browne, L.P.; Pan, Z.; Zhou, W.; Ehrlich, P.F.; Sundaram, S.S. Higher Mortality in Pediatric Liver Transplant Candidates with Sarcopenia. Liver Transplant. 2021, 27, 808–817. [Google Scholar] [CrossRef]
- Woolfson, J.P.; Perez, M.; Chavhan, G.B.; Johara, F.T.; Lurz, E.; Kamath, B.M.; Ng, V.L. Sarcopenia in Children with End-Stage Liver Disease on the Transplant Waiting List. Liver Transplant. 2021, 27, 641–651. [Google Scholar] [CrossRef]
- Cruz-Jentoft, A.J.; Baeyens, J.P.; Bauer, J.M.; Boirie, Y.; Cederholm, T.; Landi, F.; Martin, F.C.; Michel, J.-P.; Rolland, Y.; Schneider, S.; et al. Sarcopenia: European consensus on definition and diagnosis: Report of the European Working Group on Sarcopenia in Older People. Age Ageing 2010, 39, 412–423. [Google Scholar] [CrossRef] [Green Version]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Age | Males | Females |
|---|---|---|
| 3–10 years | 22.7 (Weight) + 495 | 22.5 (Weight) + 499 |
| 10–18 years | 17.5 (Weight) + 651 | 12.2 (Weight) + 746 |
| Vit | Signs and Symptoms of Deficiency | How to Monitor | Supplementation | Toxicity |
|---|---|---|---|---|
| A | Dry skin Xerophthalmia Night blindness. | Plasma retinol/retinol binding protein molar ratio > 0.8 relative dose response When serum retinol levels < 20 µg/dl, RDR test is indicative of Vit A deficiency when the plasma retinol concentration increases after exogenous administration of Vit A dose. | 3000–10,000 IU/day <10 kg: 5000 UI/day, Oral >10 kg: 10,000 UI/day, Oral 50,000 UI/1–3 monthly, IM | Hepatic and neurologic toxicity Development of long bone fractures |
| D | Hypocalcemia/hypophosphatemia/tetany Osteomalacia and rickets History of reduced intake, decreased cutaneous synthesis, altered absorption/impaired metabolism in the liver (i.e., where Vit D2 and D3 undergo 25-hydroxylation). Phenobarbital treatment. Breastfeeding | Serum 25-OH-D (Vit D deficiency < 20 ng/mL; insufficiency < 30 ng/mL) Ca, P, AP, PTH, Bone radiography/Bone densitometry used to identify osteomalacia, osteopenia or rickets | Cholecalciferol: 800–5000 IU/day, Oral 1.25-OH cholecalciferol: 0.05–0.2 µg/kg/day, Oral | Hypercalcemia leading to depression of the central nervous system and ectopic calcification. Hypercalciuria leading to nephrocalcinosis |
| E | Hypo- or a-reflexia Ataxia Impaired vibratory sensation Proximal muscle weakness Ophthalmoplegia Degenerative lesions of the retina Irreversible neurological lesions if Vit E deficiency remains untreated | Vit E/total lipids ratio (increased lipoprotein levels in cholestasis may falsely elevate serum Vit E levels in a patient with Vit E deficiency) Vit E deficiency: <0.6 mg/g (age <1 year) <0.8 mg/g (age >1 year) | Alpha-tocopherol acetate: 15–25 to 25–200 UI/kg/day, Oral TPGS (tocopheryl polyethylene glycol-1000 succinate): 15–25 UI/Kg, Oral | Potentiation of Vit K deficiency coagulopathy Diarrhea Hyperosmolality (TPGS) |
| K | Hemorrhagic disease (other risks of bleeding: portal hypertension gastrointestinal bleeds, thrombocytopenia, platelet dysfunction, reduced hepatic synthesis of other coagulation factors | Prothrombin time International normalized ratio Protein induced in Vit K absence II (PIVKA II) <3 ng/mL Deficiency can be diagnosed if these values improve after a dose of parenteral vitamin K | 2.5–5.0 mg/day from twice a week to every day 5–10 kg: 5 mg, oral >10 kg: 10 mg, oral 5–10 mg/day every two weeks, IM | Hemolytic anemia in glucose 6-phosphate dehydrogenase-deficient infants |
| Before Liver Transplant | After Liver Transplant |
|---|---|
| Energy intake 130–150% EAR | Energy intake 120% EAR |
| Carbohydrates (40% to 60% of total energy) 15–20 g/kg/day as monomers, polymers, and starch Balancing the hypoglycemia from end-stage liver disease and hyperglycemia from insulin resistance | Carbohydrates 6–8 g/kg/day as monomers, polymers, and starch Warning: Consider the diabetogenic potential of tacrolimus when it is used for immunosuppression |
| Proteins (9% of total energy) 3–4 g/kg/day BCAA-enriched formula can be used (10% of total amino acid) Low protein-diet is needed only when severe encephalopathy is present. Once encephalopathy is resolved, the patient should resume a diet with appropriate protein supply because long-term restriction <2 g/kg/day can induce endogenous muscle protein consumption | Proteins 2.5–3 g/kg/day |
| Fats (40% of total energy; 10% of which as LCPUFA) 8 g/kg/day with 30–50% as MCTs. Warning: MCT contents >80% without adequate supplementation of PUFA can lead to a deficiency of essential fatty acids | Fats 5–6 g/kg/day After liver transplantation, when bile flow is established and malabsorption is resolved, children fed with high MCT-containing supplementation pre-transplant can transition to standard formula |
| Fluids and electrolytes Fluid requirement is normal for actual weight, unless restriction is needed because of ascites or edema. Sodium intake is 1 mmol/kg/day and potassium about 2 mmol/kg/day | Fluids and electrolytes A “no added salt” diet (3 g sodium/day) is recommended to prevent water retention associated with steroid therapy |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Tessitore, M.; Sorrentino, E.; Schiano Di Cola, G.; Colucci, A.; Vajro, P.; Mandato, C. Malnutrition in Pediatric Chronic Cholestatic Disease: An Up-to-Date Overview. Nutrients 2021, 13, 2785. https://doi.org/10.3390/nu13082785
Tessitore M, Sorrentino E, Schiano Di Cola G, Colucci A, Vajro P, Mandato C. Malnutrition in Pediatric Chronic Cholestatic Disease: An Up-to-Date Overview. Nutrients. 2021; 13(8):2785. https://doi.org/10.3390/nu13082785
Chicago/Turabian StyleTessitore, Maria, Eduardo Sorrentino, Giuseppe Schiano Di Cola, Angelo Colucci, Pietro Vajro, and Claudia Mandato. 2021. "Malnutrition in Pediatric Chronic Cholestatic Disease: An Up-to-Date Overview" Nutrients 13, no. 8: 2785. https://doi.org/10.3390/nu13082785
APA StyleTessitore, M., Sorrentino, E., Schiano Di Cola, G., Colucci, A., Vajro, P., & Mandato, C. (2021). Malnutrition in Pediatric Chronic Cholestatic Disease: An Up-to-Date Overview. Nutrients, 13(8), 2785. https://doi.org/10.3390/nu13082785