
High-Fat and Resveratrol Supplemented Diets Modulate Adenosine Receptors in the Cerebral Cortex of C57BL/6J and SAMP8 Mice

 , , , and
, , , and
Abstract
:
1. Introduction
2. Materials and Methods
2.1. Animals and Diets
2.2. Plasma Membrane Isolation
2.3. 5′-Nucleotidase Activity Assay
2.4. Adenosine Receptors Quantification by Western Blotting Assay
2.5. Radioligand Binding Assays
2.6. Free Cholesterol Quantification
2.7. Statistical and Data Analysis
3. Results
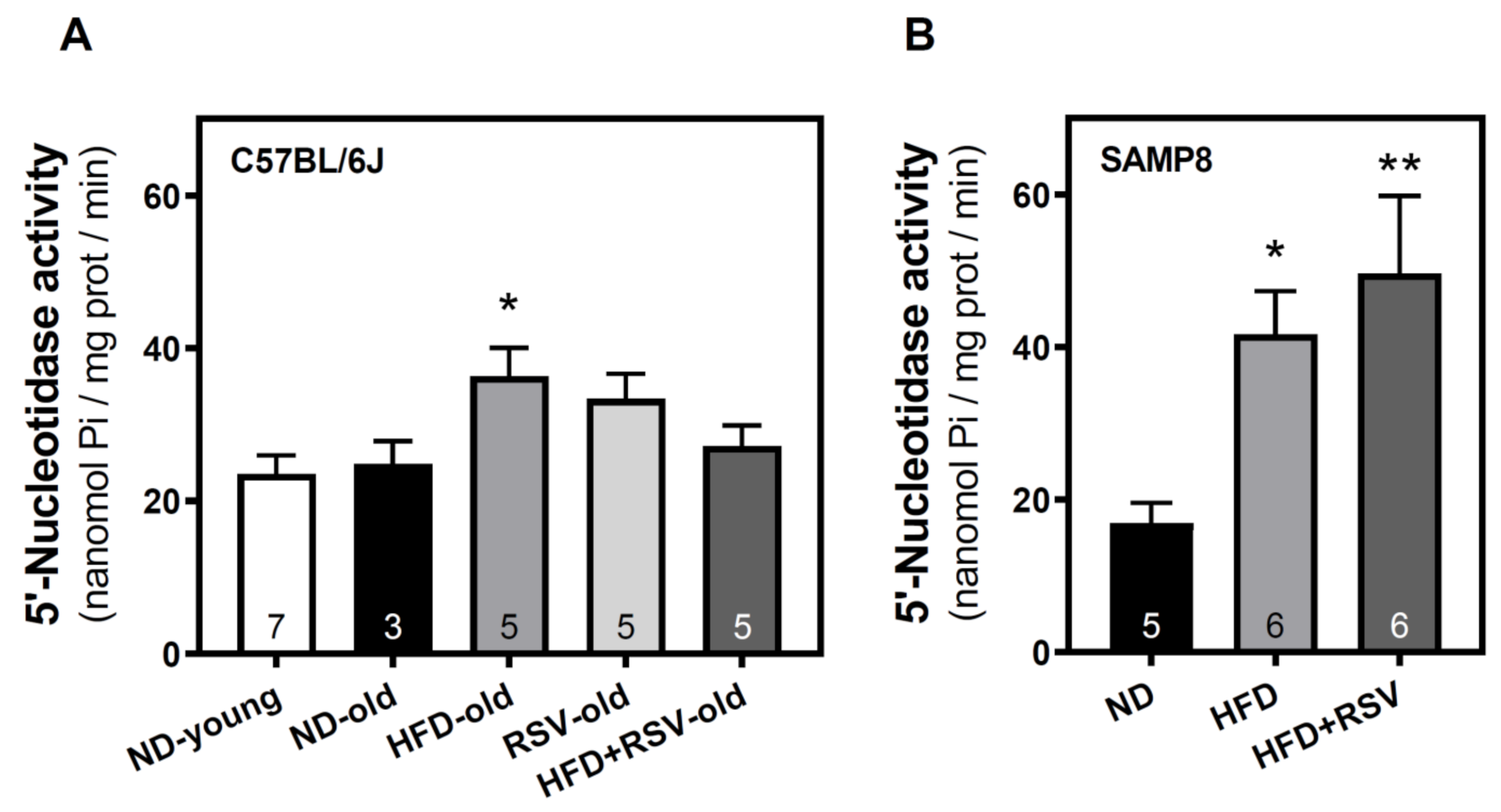
3.1. 5′-Nucleotidase Activity in the Cerebral Cortex
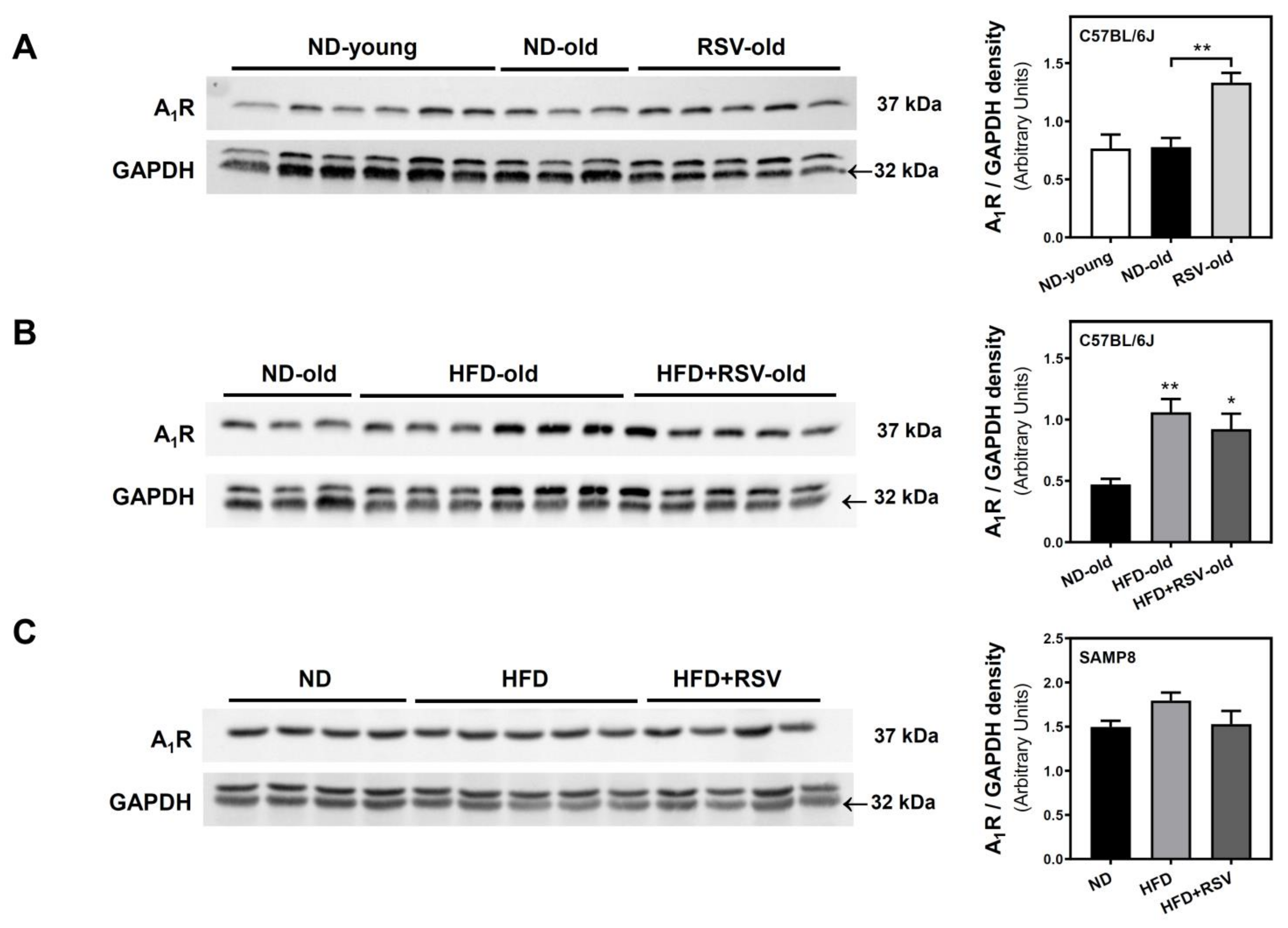
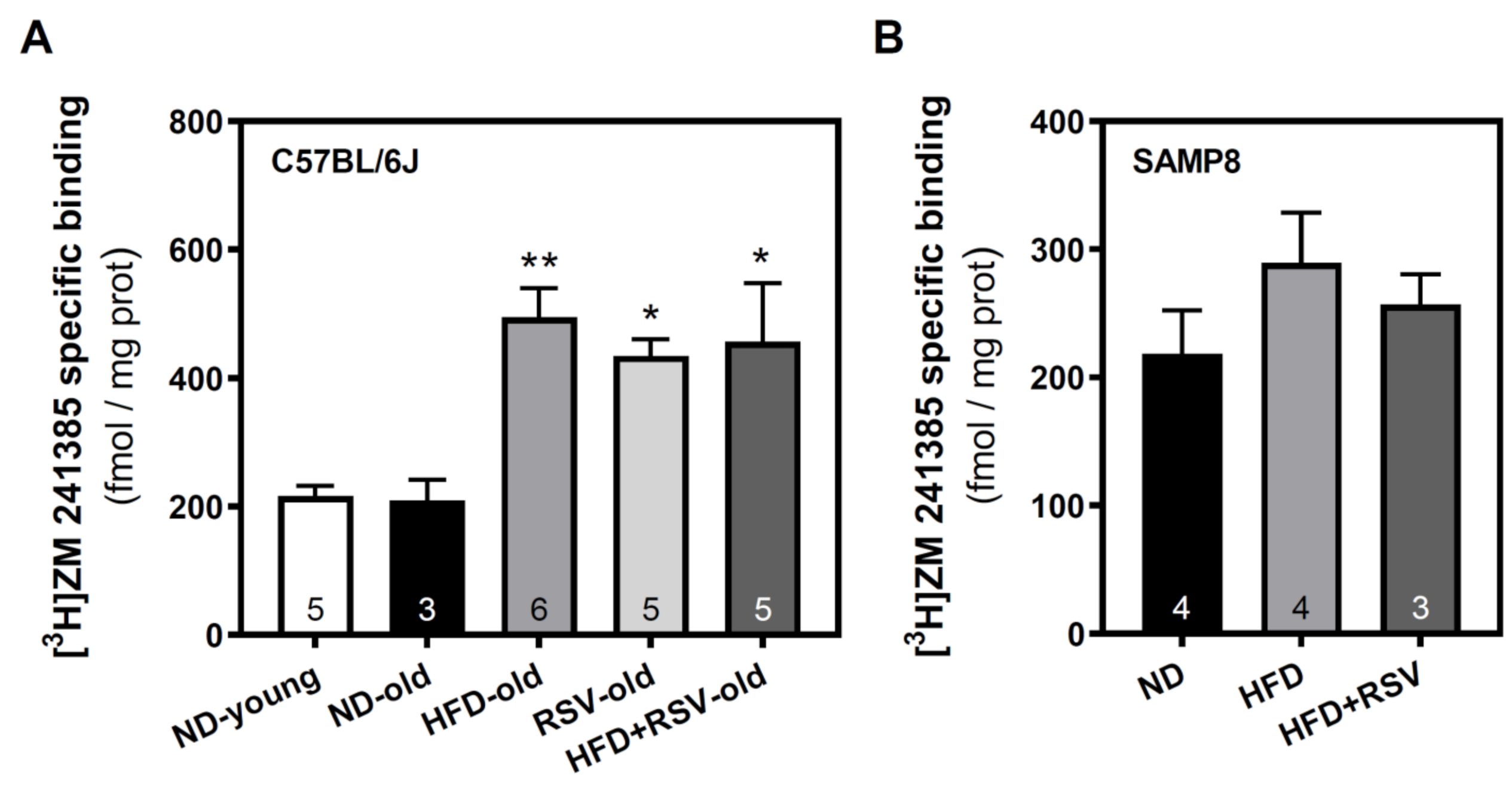
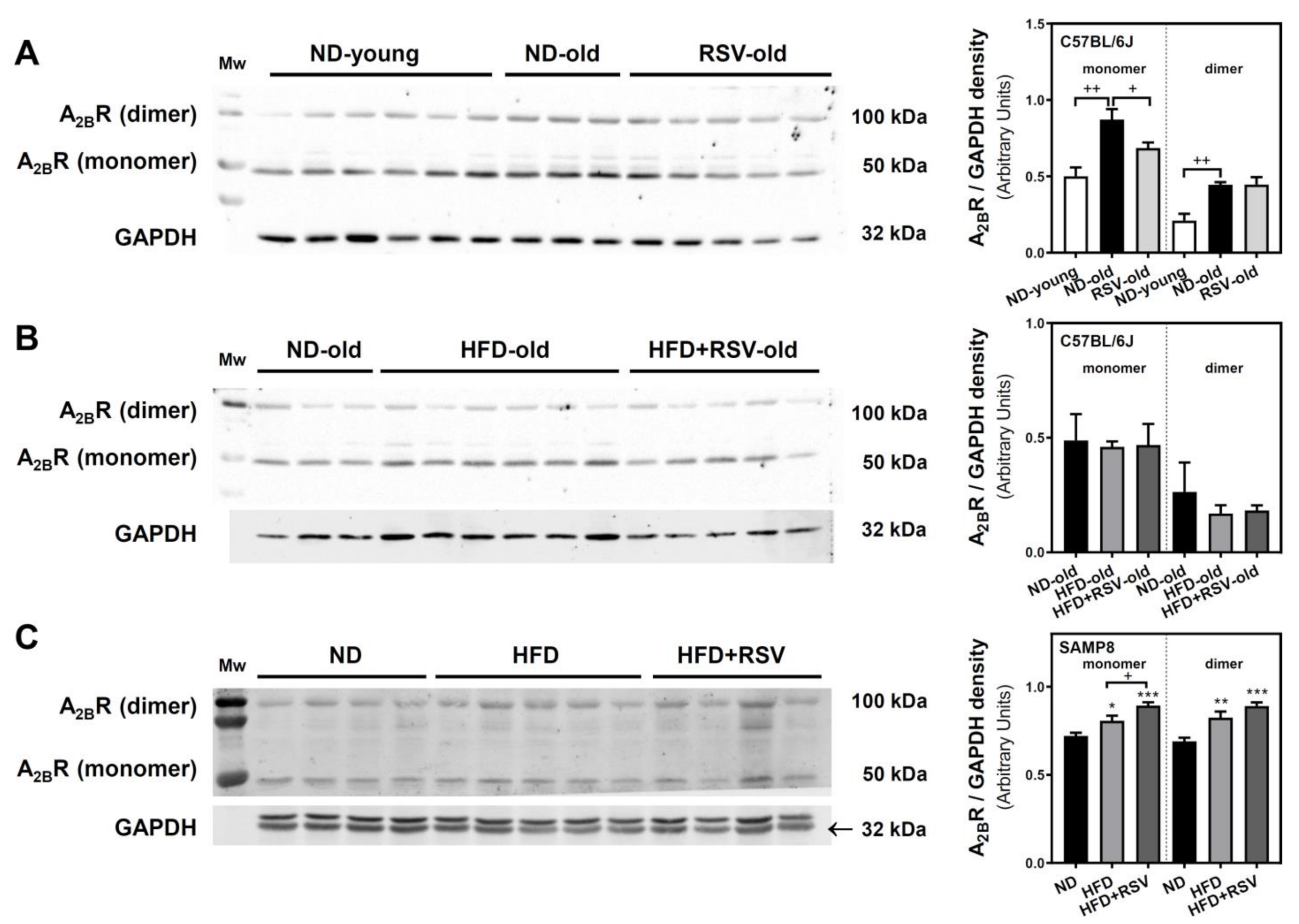
3.2. Adenosine A1, A2A and A2B Receptors Level in the Cerebral Cortex
3.3. Adenylyl Cyclase Activity in the Cerebral Cortex
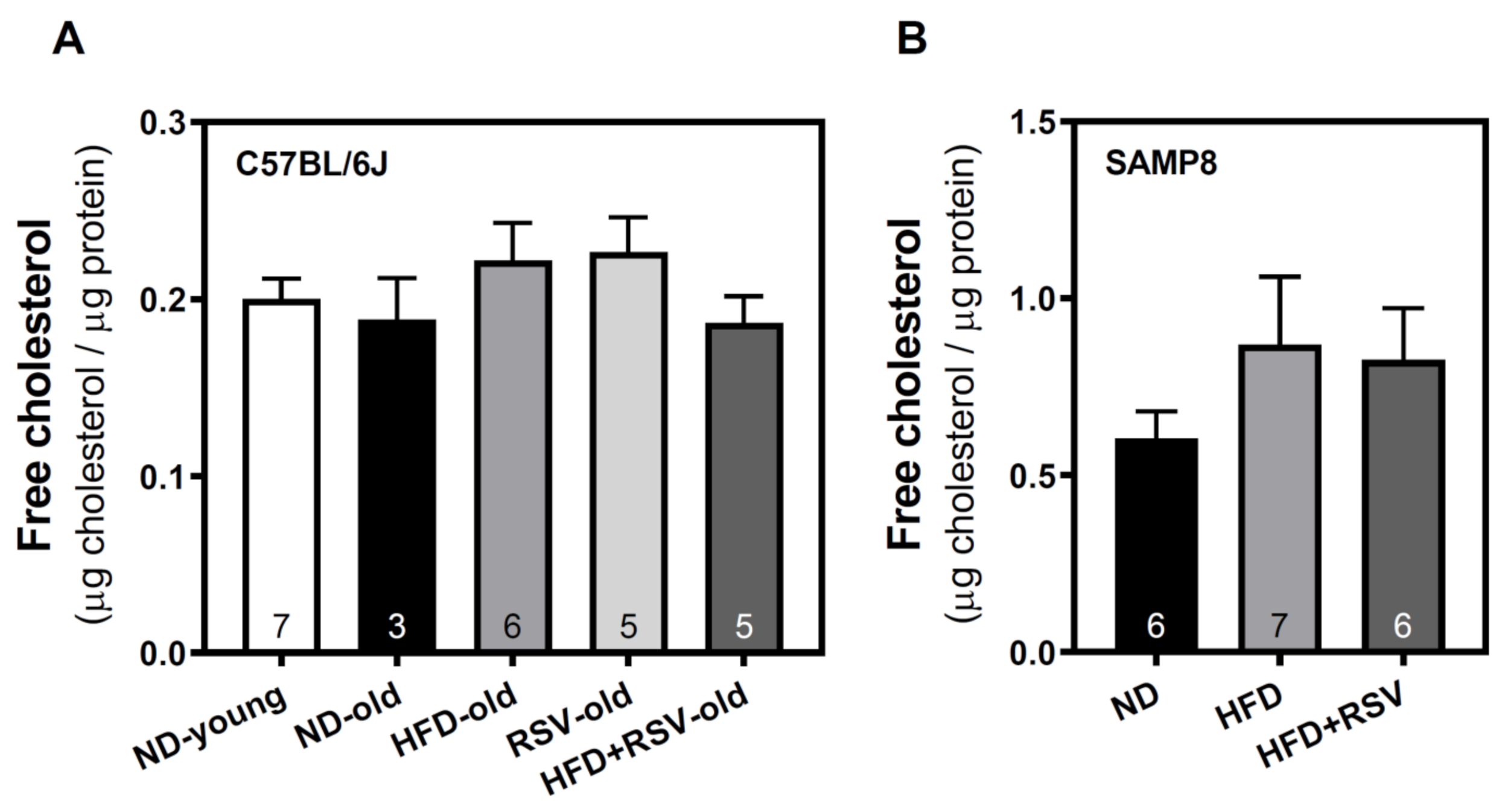
3.4. Level of Free Cholesterol in Plasma Membrane of Cortex Brain
4. Discussion
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Hou, Y.; Dan, X.; Babbar, M.; Wei, Y.; Hasselbalch, S.G.; Croteau, D.L.; Bohr, V.A. Ageing as a risk factor for neurodegenerative disease. Nat. Rev. Neurol. 2019, 15, 565–581. [Google Scholar] [CrossRef] [PubMed]
- Dye, L.; Boyle, N.B.; Champ, C.; Lawton, C. The relationship between obesity and cognitive health and decline. Proc. Nutr. Soc. 2017, 76, 443–454. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gonzalez Olmo, B.M.; Butler, M.J.; Barrientos, R.M. Evolution of the Human Diet and Its Impact on Gut Microbiota, Immune Responses, and Brain Health. Nutrients 2021, 13, 196. [Google Scholar] [CrossRef] [PubMed]
- Freeman, L.R.; Haley-Zitlin, V.; Rosenberger, D.S.; Granholm, A.C. Damaging effects of a high-fat diet to the brain and cognition: A review of proposed mechanisms. Nutr. Neurosci. 2014, 17, 241–251. [Google Scholar] [CrossRef]
- Wang, D.; Yan, J.; Chen, J.; Wu, W.; Zhu, X.; Wang, Y. Naringin Improves Neuronal Insulin Signaling, Brain Mitochondrial Function, and Cognitive Function in High-Fat Diet-Induced Obese Mice. Cell. Mol. Neurobiol. 2015, 35, 1061–1071. [Google Scholar] [CrossRef]
- Arnoldussen, I.A.C.; Wiesmann, M.; Pelgrim, C.E.; Wielemaker, E.M.; van Duyvenvoorde, W.; Amaral-Santos, P.L.; Verschuren, L.; Keijser, B.J.F.; Heerschap, A.; Kleemann, R.; et al. Butyrate restores HFD-induced adaptations in brain function and metabolism in mid-adult obese mice. Int. J. Obes. 2017, 41, 935–944. [Google Scholar] [CrossRef]
- Baxendale, S.; McGrath, K.; Donnachie, E.; Wintle, S.; Thompson, P.; Heaney, D. The role of obesity in cognitive dysfunction in people with epilepsy. Epilepsy Behav. 2015, 45, 187–190. [Google Scholar] [CrossRef] [Green Version]
- Ntlholang, O.; McCarroll, K.; Laird, E.; Molloy, A.M.; Ward, M.; McNulty, H.; Hoey, L.; Hughes, C.F.; Strain, J.J.; Casey, M.; et al. The relationship between adiposity and cognitive function in a large community-dwelling population: Data from the Trinity Ulster Department of Agriculture (TUDA) ageing cohort study. Br. J. Nutr. 2018, 120, 517–527. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hidese, S.; Ota, M.; Matsuo, J.; Ishida, I.; Hiraishi, M.; Yoshida, S.; Noda, T.; Sato, N.; Teraishi, T.; Hattori, K.; et al. Association of obesity with cognitive function and brain structure in patients with major depressive disorder. J. Affect. Disord. 2018, 225, 188–194. [Google Scholar] [CrossRef] [PubMed]
- Raine, L.; Drollette, E.; Kao, S.C.; Westfall, D.; Chaddock-Heyman, L.; Kramer, A.F.; Khan, N.; Hillman, C. The Associations between Adiposity, Cognitive Function, and Achievement in Children. Med. Sci. Sports Exerc. 2018, 50, 1868–1874. [Google Scholar] [CrossRef]
- Anderson, Y.C.; Kirkpatrick, K.; Dolan, G.M.S.; Wouldes, T.A.; Grant, C.C.; Cave, T.L.; Wild, C.E.K.; Derraik, J.G.B.; Cutfield, W.S.; Hofman, P.L. Do changes in weight status affect cognitive function in children and adolescents with obesity? A secondary analysis of a clinical trial. BMJ Open 2019, 9, e021586. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cunha, R.A. How does adenosine control neuronal dysfunction and neurodegeneration? J. Neurochem. 2016, 139, 1019–1055. [Google Scholar] [CrossRef]
- Fredholm, B.B.; AP, I.J.; Jacobson, K.A.; Linden, J.; Muller, C.E. International Union of Basic and Clinical Pharmacology. LXXXI. Nomenclature and classification of adenosine receptors—An update. Pharmacol. Rev. 2011, 63, 1–34. [Google Scholar] [CrossRef] [PubMed]
- Albasanz, J.L.; Perez, S.; Barrachina, M.; Ferrer, I.; Martin, M. Up-regulation of adenosine receptors in the frontal cortex in Alzheimer’s disease. Brain Pathol. 2008, 18, 211–219. [Google Scholar] [CrossRef]
- Villar-Menendez, I.; Porta, S.; Buira, S.P.; Pereira-Veiga, T.; Diaz-Sanchez, S.; Albasanz, J.L.; Ferrer, I.; Martin, M.; Barrachina, M. Increased striatal adenosine A2A receptor levels is an early event in Parkinson’s disease-related pathology and it is potentially regulated by miR-34b. Neurobiol. Dis. 2014, 69, 206–214. [Google Scholar] [CrossRef] [PubMed]
- Villar-Menendez, I.; Diaz-Sanchez, S.; Blanch, M.; Albasanz, J.L.; Pereira-Veiga, T.; Monje, A.; Planchat, L.M.; Ferrer, I.; Martin, M.; Barrachina, M. Reduced striatal adenosine A2A receptor levels define a molecular subgroup in schizophrenia. J. Psychiatr. Res. 2014, 51, 49–59. [Google Scholar] [CrossRef]
- Silva, A.C.; Lemos, C.; Goncalves, F.Q.; Pliassova, A.V.; Machado, N.J.; Silva, H.B.; Canas, P.M.; Cunha, R.A.; Lopes, J.P.; Agostinho, P. Blockade of adenosine A2A receptors recovers early deficits of memory and plasticity in the triple transgenic mouse model of Alzheimer’s disease. Neurobiol. Dis. 2018, 117, 72–81. [Google Scholar] [CrossRef]
- Chen, P.Z.; He, W.J.; Zhu, Z.R.; Guo-Ji, E.; Xu, G.; Chen, D.W.; Gao, Y.Q. Adenosine A2A receptor involves in neuroinflammation-mediated cognitive decline through activating microglia under acute hypobaric hypoxia. Behav. Brain Res. 2018, 347, 99–107. [Google Scholar] [CrossRef]
- Mouro, F.M.; Kofalvi, A.; Andre, L.A.; Baqi, Y.; Muller, C.E.; Ribeiro, J.A.; Sebastiao, A.M. Memory deficits induced by chronic cannabinoid exposure are prevented by adenosine A2AR receptor antagonism. Neuropharmacology 2019, 155, 10–21. [Google Scholar] [CrossRef]
- Kaster, M.P.; Machado, N.J.; Silva, H.B.; Nunes, A.; Ardais, A.P.; Santana, M.; Baqi, Y.; Muller, C.E.; Rodrigues, A.L.; Porciuncula, L.O.; et al. Caffeine acts through neuronal adenosine A2A receptors to prevent mood and memory dysfunction triggered by chronic stress. Proc. Natl. Acad. Sci. USA 2015, 112, 7833–7838. [Google Scholar] [CrossRef] [Green Version]
- Mouro, F.M.; Batalha, V.L.; Ferreira, D.G.; Coelho, J.E.; Baqi, Y.; Muller, C.E.; Lopes, L.V.; Ribeiro, J.A.; Sebastiao, A.M. Chronic and acute adenosine A2A receptor blockade prevents long-term episodic memory disruption caused by acute cannabinoid CB1 receptor activation. Neuropharmacology 2017, 117, 316–327. [Google Scholar] [CrossRef] [PubMed]
- Faivre, E.; Coelho, J.E.; Zornbach, K.; Malik, E.; Baqi, Y.; Schneider, M.; Cellai, L.; Carvalho, K.; Sebda, S.; Figeac, M.; et al. Beneficial Effect of a Selective Adenosine A2A Receptor Antagonist in the APPswe/PS1dE9 Mouse Model of Alzheimer’s Disease. Front. Mol. Neurosci. 2018, 11, 235. [Google Scholar] [CrossRef] [PubMed]
- Gnad, T.; Navarro, G.; Lahesmaa, M.; Reverte-Salisa, L.; Copperi, F.; Cordomi, A.; Naumann, J.; Hochhauser, A.; Haufs-Brusberg, S.; Wenzel, D.; et al. Adenosine/A2B Receptor Signaling Ameliorates the Effects of Aging and Counteracts Obesity. Cell Metab. 2020, 32, 56–70. [Google Scholar] [CrossRef]
- Guixa-Gonzalez, R.; Albasanz, J.L.; Rodriguez-Espigares, I.; Pastor, M.; Sanz, F.; Marti-Solano, M.; Manna, M.; Martinez-Seara, H.; Hildebrand, P.W.; Martin, M.; et al. Membrane cholesterol access into a G-protein-coupled receptor. Nat. Commun. 2017, 8, 14505. [Google Scholar] [CrossRef] [Green Version]
- Petrov, A.M.; Kasimov, M.R.; Zefirov, A.L. Brain Cholesterol Metabolism and Its Defects: Linkage to Neurodegenerative Diseases and Synaptic Dysfunction. Acta Naturae 2016, 8, 58–73. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Saeed, A.A.; Genove, G.; Li, T.; Lutjohann, D.; Olin, M.; Mast, N.; Pikuleva, I.A.; Crick, P.; Wang, Y.; Griffiths, W.; et al. Effects of a disrupted blood-brain barrier on cholesterol homeostasis in the brain. J. Biol. Chem. 2014, 289, 23712–23722. [Google Scholar] [CrossRef] [Green Version]
- Chen, H.; Du, Y.; Liu, S.; Ge, B.; Ji, Y.; Huang, G. Association between serum cholesterol levels and Alzheimer’s disease in China: A case-control study. Int. J. Food Sci. Nutr. 2019, 70, 405–411. [Google Scholar] [CrossRef]
- Marcum, Z.A.; Walker, R.; Bobb, J.F.; Sin, M.K.; Gray, S.L.; Bowen, J.D.; McCormick, W.; McCurry, S.M.; Crane, P.K.; Larson, E.B. Serum Cholesterol and Incident Alzheimer’s Disease: Findings from the Adult Changes in Thought Study. J. Am. Geriatr. Soc. 2018, 66, 2344–2352. [Google Scholar] [CrossRef]
- Chang, T.Y.; Yamauchi, Y.; Hasan, M.T.; Chang, C. Cellular cholesterol homeostasis and Alzheimer’s disease. J. Lipid. Res. 2017, 58, 2239–2254. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jeong, W.; Lee, H.; Cho, S.; Seo, J. ApoE4-Induced Cholesterol Dysregulation and Its Brain Cell Type-Specific Implications in the Pathogenesis of Alzheimer’s Disease. Mol. Cells 2019, 42, 739–746. [Google Scholar] [CrossRef] [PubMed]
- Loera-Valencia, R.; Goikolea, J.; Parrado-Fernandez, C.; Merino-Serrais, P.; Maioli, S. Alterations in cholesterol metabolism as a risk factor for developing Alzheimer’s disease: Potential novel targets for treatment. J. Steroid Biochem. Mol. Biol. 2019, 190, 104–114. [Google Scholar] [CrossRef] [PubMed]
- Sanchez-Melgar, A.; Albasanz, J.L.; Guixa-Gonzalez, R.; Saleh, N.; Selent, J.; Martin, M. The antioxidant resveratrol acts as a non-selective adenosine receptor agonist. Free Radic. Biol. Med. 2019, 135, 261–273. [Google Scholar] [CrossRef]
- Sanchez-Melgar, A.; Albasanz, J.L.; Palomera-Avalos, V.; Pallas, M.; Martin, M. Resveratrol Modulates and Reverses the Age-Related Effect on Adenosine-Mediated Signalling in SAMP8 Mice. Mol. Neurobiol. 2019, 56, 2881–2895. [Google Scholar] [CrossRef] [PubMed]
- Gong, L.; Guo, S.; Zou, Z. Resveratrol ameliorates metabolic disorders and insulin resistance in high-fat diet-fed mice. Life Sci. 2020, 242, 117212. [Google Scholar] [CrossRef]
- Hou, C.Y.; Tain, Y.L.; Yu, H.R.; Huang, L.T. The Effects of Resveratrol in the Treatment of Metabolic Syndrome. Int. J. Mol. Sci. 2019, 20, 535. [Google Scholar] [CrossRef] [Green Version]
- Lange, K.W.; Li, S. Resveratrol, pterostilbene, and dementia. Biofactors 2018, 44, 83–90. [Google Scholar] [CrossRef]
- Gomes, B.A.Q.; Silva, J.P.B.; Romeiro, C.F.R.; Dos Santos, S.M.; Rodrigues, C.A.; Goncalves, P.R.; Sakai, J.T.; Mendes, P.F.S.; Varela, E.L.P.; Monteiro, M.C. Neuroprotective Mechanisms of Resveratrol in Alzheimer’s Disease: Role of SIRT1. Oxid. Med. Cell. Longev. 2018, 2018, 8152373. [Google Scholar] [CrossRef]
- Palomera-Avalos, V.; Grinan-Ferre, C.; Izquierdo, V.; Camins, A.; Sanfeliu, C.; Pallas, M. Metabolic Stress Induces Cognitive Disturbances and Inflammation in Aged Mice: Protective Role of Resveratrol. Rejuvenation Res. 2017, 20, 202–217. [Google Scholar] [CrossRef]
- Palomera-Avalos, V.; Grinan-Ferre, C.; Puigoriol-Ilamola, D.; Camins, A.; Sanfeliu, C.; Canudas, A.M.; Pallas, M. Resveratrol Protects SAMP8 Brain Under Metabolic Stress: Focus on Mitochondrial Function and Wnt Pathway. Mol. Neurobiol. 2017, 54, 1661–1676. [Google Scholar] [CrossRef]
- Reagan-Shaw, S.; Nihal, M.; Ahmad, N. Dose translation from animal to human studies revisited. FASEB J. 2008, 22, 659–661. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Leon-Navarro, D.A.; Albasanz, J.L.; Martin, M. Hyperthermia-induced seizures alter adenosine A1 and A2A receptors and 5′-nucleotidase activity in rat cerebral cortex. J. Neurochem. 2015, 134, 395–404. [Google Scholar] [CrossRef]
- Chan, K.M.; Delfert, D.; Junger, K.D. A direct colorimetric assay for Ca2+-stimulated ATPase activity. Anal. Biochem. 1986, 157, 375–380. [Google Scholar] [CrossRef]
- Oizumi, H.; Kuriyama, N.; Imamura, S.; Tabuchi, M.; Omiya, Y.; Mizoguchi, K.; Kobayashi, H. Influence of aging on the behavioral phenotypes of C57BL/6J mice after social defeat. PLoS ONE 2019, 14, e0222076. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shoji, H.; Takao, K.; Hattori, S.; Miyakawa, T. Age-related changes in behavior in C57BL/6J mice from young adulthood to middle age. Mol. Brain 2016, 9, 11. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ahlemeyer, B.; Halupczok, S.; Rodenberg-Frank, E.; Valerius, K.P.; Baumgart-Vogt, E. Endogenous Murine Amyloid-beta Peptide Assembles into Aggregates in the Aged C57BL/6J Mouse Suggesting These Animals as a Model to Study Pathogenesis of Amyloid-beta Plaque Formation. J. Alzheimers Dis. 2018, 61, 1425–1450. [Google Scholar] [CrossRef] [PubMed]
- Akiguchi, I.; Pallas, M.; Budka, H.; Akiyama, H.; Ueno, M.; Han, J.; Yagi, H.; Nishikawa, T.; Chiba, Y.; Sugiyama, H.; et al. SAMP8 mice as a neuropathological model of accelerated brain aging and dementia: Toshio Takeda’s legacy and future directions. Neuropathology 2017, 37, 293–305. [Google Scholar] [CrossRef] [Green Version]
- Liu, B.; Liu, J.; Shi, J.S. SAMP8 Mice as a Model of Age-Related Cognition Decline with Underlying Mechanisms in Alzheimer’s Disease. J. Alzheimers Dis. 2020, 75, 385–395. [Google Scholar] [CrossRef] [PubMed]
- Pallas, M.; Camins, A.; Smith, M.A.; Perry, G.; Lee, H.G.; Casadesus, G. From aging to Alzheimer’s disease: Unveiling "the switch" with the senescence-accelerated mouse model (SAMP8). J. Alzheimers Dis. 2008, 15, 615–624. [Google Scholar] [CrossRef]
- Carter, T.A.; Greenhall, J.A.; Yoshida, S.; Fuchs, S.; Helton, R.; Swaroop, A.; Lockhart, D.J.; Barlow, C. Mechanisms of aging in senescence-accelerated mice. Genome Biol. 2005, 6, R48. [Google Scholar] [CrossRef] [Green Version]
- Takeda, T.; Hosokawa, M.; Higuchi, K. Senescence-accelerated mouse (SAM): A novel murine model of senescence. Exp. Gerontol. 1997, 32, 105–109. [Google Scholar] [CrossRef]
- Yuan, R.; Tsaih, S.W.; Petkova, S.B.; Marin de Evsikova, C.; Xing, S.; Marion, M.A.; Bogue, M.A.; Mills, K.D.; Peters, L.L.; Bult, C.J.; et al. Aging in inbred strains of mice: Study design and interim report on median lifespans and circulating IGF1 levels. Aging Cell 2009, 8, 277–287. [Google Scholar] [CrossRef] [Green Version]
- Kunstyr, I.; Leuenberger, H.G. Gerontological data of C57BL/6J mice. I. Sex differences in survival curves. J. Gerontol. 1975, 30, 157–162. [Google Scholar] [CrossRef]
- Moodley, K.K.; Chan, D. The hippocampus in neurodegenerative disease. Front. Neurol. Neurosci. 2014, 34, 95–108. [Google Scholar] [CrossRef]
- Alonso-Andres, P.; Albasanz, J.L.; Ferrer, I.; Martin, M. Purine-related metabolites and their converting enzymes are altered in frontal, parietal and temporal cortex at early stages of Alzheimer’s disease pathology. Brain Pathol. 2018, 28, 933–946. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wenk, G.L. Neuropathologic changes in Alzheimer’s disease. J. Clin. Psychiatry 2003, 64 (Suppl. 9), 7–10. [Google Scholar]
- Aarons, T.; Bradburn, S.; Robinson, A.; Payton, A.; Pendleton, N.; Murgatroyd, C. Dysregulation of BDNF in Prefrontal Cortex in Alzheimer’s Disease. J. Alzheimers Dis. 2019, 69, 1089–1097. [Google Scholar] [CrossRef] [PubMed]
- Burnstock, G.; Krugel, U.; Abbracchio, M.P.; Illes, P. Purinergic signalling: From normal behaviour to pathological brain function. Prog. Neurobiol. 2011, 95, 229–274. [Google Scholar] [CrossRef]
- Kalaria, R.N.; Sromek, S.; Wilcox, B.J.; Unnerstall, J.R. Hippocampal adenosine A1 receptors are decreased in Alzheimer’s disease. Neurosci. Lett. 1990, 118, 257–260. [Google Scholar] [CrossRef]
- Ikeda, M.; Mackay, K.B.; Dewar, D.; McCulloch, J. Differential alterations in adenosine A1 and kappa 1 opioid receptors in the striatum in Alzheimer’s disease. Brain Res. 1993, 616, 211–217. [Google Scholar] [CrossRef]
- Fukumitsu, N.; Ishii, K.; Kimura, Y.; Oda, K.; Hashimoto, M.; Suzuki, M.; Ishiwata, K. Adenosine A1 receptors using 8-dicyclopropylmethyl-1-[(11)C]methyl-3-propylxanthine PET in Alzheimer’s disease. Ann. Nucl. Med. 2008, 22, 841–847. [Google Scholar] [CrossRef]
- Angulo, E.; Casado, V.; Mallol, J.; Canela, E.I.; Vinals, F.; Ferrer, I.; Lluis, C.; Franco, R. A1 adenosine receptors accumulate in neurodegenerative structures in Alzheimer disease and mediate both amyloid precursor protein processing and tau phosphorylation and translocation. Brain Pathol. 2003, 13, 440–451. [Google Scholar] [CrossRef] [PubMed]
- Merighi, S.; Battistello, E.; Casetta, I.; Gragnaniello, D.; Poloni, T.E.; Medici, V.; Cirrincione, A.; Varani, K.; Vincenzi, F.; Borea, P.A.; et al. Upregulation of Cortical A2A Adenosine Receptors Is Reflected in Platelets of Patients with Alzheimer’s Disease. J. Alzheimers Dis. 2021, 80, 1105–1117. [Google Scholar] [CrossRef]
- Cunha, R.A. Overactivity of neuronal adenosine A2A receptors accelerates neurodegeneration. Brain 2019, 142, 3323–3324. [Google Scholar] [CrossRef] [PubMed]
- Canas, P.M.; Porciuncula, L.O.; Cunha, G.M.; Silva, C.G.; Machado, N.J.; Oliveira, J.M.; Oliveira, C.R.; Cunha, R.A. Adenosine A2A receptor blockade prevents synaptotoxicity and memory dysfunction caused by beta-amyloid peptides via p38 mitogen-activated protein kinase pathway. J. Neurosci. 2009, 29, 14741–14751. [Google Scholar] [CrossRef]
- Cunha, G.M.; Canas, P.M.; Melo, C.S.; Hockemeyer, J.; Muller, C.E.; Oliveira, C.R.; Cunha, R.A. Adenosine A2A receptor blockade prevents memory dysfunction caused by beta-amyloid peptides but not by scopolamine or MK-801. Exp. Neurol. 2008, 210, 776–781. [Google Scholar] [CrossRef]
- Akbari, A.; Khalili-Fomeshi, M.; Ashrafpour, M.; Moghadamnia, A.A.; Ghasemi-Kasman, M. Adenosine A2A receptor blockade attenuates spatial memory deficit and extent of demyelination areas in lyolecithin-induced demyelination model. Life Sci. 2018, 205, 63–72. [Google Scholar] [CrossRef]
- Sharma, S. High fat diet and its effects on cognitive health: Alterations of neuronal and vascular components of brain. Physiol. Behav. 2021, 240, 113528. [Google Scholar] [CrossRef]
- Mancini, G.; Dias, C.; Lourenco, C.F.; Laranjinha, J.; de Bem, A.; Ledo, A. A High Fat/Cholesterol Diet Recapitulates Some Alzheimer’s Disease-Like Features in Mice: Focus on Hippocampal Mitochondrial Dysfunction. J. Alzheimers Dis. 2021, 82, 1619–1633. [Google Scholar] [CrossRef] [PubMed]
- Petrov, D.; Pedros, I.; Artiach, G.; Sureda, F.X.; Barroso, E.; Pallas, M.; Casadesus, G.; Beas-Zarate, C.; Carro, E.; Ferrer, I.; et al. High-fat diet-induced deregulation of hippocampal insulin signaling and mitochondrial homeostasis deficiences contribute to Alzheimer disease pathology in rodents. Biochim. Biophys. Acta 2015, 1852, 1687–1699. [Google Scholar] [CrossRef] [Green Version]
- Busquets, O.; Ettcheto, M.; Pallas, M.; Beas-Zarate, C.; Verdaguer, E.; Auladell, C.; Folch, J.; Camins, A. Long-term exposition to a high fat diet favors the appearance of beta-amyloid depositions in the brain of C57BL/6J mice. A potential model of sporadic Alzheimer’s disease. Mech. Ageing Dev. 2017, 162, 38–45. [Google Scholar] [CrossRef]
- Augusto, E.; Matos, M.; Sevigny, J.; El-Tayeb, A.; Bynoe, M.S.; Muller, C.E.; Cunha, R.A.; Chen, J.F. Ecto-5′-nucleotidase (CD73)-mediated formation of adenosine is critical for the striatal adenosine A2A receptor functions. J. Neurosci. 2013, 33, 11390–11399. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Meng, F.; Guo, Z.; Hu, Y.; Mai, W.; Zhang, Z.; Zhang, B.; Ge, Q.; Lou, H.; Guo, F.; Chen, J.; et al. CD73-derived adenosine controls inflammation and neurodegeneration by modulating dopamine signalling. Brain 2019, 142, 700–718. [Google Scholar] [CrossRef]
- Hickman, S.; Izzy, S.; Sen, P.; Morsett, L.; El Khoury, J. Microglia in neurodegeneration. Nat. Neurosci. 2018, 21, 1359–1369. [Google Scholar] [CrossRef]
- Onyango, I.G.; Jauregui, G.V.; Carna, M.; Bennett, J.P., Jr.; Stokin, G.B. Neuroinflammation in Alzheimer’s Disease. Biomedicines 2021, 9, 524. [Google Scholar] [CrossRef] [PubMed]
- Lutshumba, J.; Nikolajczyk, B.S.; Bachstetter, A.D. Dysregulation of Systemic Immunity in Aging and Dementia. Front. Cell Neurosci. 2021, 15, 652111. [Google Scholar] [CrossRef]
- Illes, P.; Rubini, P.; Ulrich, H.; Zhao, Y.; Tang, Y. Regulation of Microglial Functions by Purinergic Mechanisms in the Healthy and Diseased CNS. Cells 2020, 9, 1108. [Google Scholar] [CrossRef] [PubMed]
- Reilly, A.M.; Tsai, A.P.; Lin, P.B.; Ericsson, A.C.; Oblak, A.L.; Ren, H. Metabolic Defects Caused by High-Fat Diet Modify Disease Risk through Inflammatory and Amyloidogenic Pathways in a Mouse Model of Alzheimer’s Disease. Nutrients 2020, 12, 2977. [Google Scholar] [CrossRef] [PubMed]
- Robison, L.S.; Gannon, O.J.; Thomas, M.A.; Salinero, A.E.; Abi-Ghanem, C.; Poitelon, Y.; Belin, S.; Zuloaga, K.L. Role of sex and high-fat diet in metabolic and hypothalamic disturbances in the 3xTg-AD mouse model of Alzheimer’s disease. J. Neuroinflammation 2020, 17, 285. [Google Scholar] [CrossRef]
- Zameer, S.; Alam, M.; Hussain, S.; Vohora, D.; Ali, J.; Najmi, A.K.; Akhtar, M. Neuroprotective role of alendronate against APP processing and neuroinflammation in mice fed a high fat diet. Brain Res. Bull. 2020, 161, 197–212. [Google Scholar] [CrossRef]
- Johnston-Cox, H.; Koupenova, M.; Yang, D.; Corkey, B.; Gokce, N.; Farb, M.G.; LeBrasseur, N.; Ravid, K. The A2b adenosine receptor modulates glucose homeostasis and obesity. PLoS ONE 2012, 7, e40584. [Google Scholar] [CrossRef] [Green Version]
- Csoka, B.; Koscso, B.; Toro, G.; Kokai, E.; Virag, L.; Nemeth, Z.H.; Pacher, P.; Bai, P.; Hasko, G. A2B adenosine receptors prevent insulin resistance by inhibiting adipose tissue inflammation via maintaining alternative macrophage activation. Diabetes 2014, 63, 850–866. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fusco, I.; Ugolini, F.; Lana, D.; Coppi, E.; Dettori, I.; Gaviano, L.; Nosi, D.; Cherchi, F.; Pedata, F.; Giovannini, M.G.; et al. The Selective Antagonism of Adenosine A2B Receptors Reduces the Synaptic Failure and Neuronal Death Induced by Oxygen and Glucose Deprivation in Rat CA1 Hippocampus in Vitro. Front. Pharmacol. 2018, 9, 399. [Google Scholar] [CrossRef] [Green Version]
- Semwal, B.C.; Garabadu, D. 5-N-ethyl Carboxamidoadenosine Stimulates Adenosine-2b Receptor-Mediated Mitogen-Activated Protein Kinase Pathway to Improve Brain Mitochondrial Function in Amyloid Beta-Induced Cognitive Deficit Mice. Neuromol. Med. 2020, 22, 542–556. [Google Scholar] [CrossRef] [PubMed]
- Mokni, M.; Elkahoui, S.; Limam, F.; Amri, M.; Aouani, E. Effect of resveratrol on antioxidant enzyme activities in the brain of healthy rat. Neurochem. Res. 2007, 32, 981–987. [Google Scholar] [CrossRef]
- Wang, Q.; Xu, J.; Rottinghaus, G.E.; Simonyi, A.; Lubahn, D.; Sun, G.Y.; Sun, A.Y. Resveratrol protects against global cerebral ischemic injury in gerbils. Brain Res. 2002, 958, 439–447. [Google Scholar] [CrossRef]
- Turner, R.S.; Thomas, R.G.; Craft, S.; van Dyck, C.H.; Mintzer, J.; Reynolds, B.A.; Brewer, J.B.; Rissman, R.A.; Raman, R.; Aisen, P.S.; et al. A randomized, double-blind, placebo-controlled trial of resveratrol for Alzheimer disease. Neurology 2015, 85, 1383–1391. [Google Scholar] [CrossRef]
- Cao, K.; Dong, Y.T.; Xiang, J.; Xu, Y.; Li, Y.; Song, H.; Yu, W.F.; Qi, X.L.; Guan, Z.Z. The neuroprotective effects of SIRT1 in mice carrying the APP/PS1 double-transgenic mutation and in SH-SY5Y cells over-expressing human APP670/671 may involve elevated levels of alpha7 nicotinic acetylcholine receptors. Aging 2020, 12, 1792–1807. [Google Scholar] [CrossRef]
- Zhao, Y.N.; Li, W.F.; Li, F.; Zhang, Z.; Dai, Y.D.; Xu, A.L.; Qi, C.; Gao, J.M.; Gao, J. Resveratrol improves learning and memory in normally aged mice through microRNA-CREB pathway. Biochem. Biophys. Res. Commun. 2013, 435, 597–602. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Izquierdo, V.; Palomera-Avalos, V.; Lopez-Ruiz, S.; Canudas, A.M.; Pallas, M.; Grinan-Ferre, C. Maternal Resveratrol Supplementation Prevents Cognitive Decline in Senescent Mice Offspring. Int. J. Mol. Sci. 2019, 20, 1134. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Corpas, R.; Grinan-Ferre, C.; Rodriguez-Farre, E.; Pallas, M.; Sanfeliu, C. Resveratrol Induces Brain Resilience Against Alzheimer Neurodegeneration Through Proteostasis Enhancement. Mol. Neurobiol. 2019, 56, 1502–1516. [Google Scholar] [CrossRef] [Green Version]
- Vance, J.E. Dysregulation of cholesterol balance in the brain: Contribution to neurodegenerative diseases. Dis. Models Mech. 2012, 5, 746–755. [Google Scholar] [CrossRef] [Green Version]
- Burlot, M.A.; Braudeau, J.; Michaelsen-Preusse, K.; Potier, B.; Ayciriex, S.; Varin, J.; Gautier, B.; Djelti, F.; Audrain, M.; Dauphinot, L.; et al. Cholesterol 24-hydroxylase defect is implicated in memory impairments associated with Alzheimer-like Tau pathology. Hum. Mol. Genet. 2015, 24, 5965–5976. [Google Scholar] [CrossRef]
- Russell, D.W.; Halford, R.W.; Ramirez, D.M.; Shah, R.; Kotti, T. Cholesterol 24-hydroxylase: An enzyme of cholesterol turnover in the brain. Annu. Rev. Biochem. 2009, 78, 1017–1040. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Leoni, V.; Solomon, A.; Kivipelto, M. Links between ApoE, brain cholesterol metabolism, tau and amyloid beta-peptide in patients with cognitive impairment. Biochem. Soc. Trans. 2010, 38, 1021–1025. [Google Scholar] [CrossRef] [PubMed]
- Umeda, T.; Tomiyama, T.; Kitajima, E.; Idomoto, T.; Nomura, S.; Lambert, M.P.; Klein, W.L.; Mori, H. Hypercholesterolemia accelerates intraneuronal accumulation of Abeta oligomers resulting in memory impairment in Alzheimer’s disease model mice. Life Sci. 2012, 91, 1169–1176. [Google Scholar] [CrossRef]
- Kiriakidi, S.; Kolocouris, A.; Liapakis, G.; Ikram, S.; Durdagi, S.; Mavromoustakos, T. Effects of Cholesterol on GPCR Function: Insights from Computational and Experimental Studies. Adv. Exp. Med. Biol. 2019, 1135, 89–103. [Google Scholar] [CrossRef] [PubMed]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| C57BL/6J | SAMP8 | |||||||
|---|---|---|---|---|---|---|---|---|
| Parameter | ND-Young | ND-Old | HFD-Old | RSV-Old | HFD + RSV-Old | ND | HFD | HFD + RSV |
| A1R | 99 | 100 | 225 ** | 171 ** | 196 * | 100 | 120 | 102 |
| A2AR | 103 | 100 | 236 ** | 208 * | 218 * | 100 | 132 | 117 |
| A2BR-monomer | 57 ** | 100 | 94 | 79 * | 96 | 100 | 112 * | 124 *** |
| A2BR-dimer | 47 ** | 100 | 64 | 100 | 69 | 100 | 119 ** | 129 *** |
| 5′-NT | 95 | 100 | 146 * | 134 | 109 | 100 | 246 * | 293 ** |
| Basal AC | 102 | 100 | 82 | 96 | 105 | -- | -- | -- |
| A1R-mediated AC inhibition | 91 | 100 | 150 * | 154 * | 190 ** | -- | -- | -- |
| cholesterol | 106 | 100 | 118 | 120 | 99 | 100 | 143 | 136 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sánchez-Melgar, A.; Izquierdo-Ramírez, P.J.; Palomera-Ávalos, V.; Pallàs, M.; Albasanz, J.L.; Martín, M. High-Fat and Resveratrol Supplemented Diets Modulate Adenosine Receptors in the Cerebral Cortex of C57BL/6J and SAMP8 Mice. Nutrients 2021, 13, 3040. https://doi.org/10.3390/nu13093040
Sánchez-Melgar A, Izquierdo-Ramírez PJ, Palomera-Ávalos V, Pallàs M, Albasanz JL, Martín M. High-Fat and Resveratrol Supplemented Diets Modulate Adenosine Receptors in the Cerebral Cortex of C57BL/6J and SAMP8 Mice. Nutrients. 2021; 13(9):3040. https://doi.org/10.3390/nu13093040
Chicago/Turabian StyleSánchez-Melgar, Alejandro, Pedro José Izquierdo-Ramírez, Verónica Palomera-Ávalos, Mercè Pallàs, José Luis Albasanz, and Mairena Martín. 2021. "High-Fat and Resveratrol Supplemented Diets Modulate Adenosine Receptors in the Cerebral Cortex of C57BL/6J and SAMP8 Mice" Nutrients 13, no. 9: 3040. https://doi.org/10.3390/nu13093040