
Maternal 3,3-Dimethyl-1-Butanol Therapy Protects Adult Male Rat Offspring against Hypertension Programmed by Perinatal TCDD Exposure

 ,
,  , , ,
, , , 
Abstract
:1. Introduction
2. Materials and Methods
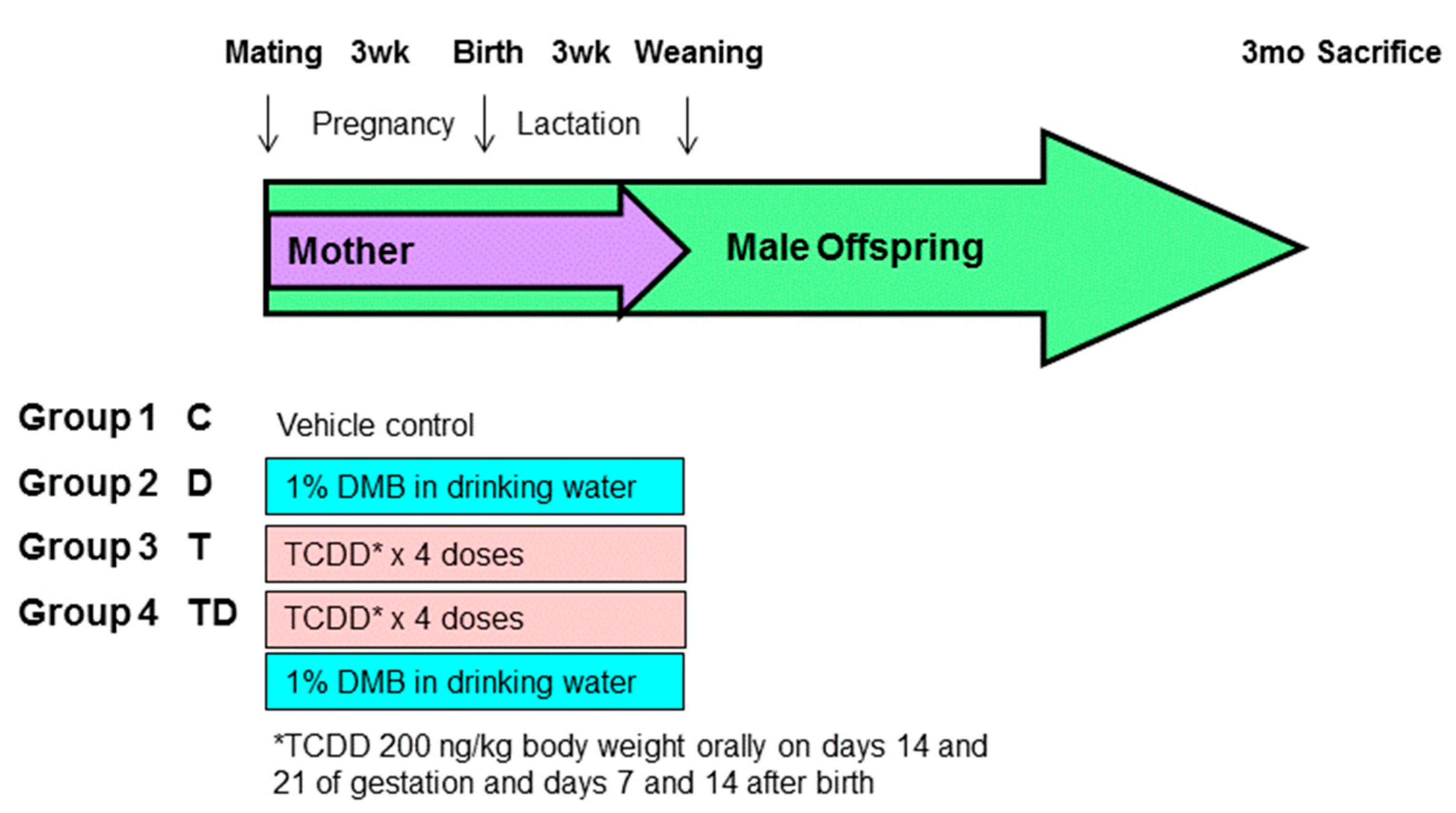
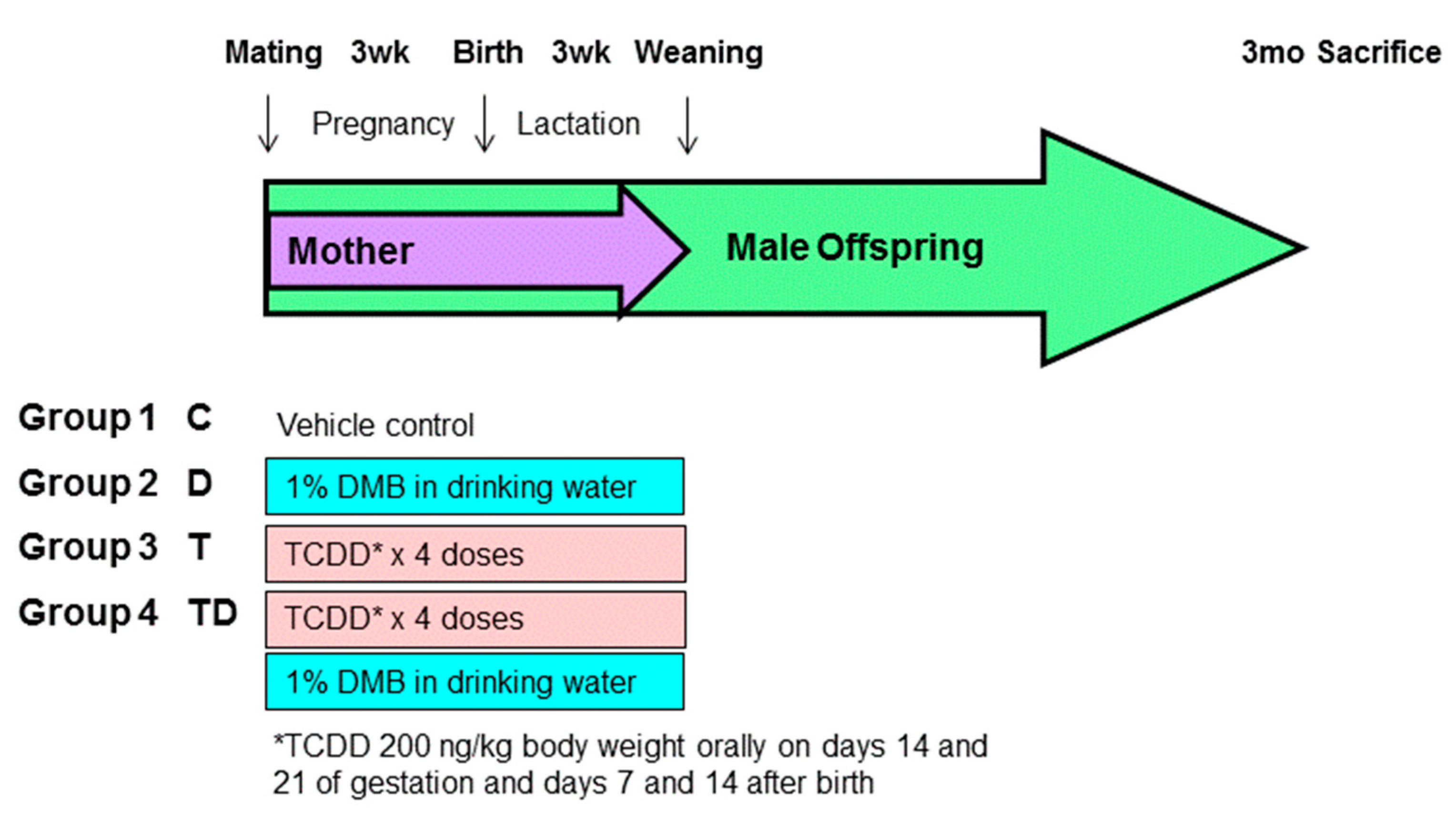
2.1. Animal Study
2.2. Liquid Chromatography–Mass Spectrometry (LC–MS) Analysis
2.3. Gas Chromatography-Mass Spectrometry (GC–MS) Analysis
2.4. Quantitative Real-Time Polymerase Chain Reaction (qPCR)
2.5. Gut Microbiota Compositions
2.6. Statistical Analysis
3. Results
3.1. Morphological Values and Blood Pressures
3.2. TMA-TMAO Pathway
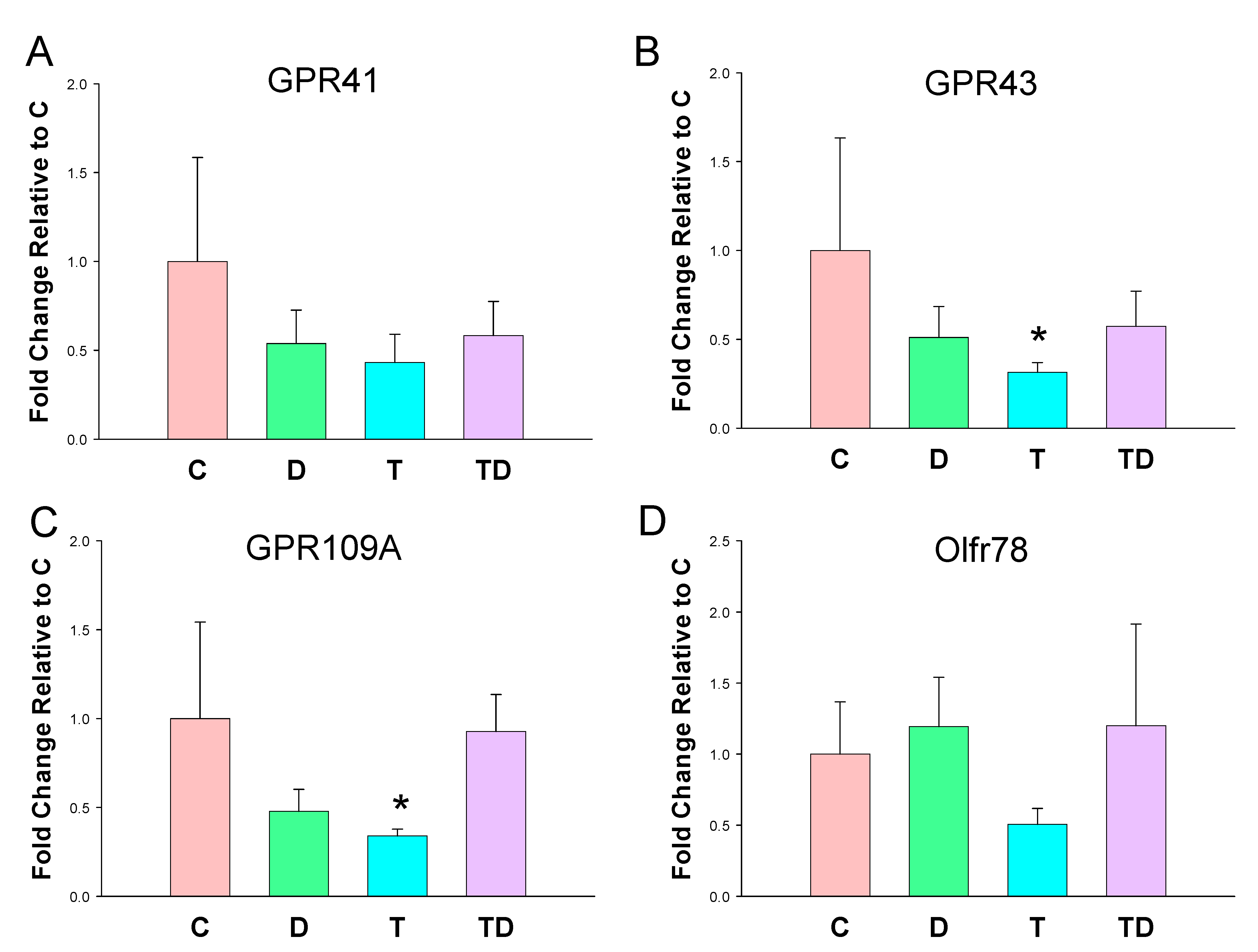
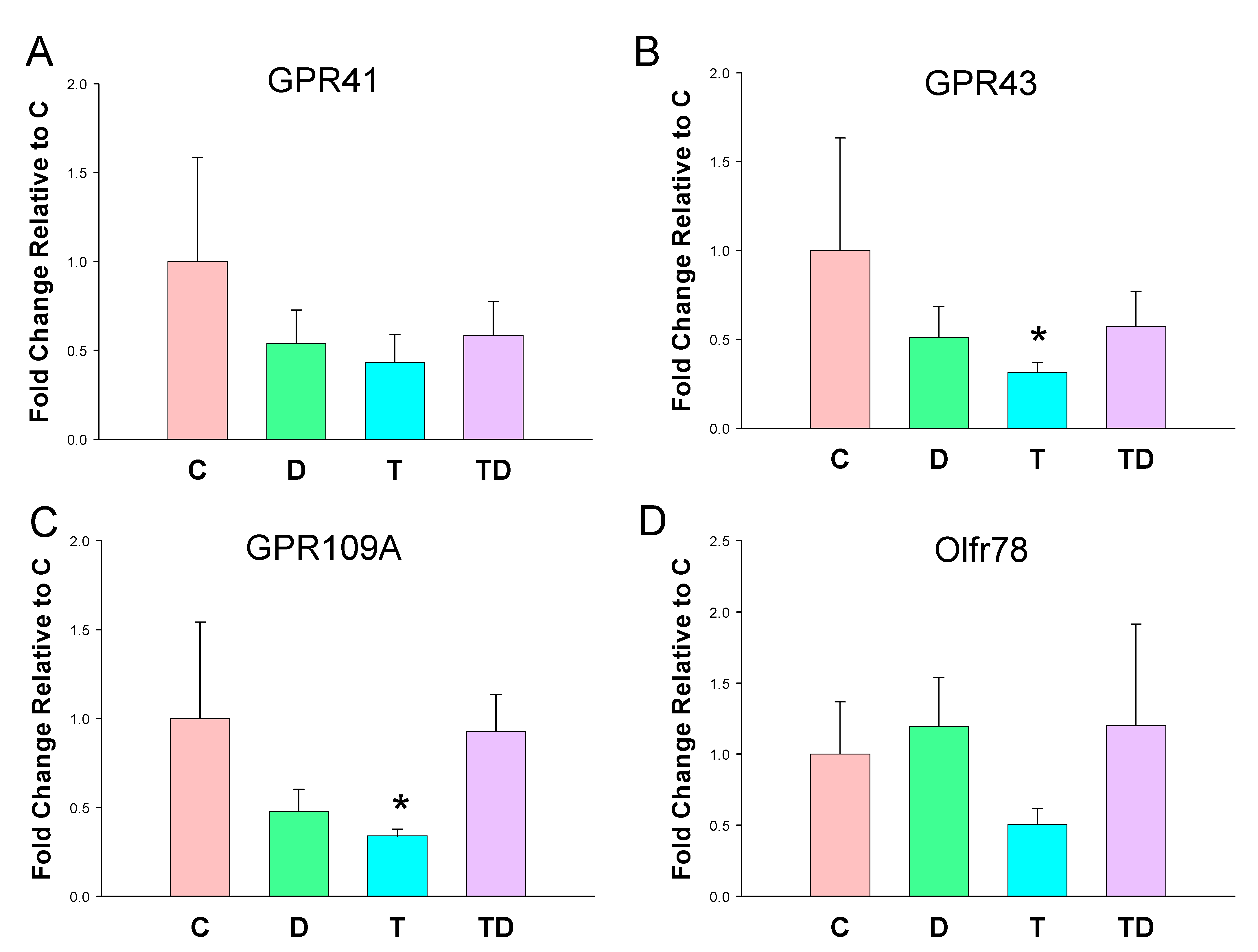
3.3. Plasma SCFA Levels and Renal SCFA Receptors
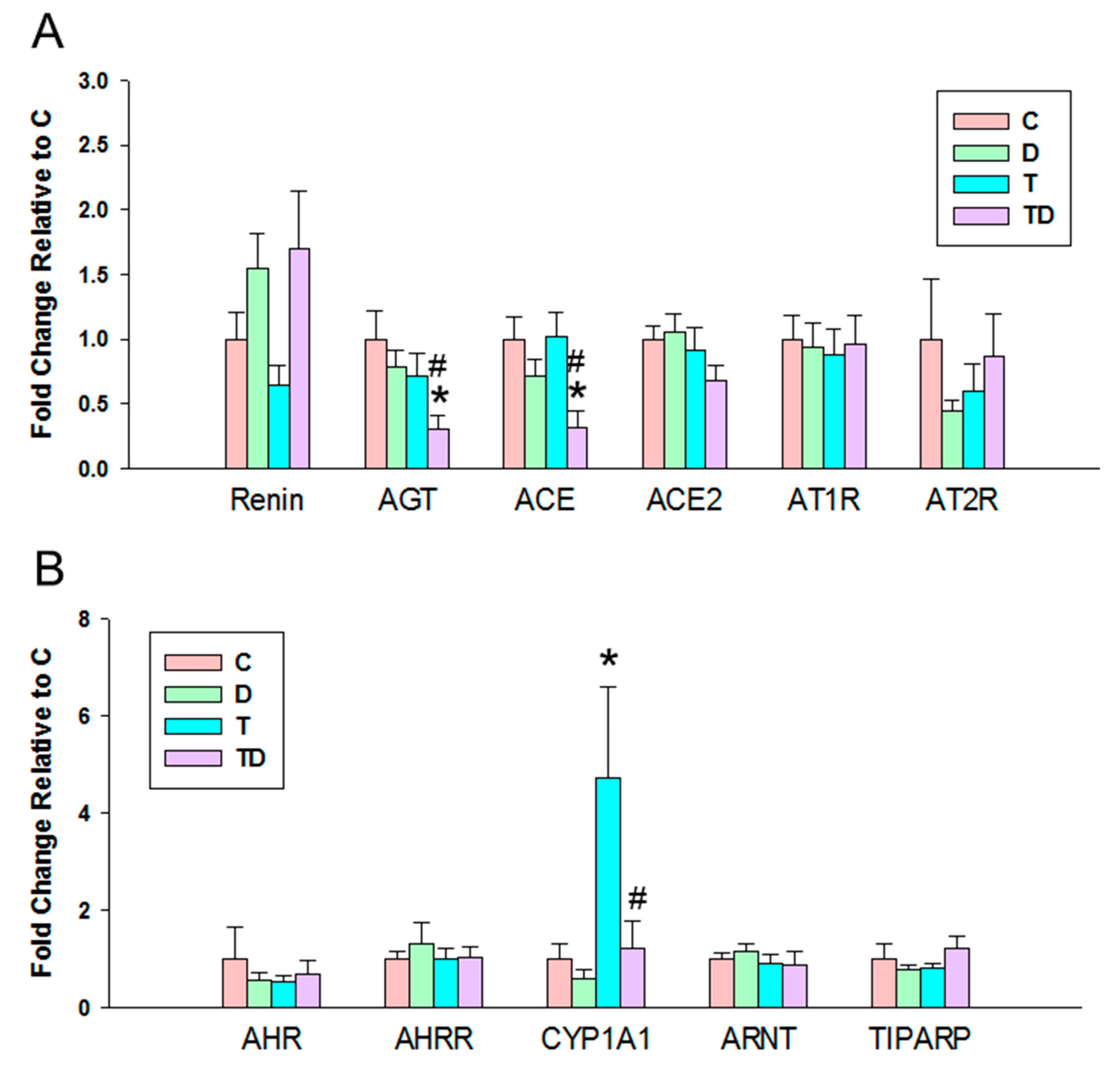
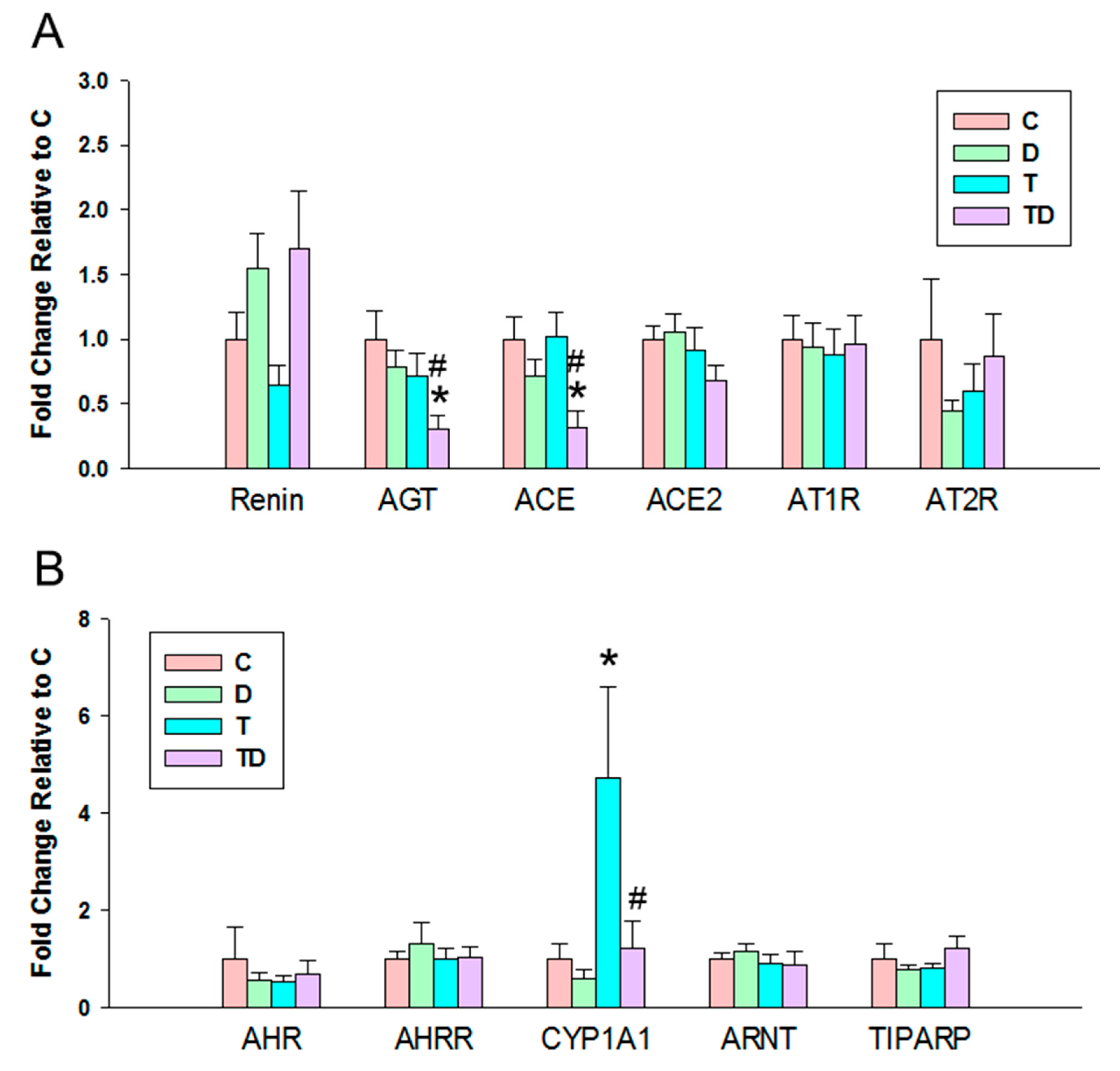
3.4. RAS and AHR Pathway
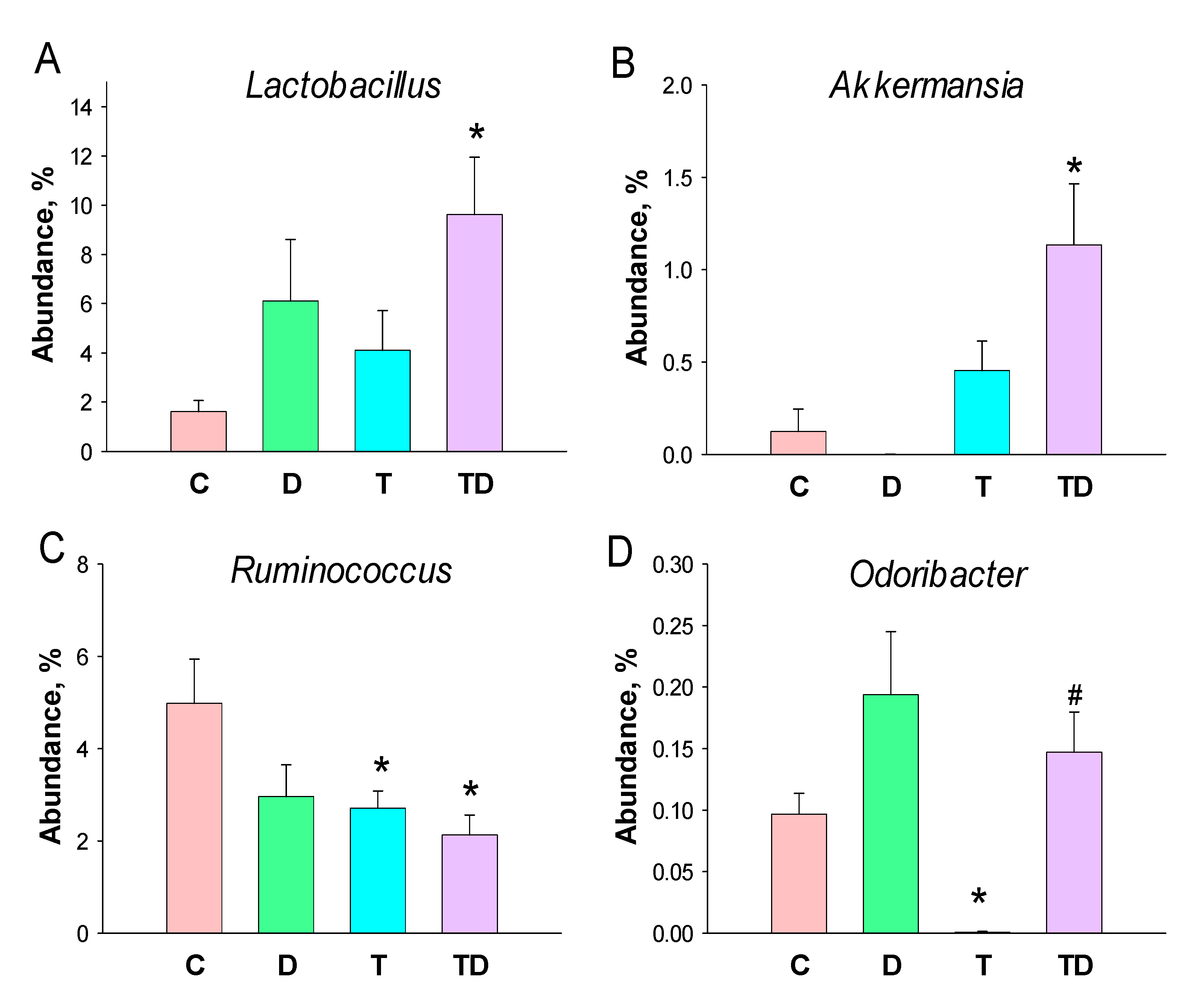
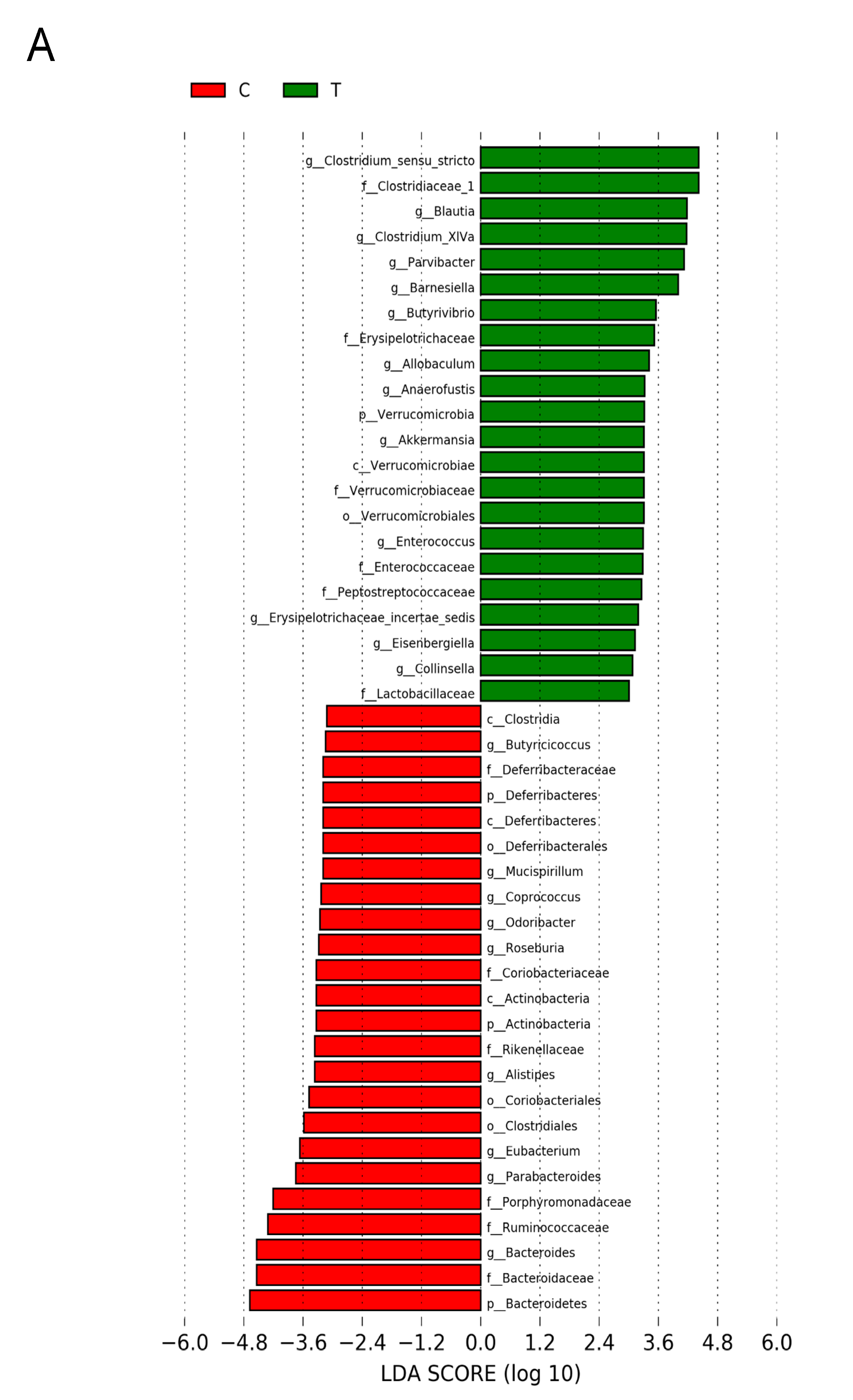
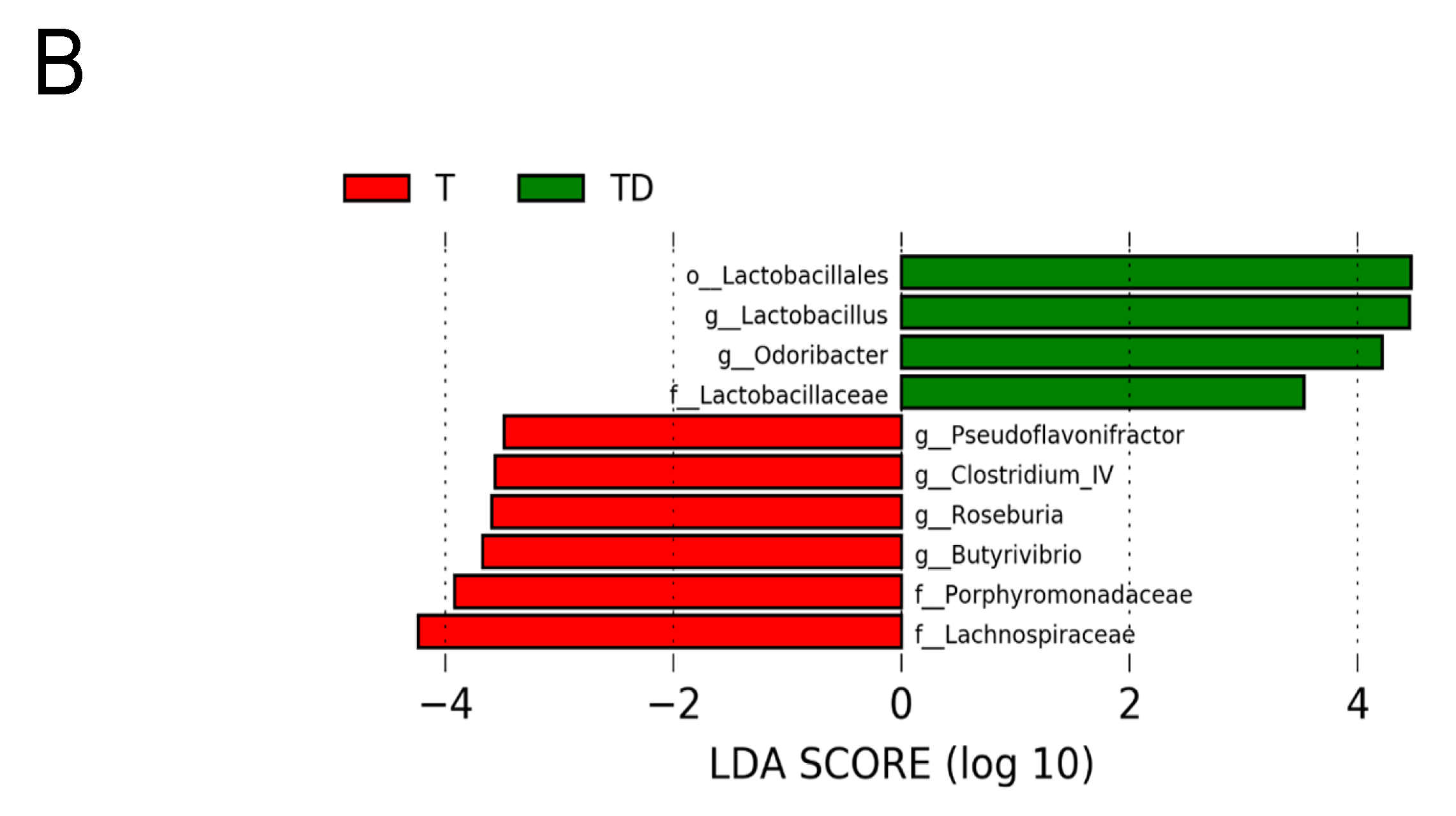
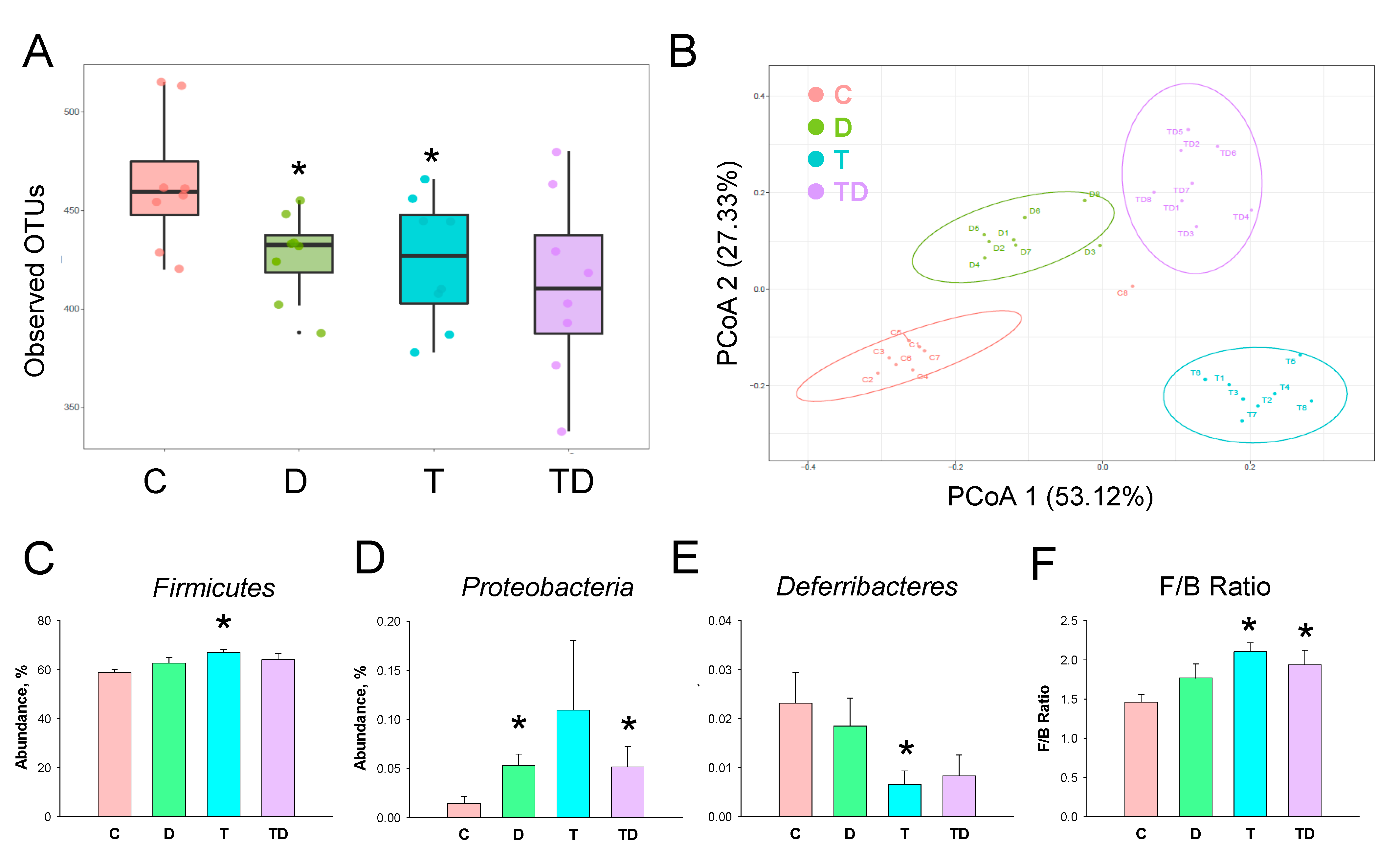
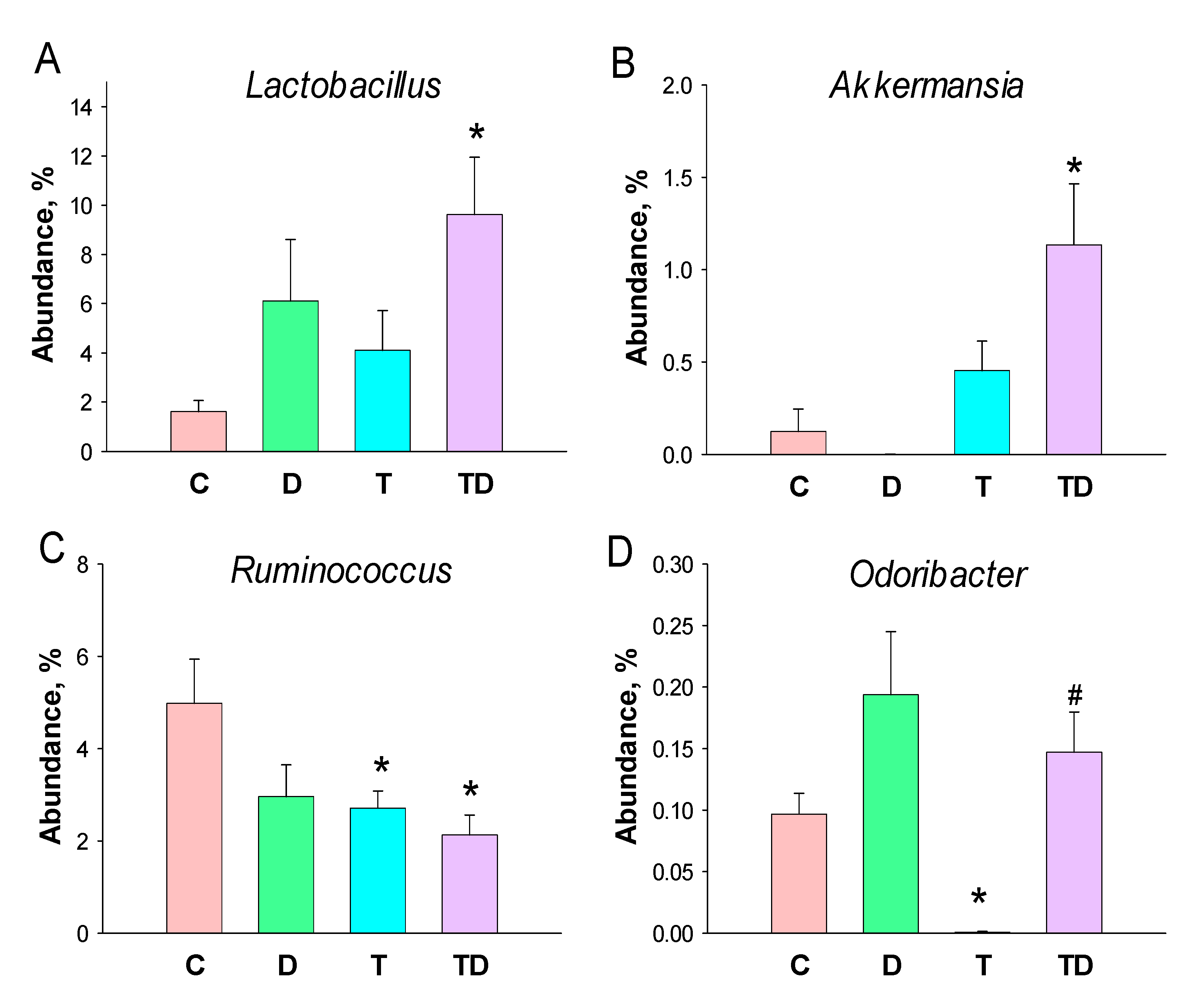
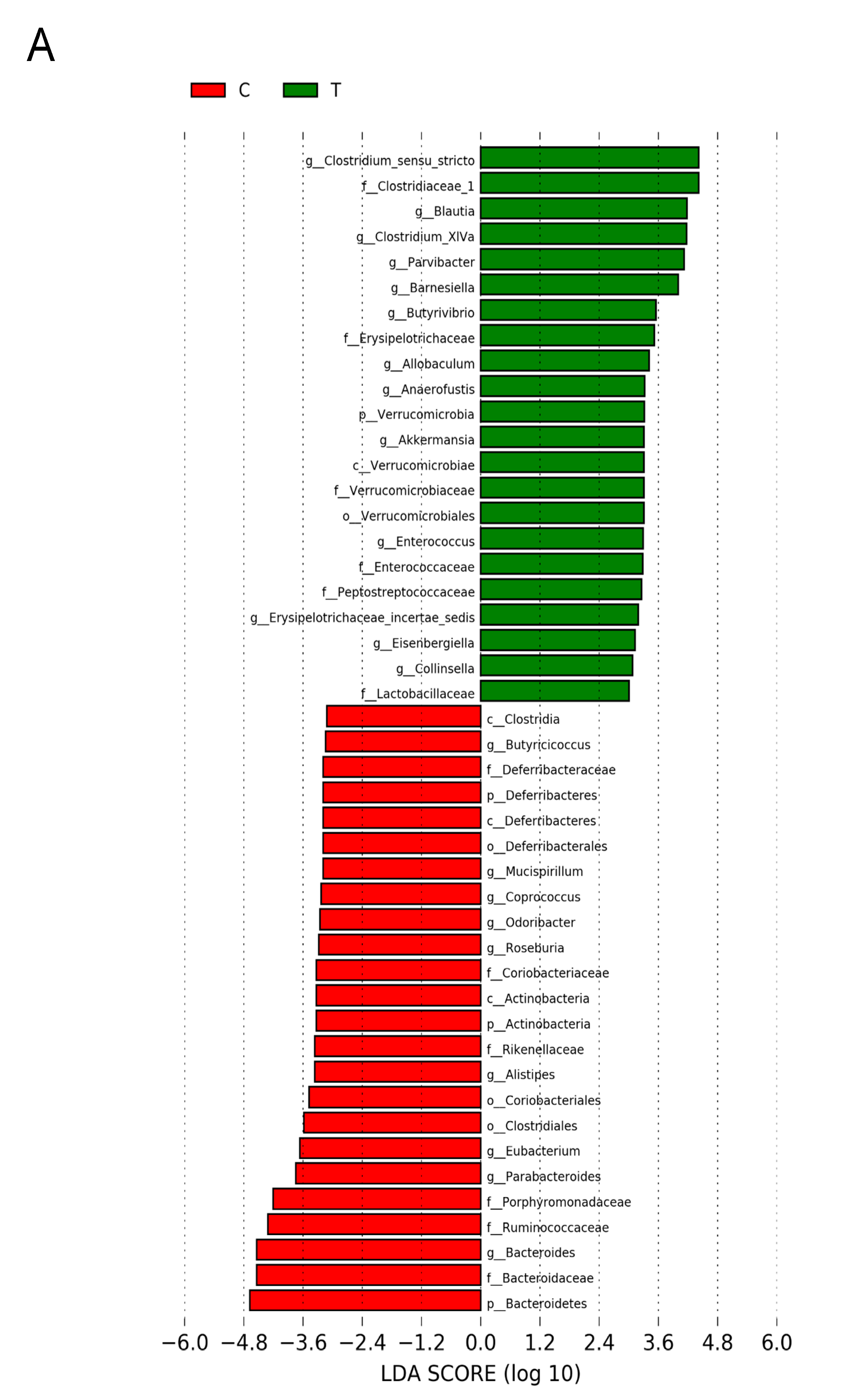
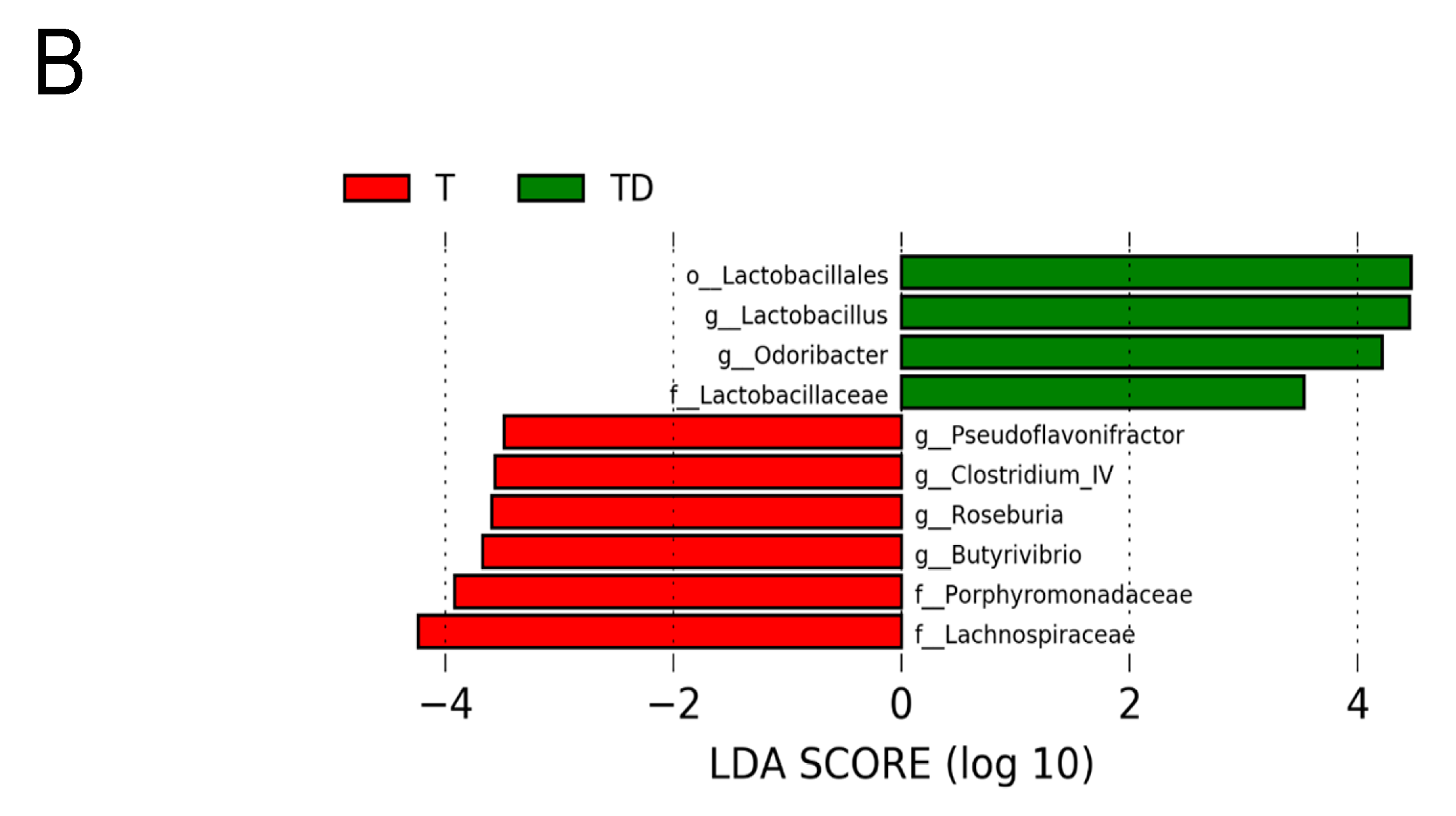
3.5. Gut Microbiota Compositions
4. Discussion
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Hsu, C.N.; Tain, Y.L. Animal Models for DOHaD Research: Focus on Hypertension of Developmental Origins. Biomedicines 2021, 9, 623. [Google Scholar] [CrossRef] [PubMed]
- Paixão, A.D.; Alexander, B.T. How the kidney is impacted by the perinatal maternal environment to develop hypertension. Biol. Reprod. 2013, 89, 144. [Google Scholar] [CrossRef] [PubMed]
- Hanson, M. The birth and future health of DOHaD. J. Dev. Orig. Health Dis. 2015, 6, 434–437. [Google Scholar] [CrossRef] [PubMed]
- Waring, R.H.; Harris, R.M.; Mitchell, S.C. In utero exposure to carcinogens: Epigenetics, developmental disruption and consequences in later life. Maturitas 2016, 86, 59–63. [Google Scholar] [CrossRef] [PubMed]
- Mulero-Navarro, S.; Fernandez-Salguero, P.M. New trends in aryl hydrocarbon receptor biology. Front. Cell Dev. Biol. 2016, 4, 45. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Aragon, A.C.; Goens, M.B.; Carbett, E.; Walker, M.K. Perinatal 2,3,7,8-tetrachlorodibenzo-p-dioxin exposure sensitizes offspring to angiotensin II-induced hypertension. Cardiovasc. Toxicol. 2008, 8, 145–154. [Google Scholar] [CrossRef] [Green Version]
- Hsu, C.N.; Lin, Y.J.; Lu, P.C.; Tain, Y.L. Maternal resveratrol therapy protects male rat offspring against programmed hypertension induced by TCDD and dexamethasone exposures: Is it relevant to aryl hydrocarbon receptor? Int. J. Mol. Sci. 2018, 19, 2459. [Google Scholar] [CrossRef] [Green Version]
- Hsu, C.N.; Chan, J.Y.H.; Yu, H.R.; Lee, W.C.; Wu, K.L.H.; Chang-Chien, G.P.; Lin, S.; Hou, C.Y.; Tain, Y.L. Targeting on Gut Microbiota-Derived Metabolite Trimethylamine to Protect Adult Male Rat Offspring against Hypertension Programmed by Combined Maternal High-Fructose Intake and Dioxin Exposure. Int. J. Mol. Sci. 2020, 21, 5488. [Google Scholar] [CrossRef]
- Yang, T.; Santisteban, M.M.; Rodriguez, V.; Li, E.; Ahmari, N.; Carvajal, J.M.; Zadeh, M.; Gong, M.; Qi, Y.; Zubcevic, J.; et al. Gut dysbiosis is linked to hypertension. Hypertension 2015, 65, 1331–1340. [Google Scholar] [CrossRef] [Green Version]
- Richards, E.M.; Pepine, C.J.; Raizada, M.K.; Kim, S. The Gut, Its Microbiome, and Hypertension. Curr. Hypertens. Rep. 2017, 19, 36. [Google Scholar] [CrossRef] [Green Version]
- Hsu, C.N.; Hou, C.Y.; Hsu, W.H.; Tain, Y.L. Cardiovascular Diseases of Developmental Origins: Preventive Aspects of Gut Microbiota-Targeted Therapy. Nutrients 2021, 13, 2290. [Google Scholar] [CrossRef]
- Schiattarella, G.G.; Sannino, A.; Toscano, E.; Giugliano, G.; Gargiulo, G.; Franzone, A.; Trimarco, B.; Esposito, G.; Perrino, C. Gut microbe-generated metabolite trimethylamine-N-oxide as cardiovascular risk biomarker: A systematic review and dose-response meta-analysis. Eur. Heart J. 2017, 38, 2948–2956. [Google Scholar] [CrossRef] [Green Version]
- Pluznick, J.L. Microbial Short-Chain Fatty Acids and Blood Pressure Regulation. Curr. Hypertens. Rep. 2017, 19, 25. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Velasquez, M.T.; Ramezani, A.; Manal, A.; Raj, D.S. Trimethylamine N-Oxide: The Good, the Bad and the Unknown. Toxins 2016, 8, 326. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Petriello, M.C.; Charnigo, R.; Sunkara, M.; Soman, S.; Pavuk, M.; Birnbaum, L.; Morris, A.J.; Hennig, B. Relationship between serum trimethylamine N-oxide and exposure to dioxin-like pollutants. Environ. Res. 2018, 162, 211–218. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.; Roberts, A.B.; Buffa, J.A.; Levison, B.S.; Zhu, W.; Org, E.; Gu, X.; Huang, Y.; Zamanian-Daryoush, M.; Culley, M.K.; et al. Non-lethal Inhibition of Gut Microbial Trimethylamine Production for the Treatment of Atherosclerosis. Cell 2015, 163, 1585–1595. [Google Scholar] [CrossRef] [Green Version]
- Hsu, C.N.; Chang-Chien, G.P.; Lin, S.; Hou, C.Y.; Tain, Y.L. Targeting on Gut Microbial Metabolite Trimethylamine-N-Oxide and Short-Chain Fatty Acid to Prevent Maternal High-Fructose-Diet-Induced Developmental Programming of Hypertension in Adult Male Offspring. Mol. Nutr. Food Res. 2019, 63, 1900073. [Google Scholar] [CrossRef] [PubMed]
- Hsu, C.N.; Tain, Y.L. Targeting the Renin-Angiotensin-Aldosterone System to Prevent Hypertension and Kidney Disease of Developmental Origins. Int. J. Mol. Sci. 2021, 22, 2298. [Google Scholar] [CrossRef] [PubMed]
- Reckelhoff, J.F. Gender differences in the regulation of blood pressure. Hypertension 2001, 7, 1199–1208. [Google Scholar] [CrossRef] [Green Version]
- Franczak, A.; Nynca, A.; Valdez, K.E.; Mizinga, K.M.; Petroff, B.K. Effects of acute and chronic exposure to the aryl hydrocarbon receptor agonist 2,3,7,8-tetrachlorodibenzo-p-dioxin on the transition to reproductive senescence in female Sprague-Dawley rats. Biol. Reprod. 2006, 74, 125–130. [Google Scholar] [CrossRef] [Green Version]
- Clarke, K.R.; Green, R.H. Statistical design and analysis for a ‘biological effects’ study. Mar. Ecol. Prog. Ser. 1988, 46, 213–226. [Google Scholar] [CrossRef]
- Wagner, B.D.; Grunwald, G.K.; Zerbe, G.O.; Mikulich-Gilbertson, S.K.; Robertson, C.E.; Zemanick, E.T.; Harris, J.K. On the use of diversity measures in longitudinal sequencing studies of microbial communities. Front. Microbiol. 2018, 9, 1037. [Google Scholar] [CrossRef] [Green Version]
- Pluznick, J.L. Renal and cardiovascular sensory receptors and blood pressure regulation. Am. J. Physiol. Ren. Physiol. 2013, 305, F439–F444. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, N. The role of endogenous aryl hydrocarbon receptor signaling in cardiovascular physiology. J. Cardiovasc. Dis. Res. 2011, 2, 91–95. [Google Scholar] [CrossRef] [Green Version]
- Sugai, E.; Yoshioka, W.; Kakeyama, M.; Ohsako, S.; Tohyama, C. In utero and lactational exposure to 2,3,7,8-tetrachlorodibenzo-p-dioxin modulates dysregulation of the lipid metabolism in mouse offspring fed a high-calorie diet. J. Appl. Toxicol. 2014, 34, 296–306. [Google Scholar] [CrossRef] [PubMed]
- Yang, T.; Richards, E.M.; Pepine, C.J.; Raizada, M.K. The gut microbiota and the brain-gut-kidney axis in hypertension and chronic kidney disease. Nat. Rev. Nephrol. 2018, 14, 442–456. [Google Scholar] [CrossRef]
- Yan, Q.; Gu, Y.; Li, X.; Yang, W.; Jia, L.; Chen, C.; Han, X.; Huang, Y.; Zhao, L.; Li, P.; et al. Alterations of the Gut Microbiome in Hypertension. Front. Cell Infect. Microbiol. 2017, 7, 381. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cani, P.D.; de Vos, W.M. Next-Generation Beneficial Microbes: The Case of Akkermansia muciniphila. Front. Microbiol. 2017, 8, 1765. [Google Scholar] [CrossRef] [PubMed]
- Papandreou, C.; Moré, M.; Bellamine, A. Trimethylamine N-Oxide in Relation to Cardiometabolic Health-Cause or Effect? Nutrients 2020, 12, 1330. [Google Scholar] [CrossRef]
- Romano, K.A.; Vivas, E.I.; Amador-Noguez, D.; Rey, F.E. Intestinal microbiota composition modulates choline bioavailability from diet and accumulation of the proatherogenic metabolite trimethylamine-N-oxide. mBio 2015, 6, e02481. [Google Scholar] [CrossRef] [Green Version]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Gene | Forward | Reverse |
|---|---|---|
| GPR41 | TCTTCACCACCGTCTATCTCAC | CACAAGTCCTGCCACCCTC |
| GPR43 | CTGCCTGGGATCGTCTGTG | CATACCCTCGGCCTTCTGG |
| GPR109A | CGGTGGTCTACTATTTCTCC | CCCCTGGAATACTTCTGATT |
| Olfr78 | GAGGAAGCTCACTTTTGGTTTGG | CAGCTTCAATGTCCTTGTCACAG |
| Renin | AACATTACCAGGGCAACTTTCACT | ACCCCCTTCATGGTGATCTG |
| AGT | GCCCAGGTCGCGATGAT | TGTACAAGATGCTGAGTGAGGCAA |
| ACE | CACCGGCAAGGTCTGCTT | CTTGGCATAGTTTCGTGAGGAA |
| ACE2 | ACCCTTCTTACATCAGCCCTACTG | TGTCCAAAACCTACCCCACATAT |
| AT1R | GCTGGGCAACGAGTTTGTCT | CAGTCCTTCAGCTGGATCTTCA |
| AT2R | CAATCTGGCTGTGGCTGACTT | TGCACATCACAGGTCCAAAGA |
| AHR | GTCCTCAGCAGGAACGAAAG | CCAGGGAAGTCCAACTGTGT |
| AHRR | CAGCAACATGGCTTCTTTCA | TGAAGCACTGCATTCCAGAC |
| CYP1A1 | GCACTCTGGACAAACACCTG | ATATCCACCTTCTCGCCTGG |
| ARNT | GTCTCCCTCCCAGATGATGA | GCTGGTAGCCAACAGTAGCC |
| TIPARP | GTTGAGGGCCAATTACCAGA | GCTCCTGGCACATAATCCAT |
| R18S | GCCGCGGTAATTCCAGCTCCA | CCCGCCCGCTCCCAAGATC |
| Groups | C | D | T | TD |
|---|---|---|---|---|
| Body weight (BW) (g) | 360 ± 13 | 311 ± 8 * | 309 ± 7 * | 286 ± 14 * |
| Left kidney weight (g) | 1.57 ± 0.08 | 1.2 ± 0.07 * | 1.51 ± 0.07 | 1.37 ± 0.08 |
| Left kidney weight/100g BW | 0.44 ± 0.01 | 0.49 ± 0.01 | 0.49 ± 0.02 | 0.48 ± 0.01 |
| Systolic BP (mmHg) | 133 ± 1 | 133 ± 1 | 143 ± 1 * | 135 ± 1 # |
| Diastolic BP (mmHg) | 90 ± 2 | 86 ± 2 | 91 ± 2 | 86 ± 2 # |
| MAP (mmHg) | 104 ± 1 | 102 ± 2 | 108 ± 1 * | 103 ± 1 |
| Creatinine (μM) | 16.4 ± 0.5 | 17.4 ± 0.9 | 15 ± 0.7 | 16.2 ± 0.6 |
| Groups | C | D | T | TD |
|---|---|---|---|---|
| DMA (ng/mL) | 117 ± 10 | 132 ± 11 | 118 ± 9 | 146 ± 15 |
| TMA (ng/mL) | 564 ± 23 | 810 ± 29 * | 593 ± 28 | 844 ± 57 * |
| TMAO (ng/mL) | 440 ± 23 | 444 ± 20 | 387 ± 20 | 429 ± 35 |
| TMAO-to-TMA ratio | 0.78 ± 0.02 | 0.55 ± 0.03 * | 0.66 ± 0.04 | 0.53 ± 0.06 * |
| DMA-to-TMAO ratio | 0.27 ± 0.02 | 0.3 ± 0.03 | 0.31 ± 0.03 | 0.35 ± 0.03 |
| Groups | C | D | T | TD |
|---|---|---|---|---|
| Acetic acid (ng/mL) | 169.9 ± 10 | 232.3 ± 6 * | 214.7 ± 10.1 * | 288.5 ± 16.4 *,# |
| Propionic acid (ng/mL) | 1.43 ± 0.06 | 1.4 ± 0.16 | 1.24 ± 0.23 | 1.36 ± 0.09 |
| Butyric acid (ng/mL) | 1.44 ± 0.13 | 1.28 ± 0.05 | 1.38 ± 0.07 | 1.32 ± 0.08 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hsu, C.-N.; Hou, C.-Y.; Lee, C.-T.; Chang-Chien, G.-P.; Lin, S.; Tain, Y.-L. Maternal 3,3-Dimethyl-1-Butanol Therapy Protects Adult Male Rat Offspring against Hypertension Programmed by Perinatal TCDD Exposure. Nutrients 2021, 13, 3041. https://doi.org/10.3390/nu13093041
Hsu C-N, Hou C-Y, Lee C-T, Chang-Chien G-P, Lin S, Tain Y-L. Maternal 3,3-Dimethyl-1-Butanol Therapy Protects Adult Male Rat Offspring against Hypertension Programmed by Perinatal TCDD Exposure. Nutrients. 2021; 13(9):3041. https://doi.org/10.3390/nu13093041
Chicago/Turabian StyleHsu, Chien-Ning, Chih-Yao Hou, Chien-Te Lee, Guo-Ping Chang-Chien, Sufan Lin, and You-Lin Tain. 2021. "Maternal 3,3-Dimethyl-1-Butanol Therapy Protects Adult Male Rat Offspring against Hypertension Programmed by Perinatal TCDD Exposure" Nutrients 13, no. 9: 3041. https://doi.org/10.3390/nu13093041
APA StyleHsu, C.-N., Hou, C.-Y., Lee, C.-T., Chang-Chien, G.-P., Lin, S., & Tain, Y.-L. (2021). Maternal 3,3-Dimethyl-1-Butanol Therapy Protects Adult Male Rat Offspring against Hypertension Programmed by Perinatal TCDD Exposure. Nutrients, 13(9), 3041. https://doi.org/10.3390/nu13093041