
Casein Glycomacropeptide: An Alternative Protein Substitute in Tyrosinemia Type I



Abstract
:1. Introduction
2. Materials and Methods
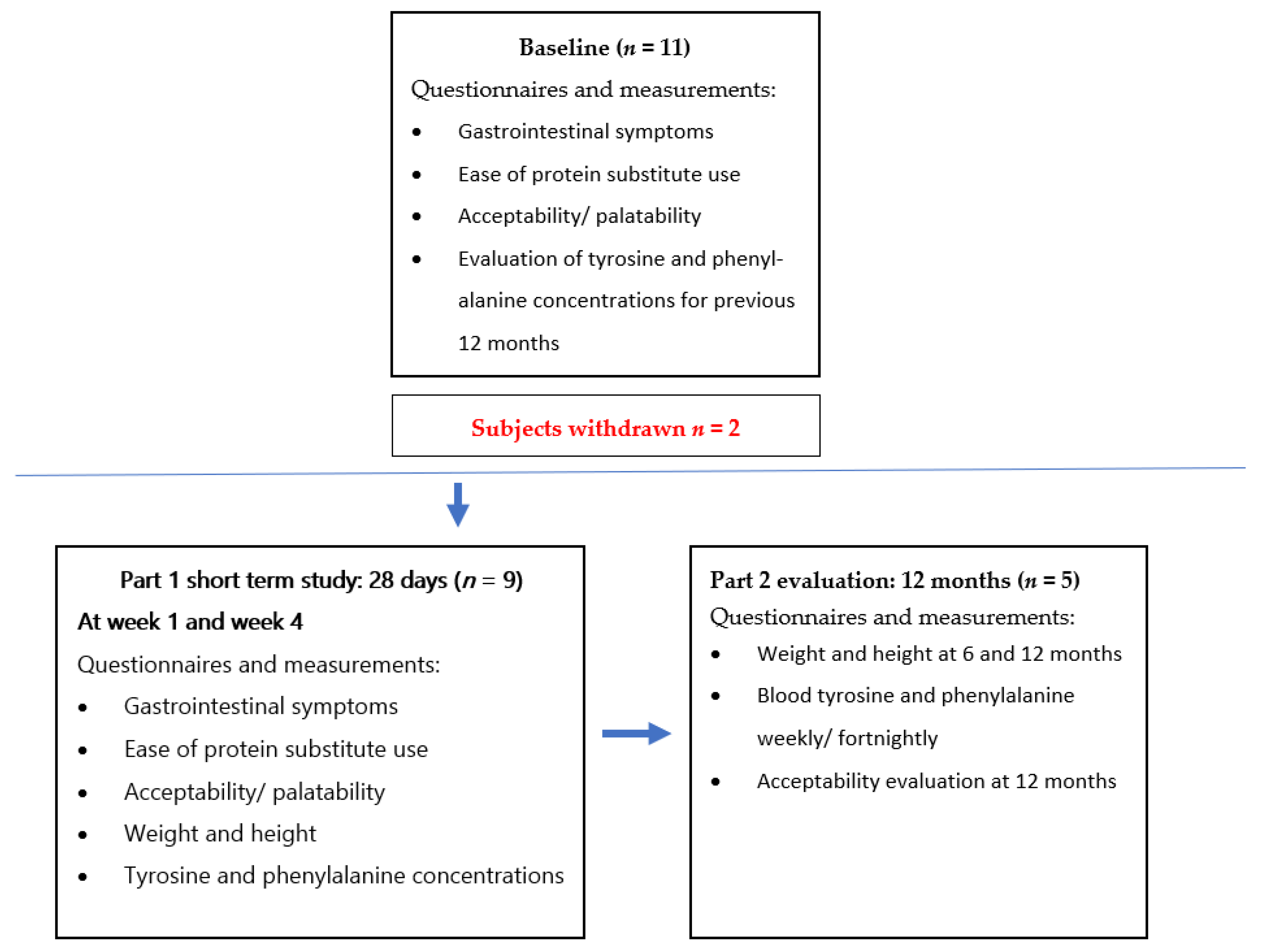
2.1. Study Design
2.2. Subjects
2.3. Protein Substitutes Used in the Study
2.4. Anthropometry
2.5. Blood Tyrosine, Phenylalanine, Succinylacetone and NTBC Concentrations
2.6. Natural Protein Intake
2.7. Statistical Analysis
2.8. Ethical Approval
3. Results
3.1. Subjects
3.2. Subject Withdrawal
3.3. Protein Substitute Type
Median Protein Substitute Intake in Part 1 and 2
3.4. NTBC and Concurrent Medications
3.5. Tyrosine and Phenylalanine Blood Concentrations
3.6. Routine Biochemistry
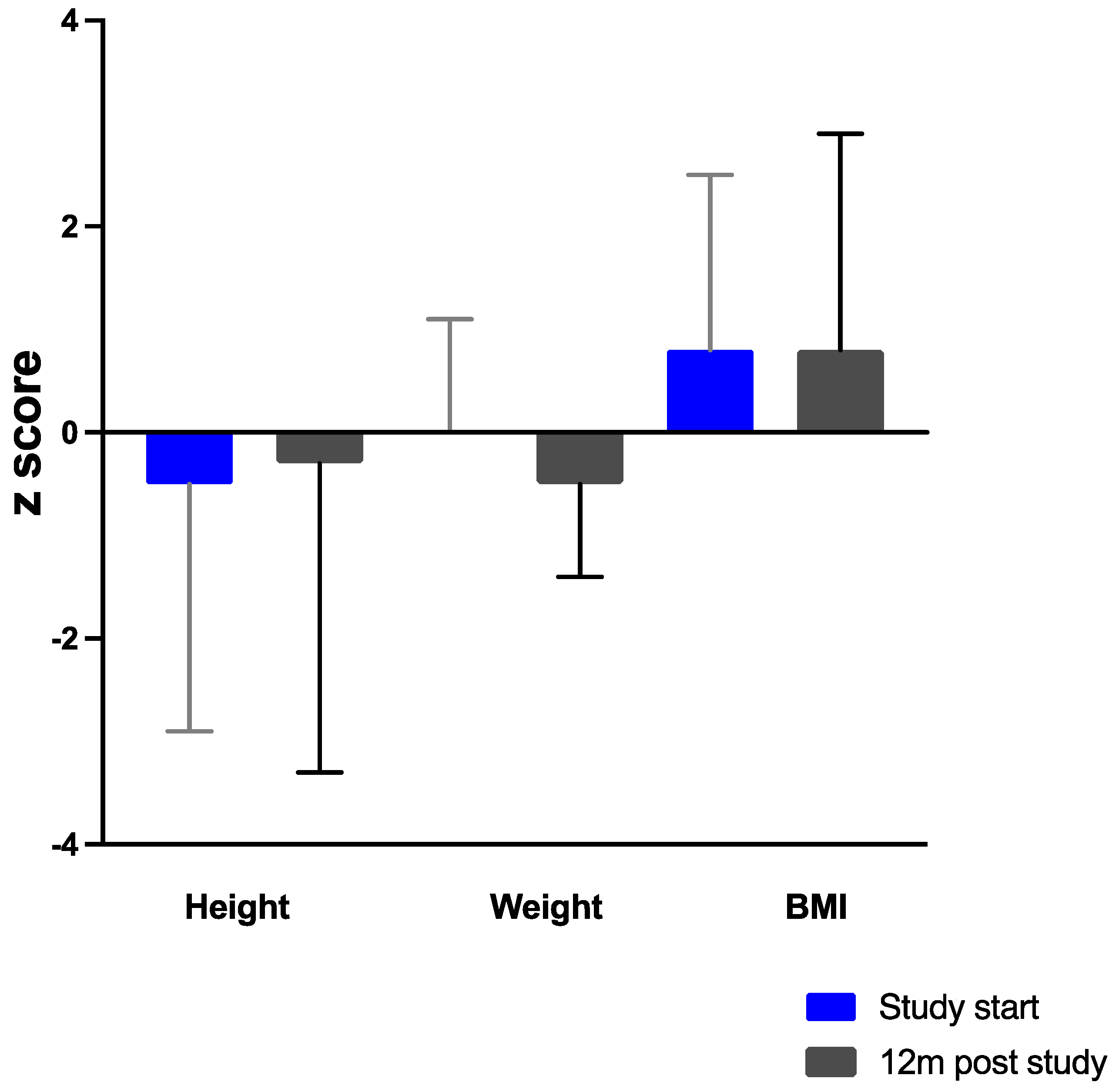
3.7. Anthropometry
3.8. Gastrointestinal Tolerance
3.9. Palatability
3.10. Ease of Preparation
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Baber, M.D. A case of congenital cirrhosis of the liver with renal tubular defects akin to those in the Fanconi syndrome. Arch. Dis. Child. 1956, 31, 335–339. [Google Scholar] [CrossRef] [Green Version]
- Van Spronsen, F.J.; van Rijn, M.; Meyer, U.; Das, A.M. Dietary Considerations in Tyrosinemia Type I. Adv. Exp. Med. Biol 2017, 959, 197–204. [Google Scholar] [CrossRef]
- Chakarapani, A.; Gissen, P.; McKiernan, P. Disorders of Tyrosine Metabolism. In Inborn Metabolic Diseases, 5th ed.; Saudubray, J.M., van den Berghe, G., Walter, J., Eds.; Springer: Berlin/Heidelberg, Germany, 2012; pp. 275–276. [Google Scholar]
- Van Ginkel, W.G.; van Reemst, H.E.; Kienstra, N.S.; Daly, A.; Rodenburg, I.L.; MacDonald, A.; Burgerhof, J.G.M.; de Blaauw, P.; van de Krogt, J.; Santra, S.; et al. The Effect of Various Doses of Phenylalanine Supplementation on Blood Phenylalanine and Tyrosine Concentrations in Tyrosinemia Type 1 Patients. Nutrients 2019, 11, 2816. [Google Scholar] [CrossRef] [Green Version]
- Wilson, C.J.; Van Wyk, K.G.; Leonard, J.V.; Clayton, P.T. Phenylalanine supplementation improves the phenylalanine profile in tyrosinaemia. J. Inherit. Metab. Dis. 2000, 23, 677–683. [Google Scholar] [CrossRef] [PubMed]
- MacDonald, A.; van Rijn, M.; Feillet, F.; Lund, A.M.; Bernstein, L.; Bosch, A.M.; Gizewska, M.; van Spronsen, F.J. Adherence issues in inherited metabolic disorders treated by low natural protein diets. Ann. Nutr. Metab. 2012, 61, 289–295. [Google Scholar] [CrossRef] [PubMed]
- Green, B.; Browne, R.; Firman, S.; Hill, M.; Rahman, Y.; Kaalund Hansen, K.; Adam, S.; Skeath, R.; Hallam, P.; Herlihy, I.; et al. Nutritional and Metabolic Characteristics of UK Adult Phenylketonuria Patients with Varying Dietary Adherence. Nutrients 2019, 11, 2459. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, B.; Brand-Miller, J. The role and potential of sialic acid in human nutrition. Eur. J. Clin. Nutr. 2003, 57, 1351–1369. [Google Scholar] [CrossRef] [Green Version]
- Ntemiri, A.; Chonchuir, F.N.; O’Callaghan, T.F.; Stanton, C.; Ross, R.P.; O’Toole, P.W. Glycomacropeptide Sustains Microbiota Diversity and Promotes Specific Taxa in an Artificial Colon Model of Elderly Gut Microbiota. J. Agric. Food Chem. 2017, 65, 1836–1846. [Google Scholar] [CrossRef]
- Ortega-Gonzalez, M.; Capitan-Canadas, F.; Requena, P.; Ocon, B.; Romero-Calvo, I.; Aranda, C.; Suarez, M.D.; Zarzuelo, A.; Sanchez de Medina, F.; Martinez-Augustin, O. Validation of bovine glycomacropeptide as an intestinal anti-inflammatory nutraceutical in the lymphocyte-transfer model of colitis. Br. J. Nutr. 2014, 111, 1202–1212. [Google Scholar] [CrossRef]
- Otani, H.; Monnai, M.; Kawasaki, Y.; Kawakami, H.; Tanimoto, M. Inhibition of mitogen-induced proliferative responses of lymphocytes by bovine kappa-caseinoglycopeptides having different carbohydrate chains. J. Dairy Res. 1995, 62, 349–357. [Google Scholar] [CrossRef]
- Aimutis, W.R. Bioactive properties of milk proteins with particular focus on anticariogenesis. J. Nutr. 2004, 134, 989S–995S. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lim, K.; van Calcar, S.C.; Nelson, K.L.; Gleason, S.T.; Ney, D.M. Acceptable low-phenylalanine foods and beverages can be made with glycomacropeptide from cheese whey for individuals with PKU. Mol. Genet. Metab. 2007, 92, 176–178. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ney, D.M.; Gleason, S.T.; van Calcar, S.C.; MacLeod, E.L.; Nelson, K.L.; Etzel, M.R.; Rice, G.M.; Wolff, J.A. Nutritional management of PKU with glycomacropeptide from cheese whey. J. Inherit. Metab. Dis. 2009, 32, 32–39. [Google Scholar] [CrossRef] [Green Version]
- Pena, M.J.; Pinto, A.; Daly, A.; MacDonald, A.; Azevedo, L.; Rocha, J.C.; Borges, N. The Use of Glycomacropeptide in Patients with Phenylketonuria: A Systematic Review and Meta-Analysis. Nutrients 2018, 10, 1794. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Daly, A.; Hogler, W.; Crabtree, N.; Shaw, N.; Evans, S.; Pinto, A.; Jackson, R.; Strauss, B.J.; Wilcox, G.; Rocha, J.C.; et al. Growth and Body Composition in PKU Children-A Three-Year Prospective Study Comparing the Effects of L-Amino Acid to Glycomacropeptide Protein Substitutes. Nutrients 2021, 13, 1323. [Google Scholar] [CrossRef]
- Daly, A.; Evans, S.; Chahal, S.; Santra, S.; MacDonald, A. Glycomacropeptide in children with phenylketonuria: Does its phenylalanine content affect blood phenylalanine control? J. Hum. Nutr. Diet. 2017, 30, 515–523. [Google Scholar] [CrossRef]
- Chinsky, J.M.; Singh, R.; Ficicioglu, C.; van Karnebeek, C.D.M.; Grompe, M.; Mitchell, G.; Waisbren, S.E.; Gucsavas-Calikoglu, M.; Wasserstein, M.P.; Coakley, K.; et al. Diagnosis and treatment of tyrosinemia type I: A US and Canadian consensus group review and recommendations. Genet. Med. 2017, 19, 1380. [Google Scholar] [CrossRef] [Green Version]
- Van Dam, E.; Daly, A.; Venema-Liefaard, G.; van Rijn, M.; Derks, T.G.J.; McKiernan, P.J.; Rebecca Heiner-Fokkema, M.; MacDonald, A.; van Spronsen, F.J. What Is the Best Blood Sampling Time for Metabolic Control of Phenylalanine and Tyrosine Concentrations in Tyrosinemia Type 1 Patients? JIMD Rep. 2017, 36, 49–57. [Google Scholar] [CrossRef] [Green Version]
- Sawin, E.A.; De Wolfe, T.J.; Aktas, B.; Stroup, B.M.; Murali, S.G.; Steele, J.L.; Ney, D.M. Glycomacropeptide is a prebiotic that reduces Desulfovibrio bacteria, increases cecal short-chain fatty acids, and is anti-inflammatory in mice. Am. J. Physiol. Gastrointest. Liver Physiol. 2015, 309, G590–G601. [Google Scholar] [CrossRef] [Green Version]
- Friedman, M.; Levin, C.E. Nutritional and medicinal aspects of D-amino acids. Amino Acids 2012, 42, 1553–1582. [Google Scholar] [CrossRef]
- Acosta, P.B.; Yannicelli, S.; Singh, R.; Mofidi, S.; Steiner, R.; DeVincentis, E.; Jurecki, E.; Bernstein, L.; Gleason, S.; Chetty, M.; et al. Nutrient intakes and physical growth of children with phenylketonuria undergoing nutrition therapy. J. Am. Diet. Assoc. 2003, 103, 1167–1173. [Google Scholar] [CrossRef]
- Pardridge, W.M. Blood-brain barrier carrier-mediated transport and brain metabolism of amino acids. Neurochem. Res. 1998, 23, 635–644. [Google Scholar] [CrossRef]
- Weglage, J.; Bramswig, J.H.; Koch, H.G.; Karassalidou, S.; Ullrich, K. Growth in patients with phenylketonuria. Eur. J. Pediatr. 1994, 153, 537–538. [Google Scholar] [CrossRef]
- Van Ginkel, W.G.; van Vliet, D.; van der Goot, E.; Faassen, M.; Vogel, A.; Heiner-Fokkema, M.R.; van der Zee, E.A.; van Spronsen, F.J. Blood and Brain Biochemistry and Behaviour in NTBC and Dietary Treated Tyrosinemia Type 1 Mice. Nutrients 2019, 11, 2486. [Google Scholar] [CrossRef] [Green Version]
- Masurel-Paulet, A.; Poggi-Bach, J.; Rolland, M.O.; Bernard, O.; Guffon, N.; Dobbelaere, D.; Sarles, J.; de Baulny, H.O.; Touati, G. NTBC treatment in tyrosinaemia type I: Long-term outcome in French patients. J. Inherit. Metab. Dis. 2008, 31, 81–87. [Google Scholar] [CrossRef]
- Thimm, E.; Herebian, D.; Assmann, B.; Klee, D.; Mayatepek, E.; Spiekerkoetter, U. Increase of CSF tyrosine and impaired serotonin turnover in tyrosinemia type I. Mol. Genet. Metab. 2011, 102, 122–125. [Google Scholar] [CrossRef] [PubMed]
- Van Ginkel, W.G.; Jahja, R.; Huijbregts, S.C.; Daly, A.; MacDonald, A.; De Laet, C.; Cassiman, D.; Eyskens, F.; Korver-Keularts, I.M.; Goyens, P.J.; et al. Neurocognitive outcome in tyrosinemia type 1 patients compared to healthy controls. Orphanet J. Rare Dis. 2016, 11, 87. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Van Ginkel, W.G.; Rodenburg, I.L.; Harding, C.O.; Hollak, C.E.M.; Heiner-Fokkema, M.R.; van Spronsen, F.J. Long-Term Outcomes and Practical Considerations in the Pharmacological Management of Tyrosinemia Type 1. Paediatr. Drugs 2019, 21, 413–426. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Naoi, M.; Maruyama, W.; Takahashi, T.; Ota, M.; Parvez, H. Inhibition of tryptophan hydroxylase by dopamine and the precursor amino acids. Biochem. Pharm. 1994, 48, 207–211. [Google Scholar] [CrossRef] [PubMed]
- Aarela, L.; Hiltunen, P.; Soini, T.; Vuorela, N.; Huhtala, H.; Nevalainen, P.I.; Heikinheimo, M.; Kivela, L.; Kurppa, K. Type 1 tyrosinemia in Finland: A nationwide study. Orphanet J. Rare Dis. 2020, 15, 281. [Google Scholar] [CrossRef] [PubMed]
- Yilmaz, O.; Daly, A.; Pinto, A.; Ashmore, C.; Evans, S.; Gupte, G.; Santra, S.; Preece, M.A.; McKiernan, P.; Kitchen, S.; et al. Natural Protein Tolerance and Metabolic Control in Patients with Hereditary Tyrosinaemia Type 1. Nutrients 2020, 12, 1148. [Google Scholar] [CrossRef] [Green Version]
- Van Vliet, D.; van Dam, E.; van Rijn, M.; Derks, T.G.; Venema-Liefaard, G.; Hitzert, M.M.; Lunsing, R.J.; Heiner-Fokkema, M.R.; van Spronsen, F.J. Infants with Tyrosinemia Type 1: Should phenylalanine be supplemented? JIMD Rep. 2015, 18, 117–124. [Google Scholar] [CrossRef] [PubMed] [Green Version]

{kind=link}
{kind=link}
| Low Phe/TyrCGMP | Phenylalanine/Tyrosine-Free Amino Acid Supplements (L-AA) | ||||
|---|---|---|---|---|---|
| Units | CGMP PS per 20 g PE | Tyr Cooler 20 g PE | Tyr Express 20 g PE | Tyr Shake and Go 20 g PE | |
| Manufacturer | Vitaflo International | Vitaflo– International | Vitaflo– International | Galen Ltd | |
| Macronutrients | |||||
| Energy | kJ | 508 | 549 | 429 | 693 |
| kcal | 120 | 130 | 101 | 163 | |
| Fat | g | 1.6 | 1.6 | 0.07 | <0.5 |
| of which saturates | g | 0.35 | 0.3 | 0 | <0.5 |
| of which DHA | mg | 110 | 134 | NA | NA |
| Carbohydrate | g | 6.3 | 8.9 | 4.7 | 17 |
| of which sugars | g | 1.4 | 5.9 | 0.33 | <12 |
| Protein equivalent | g | 20 | 20 | 20 | 20 |
| Fiber | g | 0 | 0 | 0 | 1.7 |
| Salt | g | 0.71 | 0.26 | 0.44 | 0.4 |
| Vitamins, minerals and micronutrients | |||||
| Vitamin A | µg RE | 259 | 261 | 283 | 241 |
| Vitamin D | µg | 5.0 | 10 | 4.5 | 4.7 |
| Vitamin E | mg αTE | 5.3 | 5.2 | 5.3 | 5.0 |
| Vitamin C | mg | 26 | 37 | 36.7 | 30.5 |
| Vitamin K | µg | 23 | 24 | 34 | 33 |
| Thiamin | mg | 0.6 | 0.7 | 0.7 | 0.7 |
| Riboflavin | mg | 0.6 | 0.8 | 0.8 | 0.8 |
| Niacin | mg | 3.2 | 3.5 | 8.4 | 8.6 |
| Vitamin B6 | mg | 0.6 | 0.9 | 1.0 | 0.8 |
| Folic Acid | µg | 102 | 101 | 136 | 103 |
| Vitamin B12 | µg | 1.6 | 1.6 | 1.6 | 1.3 |
| Biotin | µg | 13 | 13 | 64 | 52 |
| Pantothenic acid | mg | 2.0 | 1.9 | 2.7 | 2.4 |
| Choline | mg | 200 | 200 | 204 | 198 |
| Sodium | mmol | 12 | 4.5 | 7.5 | 7.9 |
| Potassium | mmol | 5.9 | 6.1 | 8.0 | 8.9 |
| Chloride | mmol | 0.2 | 3.9 | 6.9 | 7.9 |
| Calcium | mg | 399 | 400 | 407 | 371 |
| Phosphorus | mg | 413 | 357 | 363 | 293 |
| Magnesium | mg | 115 | 110 | 128 | 103 |
| Iron | mg | 7.4 | 7.3 | 7.3 | 6.9 |
| Copper | µg | 0.6 | 0.7 | 0.8 | 0.6 |
| Zinc | mg | 7.4 | 5.6 | 7.3 | 5.7 |
| Manganese | mg | 0.4 | 0.5 | 1.1 | 1.1 |
| Iodine | µg | 84 | 85 | 86 | 81 |
| Molybdenum | µg | 20 | 23 | 49 | 60 |
| Selenium | µg | 30 | 26 | 30 | 28 |
| Chromium | µg | 12 | 14 | 30 | 24 |
| Amino acids g/20 g protein equivalent | |||||
| L-Alanine | g | 0.83 | 1.62 | 1.44 | 1.32 |
| L-Arginine | g | 1.70 | 1.98 | 1.85 | 1.58 |
| L-Aspartic Acid | g | 1.31 | 3.06 | 2.86 | 2.75 |
| L-Cystine | g | 0.24 | 0.73 | 0.69 | 0.74 |
| L-Glutamine | g | 2.70 | 0.00 | 1.83 | 1.8 |
| Glycine | g | 2.48 | 1.62 | 1.50 | 2.46 |
| L-Histidine | g | 0.70 | 1.08 | 1.01 | 0.86 |
| L-Isoleucine | g | 1.42 | 1.79 | 1.68 | 1.48 |
| L-Leucine | g | 3.00 | 2.89 | 2.69 | 2.57 |
| L-Lysine | g | 0.95 | 2.05 | 1.91 | 1.58 |
| L-Methionine | g | 0.28 | 0.47 | 0.43 | 0.40 |
| L-Phenylalanine | g | 0.036 (36 mg) | 0.00 | 0.00 | 0.00 |
| L-Proline | g | 1.60 | 1.65 | 1.55 | 1.72 |
| L-Serine | g | 1.01 | 1.27 | 1.18 | 1.00 |
| L-Threonine | g | 2.29 | 1.39 | 1.29 | 1.72 |
| L-Tryptophan | g | 0.40 | 0.57 | 0.54 | 0.46 |
| L-Tyrosine | g | <0.011 (11 mg) | 0.00 | 0.00 | 0.00 |
| L-Valine | g | 1.14 | 1.97 | 1.83 | 1.75 |
| PART 1 SUBJECTS SHORT TERM EVALUATION OVER 28 DAYS | |||||||
|---|---|---|---|---|---|---|---|
| Subject | Age (years) | Sex | Number of Estimated Protein Exchanges | Ethnicity | % (Number) of Days Completed in Part 1 | Subjects Completing Part 2 | % of Protein Equivalent from CGMP |
| 1 | 17.1 | M | 28 | British Asian | 89% (25) | No | 100% |
| 2 | 10.5 | M | 25 | British Asian | 86% (24) | No | 33% |
| 3 | 17.7 | F | 26 | Caucasian | 100% (28) | No | 33% |
| 4 | 14.7 | M | 16 | Caucasian | 64% (18) | No | 33% |
| 5 | 5.3 | F | 12 | Caucasian | 1 Withdrawn | No | 44% |
| 6 | 15.5 | M | 25 | British Asian | 17 Withdrawn | No | 33% |
| PART 2 SUBJECTS FOLLOWED UP FOR 12 MONTHS | |||||||
| 7 | 15.3 | F | 24 | Caucasian | 100% (28) | Yes | 100% |
| 8 | 15.4 | M | 30 | Arabic | 100% (28) | Yes | 100% |
| 9 | 13.9 | M | 20 | British Asian | 100% (28) | Yes | 100% |
| 10 | 15.0 | F | 20 | British Asian | 100% (28) | Yes | 100% |
| 11 | 8.6 | F | 20 | British Asian | 100% (28) | Yes | 100% |
| Subjects n = 9 | Median Tyr (Range) μmol/L 12 Months Pre-Study | Median Tyr (Range) μmol/L During 28 Day Study | Median Tyr (Range) μmol/L for 12 Months Using Study Product | Median Phe (Range) μmol/L 12 Months Pre-Study | Median Phe (Range) μmol/L during 28 Day Study | Median Phe (Range) μmol/L for 12 Months Using Study Product |
|---|---|---|---|---|---|---|
| 1 | 312 (200–400) | 330 (200–400) | - | 45 (30–100) | 40 (30–100) | - |
| 2 | 338 (200–400) | 375 (200–400) | - | 30 (30–60) | 40 (30–60) | - |
| 3 | 315 (290–400) | 324 (290–400) | - | 45 (30–60) | 55 (50–60) | - |
| 4 | 428 (200–440) | 415 (200–430) | - | 40 (30–60) | 40 (30–60) | - |
| 7 | 393 (200–400) | 375 (200–400) | 430 (290–660) | 55 (30–60) | 40 (30–60) | 40 (30–50) |
| 8 | 490 (200–500) | 460 (200–490) | 430 (270–710) | 50 (30–100) | 50 (30–100) | 40 (30–50) |
| 9 | 535 (200–600) | 495 (200–600) | 330 (270–380) | 55 (30–60) | 50 (30–60) | 30 (30–40) |
| 10 | 370 (200–600) | 445 (200–600) | 570 (320–940) | 30 (30–60) | 30 (30–60) | 50 (30–70) |
| 11 | 525 (200–600) | 385 (200–400) | 490 (290–830) | 50 (30–70) | 60 (30–70) | 30 (20–60) |
| Median | 393 (200–600) | 385 (200–600) | 430 (270 to 940) | 45 (30–100) | 40 (30–100) | 40 (20 to 70) |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Daly, A.; Evans, S.; Pinto, A.; Ashmore, C.; MacDonald, A. Casein Glycomacropeptide: An Alternative Protein Substitute in Tyrosinemia Type I. Nutrients 2021, 13, 3224. https://doi.org/10.3390/nu13093224
Daly A, Evans S, Pinto A, Ashmore C, MacDonald A. Casein Glycomacropeptide: An Alternative Protein Substitute in Tyrosinemia Type I. Nutrients. 2021; 13(9):3224. https://doi.org/10.3390/nu13093224
Chicago/Turabian StyleDaly, Anne, Sharon Evans, Alex Pinto, Catherine Ashmore, and Anita MacDonald. 2021. "Casein Glycomacropeptide: An Alternative Protein Substitute in Tyrosinemia Type I" Nutrients 13, no. 9: 3224. https://doi.org/10.3390/nu13093224
APA StyleDaly, A., Evans, S., Pinto, A., Ashmore, C., & MacDonald, A. (2021). Casein Glycomacropeptide: An Alternative Protein Substitute in Tyrosinemia Type I. Nutrients, 13(9), 3224. https://doi.org/10.3390/nu13093224