
The Timing and Duration of Folate Restriction Differentially Impacts Colon Carcinogenesis

 , ,
, , {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
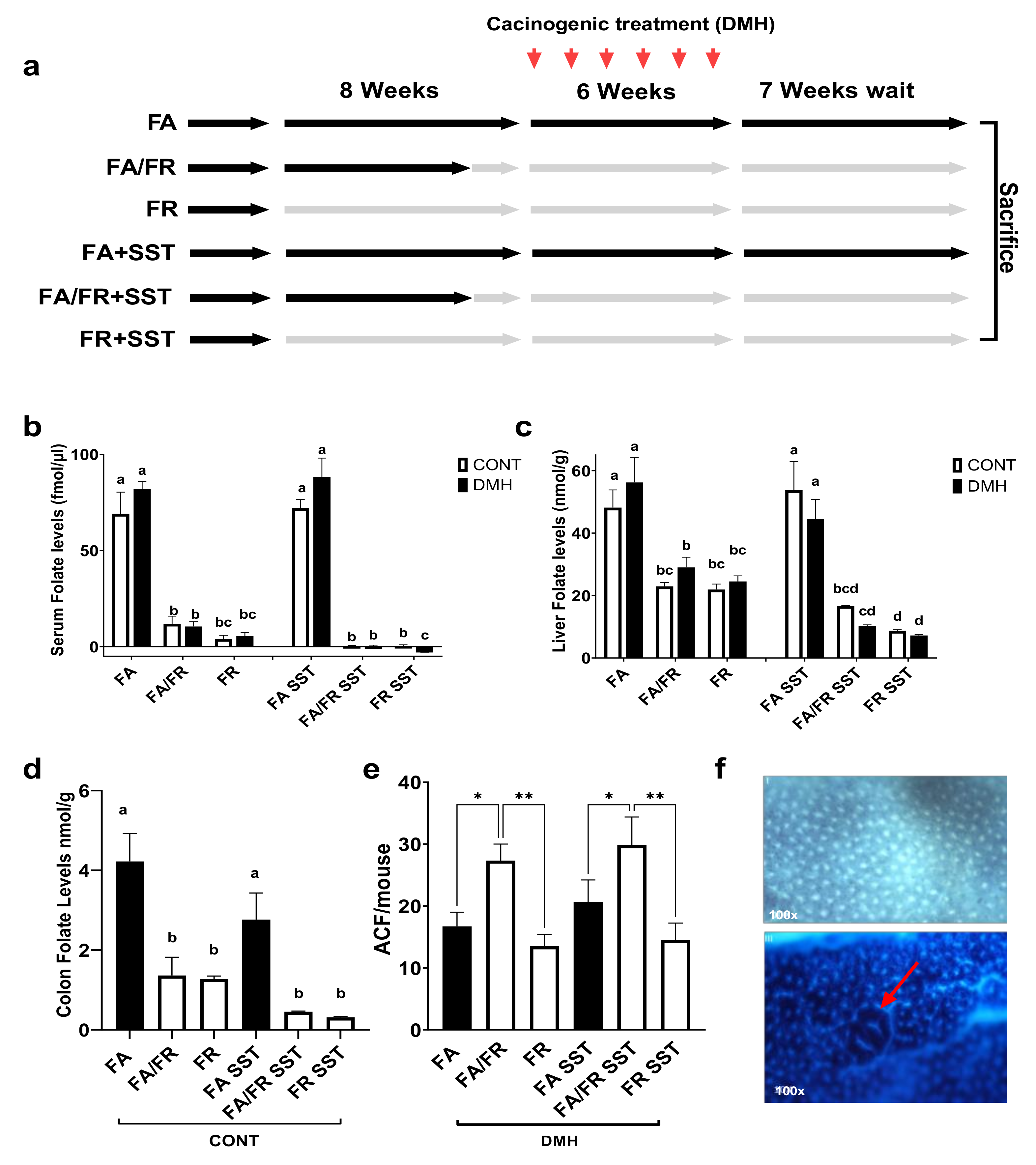
2.1. Study Design
2.2. Animals
2.3. Diets
2.4. Food Intake and Weights
2.5. Carcinogen Treatment
2.6. Aberrant Crypt Foci (ACF) Analysis
2.7. Folate Assay
2.8. Isolation of Whole Cell Extract and Sample Preparation for Western Blotting
2.9. Western Blot Analysis
2.10. Gene Expression Profiling
2.11. ATP Assay
2.12. NAD+/NADH Assay
2.13. Statistical Analysis
3. Results
3.1. Effects of Folate Restriction on the Folate Level and Weight
3.2. Adaptations to Folate Restriction Protects against ACF Formation
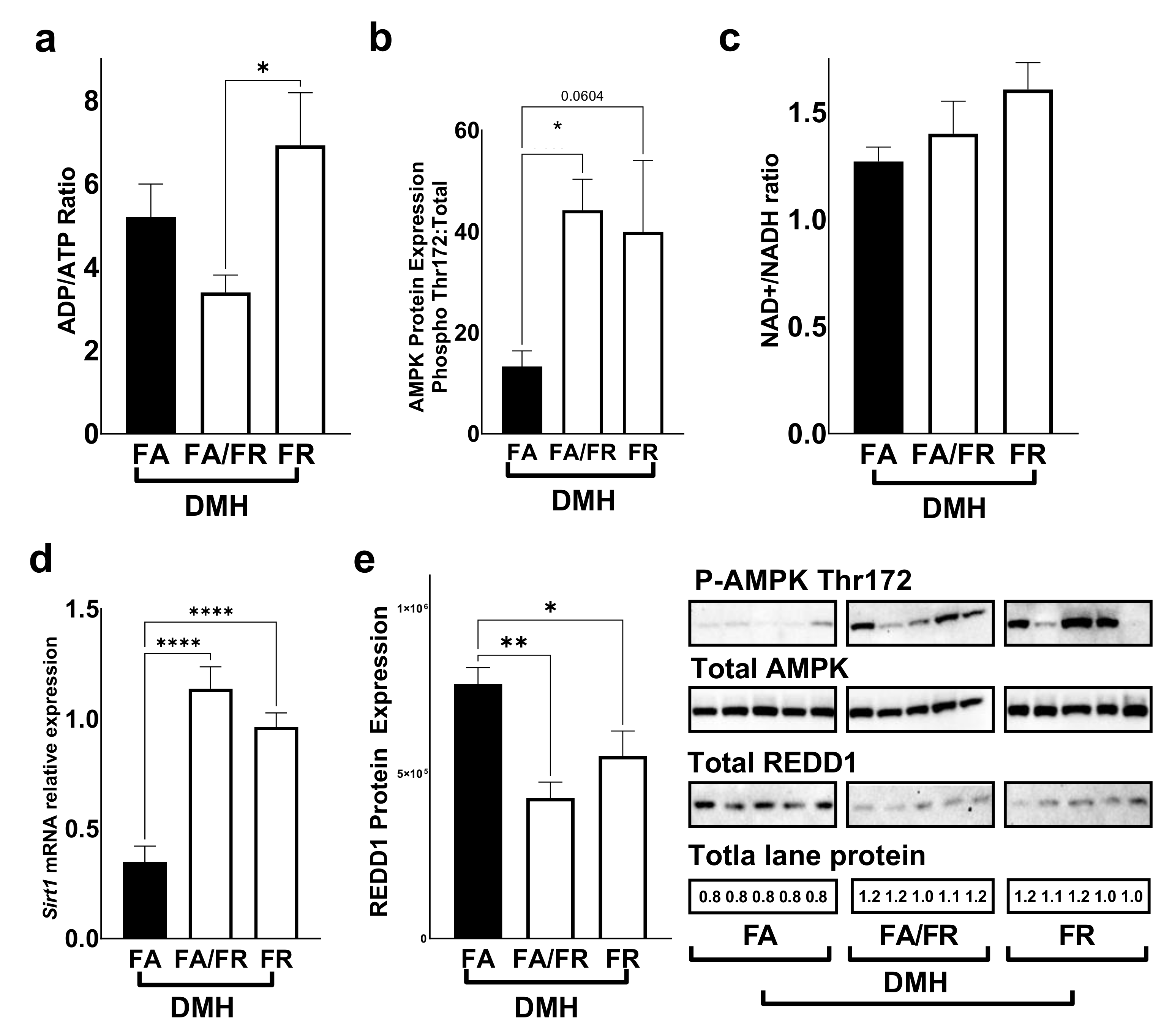
3.3. Folate Restriction Modulates the mTOR Pathway
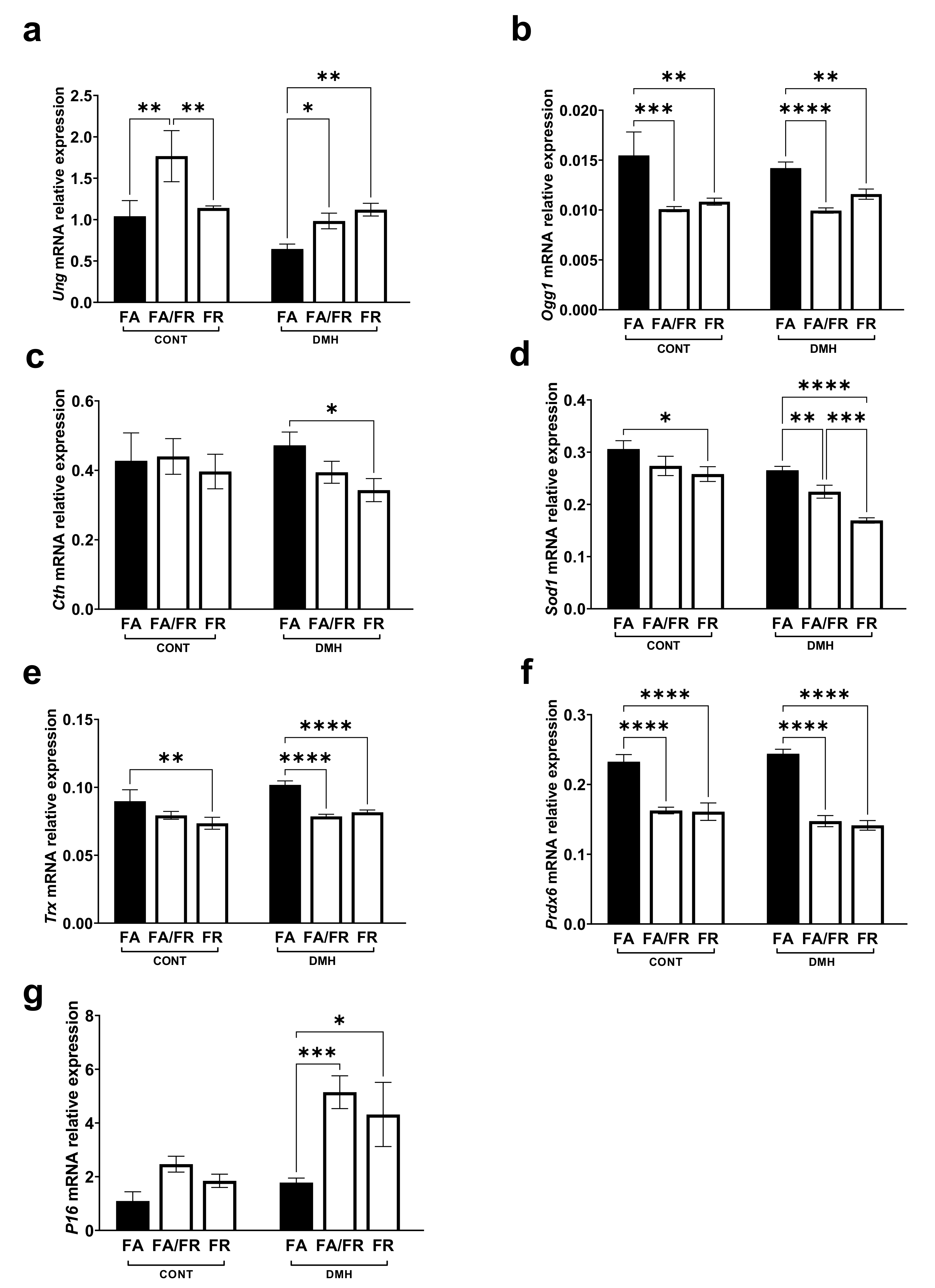
3.4. Folate Restriction Reduces the Expression of Methylation Genes
3.5. Folate Restriction Impacts DNA Repair, Antioxidants, and Tumor Suppressor Genes
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Conflicts of Interest
References
- Shulpekova, Y.; Nechaev, V.; Kardasheva, S.; Sedova, A.; Kurbatova, A.; Bueverova, E.; Kopylov, A.; Malsagova, K.; Dlamini, J.; Ivashkin, V. The Concept of Folic Acid in Health and Disease. Molecules 2021, 26, 3731. [Google Scholar] [CrossRef]
- Mason, J.B.; Tang, S.Y. Folate status and colorectal cancer risk: A 2016 update. Mol. Asp. Med. 2016, 53, 73–79. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mason, J.B. Unraveling the complex relationship between folate and cancer risk. BioFactors 2011, 37, 253–260. [Google Scholar] [CrossRef] [PubMed]
- Field, M.; Stover, P.J. Safety of folic acid. Ann. N. Y. Acad. Sci. 2017, 1414, 59–71. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Smith, A.D.; Kim, Y.-I.; Refsum, H. Is folic acid good for everyone? Am. J. Clin. Nutr. 2008, 87, 517–533. [Google Scholar] [CrossRef] [Green Version]
- Ulrich, C.M.; Potter, J.D. Folate supplementation: Too much of a good thing? Cancer Epidemiol. Biomark. Prev. 2006, 15, 189–193. [Google Scholar] [CrossRef]
- Kim, Y.-I. Folic Acid Fortification and Supplementation—Good for Some but Not So Good for Others. Nutr. Rev. 2007, 65. [Google Scholar] [CrossRef]
- Sanderson, P.; Stone, E.; Kim, Y.-I.; Mathers, J.C.; Kampman, E.; Downes, C.S.; Muir, K.R.; Baron, J.A. Folate and colo-rectal cancer risk. Br. J. Nutr. 2007, 98, 1299–1304. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Weißenborn, A.; Ehlers, A.; Hirsch-Ernst, K.-I.; Lampen, A.; Niemann, B. Ein Vitamin mit zwei Gesichtern. Bundesgesundheitsblatt Gesundheitsforschung Gesundheitsschutz 2017, 60, 332–340. [Google Scholar] [CrossRef]
- Knock, E.; Deng, L.; Wu, Q.; Leclerc, D.; Wang, X.-L.; Rozen, R. Low Dietary Folate Initiates Intestinal Tumors in Mice, with Altered Expression of G2-M Checkpoint Regulators Polo-Like Kinase 1 and Cell Division Cycle 25c. Cancer Res. 2006, 66, 10349–10356. [Google Scholar] [CrossRef] [Green Version]
- Cravo, M.; Mason, J.B.; Dayal, Y.; Hutchinson, M.; Smith, D.; Selhub, J.; Rosenberg, I.H. Folate deficiency enhances the development of colonic neoplasia in dimethylhydrazine-treated rats. Cancer Res. 1992, 52, 5002–5006. [Google Scholar] [PubMed]
- Song, J.; Medline, A.; Mason, J.B.; Gallinger, S.; Kim, Y.I. Effects of dietary folate on intestinal tumorigenesis in the apcMin mouse. Cancer Res. 2000, 60, 5434–5440. [Google Scholar] [CrossRef]
- Liu, Z.; Ciappio, E.D.; Crott, J.W.; Brooks, R.S.; Nesvet, J.; Smith, D.E.; Choi, S.; Mason, J.B. Combined inadequacies of multiple B vitamins amplify colonic Wnt signaling and promote intestinal tumorigenesis in BAT-LacZ × Apc1638N mice. FASEB J. 2011, 25, 3136–3145. [Google Scholar] [CrossRef] [Green Version]
- Davis, C.D.; Uthus, E.O. Dietary Folate and Selenium Affect Dimethylhydrazine-Induced Aberrant Crypt Formation, Global DNA Methylation and One-Carbon Metabolism in Rats. J. Nutr. 2003, 133, 2907–2914. [Google Scholar] [CrossRef]
- Lawrance, A.K.; Deng, L.; Rozen, R. Methylenetetrahydrofolate reductase deficiency and low dietary folate reduce tumorigenesis in Apcmin/+ mice. Gut 2009, 58, 805–811. [Google Scholar] [CrossRef]
- Hanley, M.P.; Kadaveru, K.; Perret, C.; Giardina, C.; Rosenberg, D.W. Dietary Methyl Donor Depletion Suppresses Intestinal Adenoma Development. Cancer Prev. Res. 2016, 9, 812–820. [Google Scholar] [CrossRef] [Green Version]
- Kadaveru, K.; Protiva, P.; Greenspan, E.J.; Kim, Y.-I.; Rosenberg, D.W. Dietary Methyl Donor Depletion Protects Against Intestinal Tumorigenesis in ApcMin/+ Mice. Cancer Prev. Res. 2012, 5, 911–920. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Le Leu, R.K.; Young, G.; McIntosh, G.H. Folate deficiency reduces the development of colorectal cancer in rats. Carcinogenesis 2000, 21, 2261–2265. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- I Kim, Y.; Salomon, R.N.; Graeme-Cook, F.; Choi, S.W.; E Smith, D.; E Dallal, G.; Mason, J.B. Dietary folate protects against the development of macroscopic colonic neoplasia in a dose responsive manner in rats. Gut 1996, 39, 732–740. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Branda, R.F.; Hacker, M.; Lafayette, A.; Nigels, E.; Sullivan, L.; Nicklas, J.A.; O’Neill, J.P. Nutritional folate deficiency augments the in vivo mutagenic and lymphocytotoxic activities of alkylating agents. Environ. Mol. Mutagen. 1998, 32, 33–38. [Google Scholar] [CrossRef]
- Ciappio, E.D.; Liu, Z.; Brooks, R.S.; Mason, J.B.; Bronson, R.T.; Crott, J.W. Maternal B vitamin supplementation from preconception through weaning suppresses intestinal tumorigenesis in Apc1638N mouse offspring. Gut 2011, 60, 1695–1702. [Google Scholar] [CrossRef] [Green Version]
- Lindzon, G.M.; Medline, A.; Sohn, K.-J.; Depeint, F.; Croxford, R.; Kim, Y.-I. Effect of folic acid supplementation on the progression of colorectal aberrant crypt foci. Carcinogenesis 2009, 30, 1536–1543. [Google Scholar] [CrossRef]
- Le Leu, R.K.; Young, G.; McIntosh, G.H. Folate deficiency diminishes the occurrence of aberrant crypt foci in the rat colon but does not alter global DNA methylation status. J. Gastroenterol. Hepatol. 2000, 15, 1158–1164. [Google Scholar] [CrossRef] [PubMed]
- MacFarlane, A.J.; Perry, C.A.; McEntee, M.F.; Lin, D.M.; Stover, P.J. Shmt1Heterozygosity Impairs Folate-Dependent Thymidylate Synthesis Capacity and Modifies Risk ofApcmin-Mediated Intestinal Cancer Risk. Cancer Res. 2011, 71, 2098–2107. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Al-Numair, K.S.; Waly, M.; Ali, A.; Essa, M.M.; Farhat, M.F.; A Alsaif, M. Dietary folate protects against azoxymethane-induced aberrant crypt foci development and oxidative stress in rat colon. Exp. Biol. Med. 2011, 236, 1005–1011. [Google Scholar] [CrossRef] [PubMed]
- Rosati, R.; Ma, H.; Cabelof, D.C. Folate and Colorectal Cancer in Rodents: A Model of DNA Repair Deficiency. J. Oncol. 2012, 2012, 1–17. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- National Research Council (US) Subcommittee on Laboratory Animal Nutrition. Nutrient Requirements of Laboratory Animals, 4th ed.; National Academies Press: Washington, DC, USA, 1995. [Google Scholar] [CrossRef]
- Reeves, P.G. Components of the AIN-93 Diets as Improvements in the AIN-76A Diet. J. Nutr. 1997, 127, 838S–841S. [Google Scholar] [CrossRef]
- Wright, A.J.A.; Dainty, J.R.; Finglas, P. Folic acid metabolism in human subjects revisited: Potential implications for proposed mandatory folic acid fortification in the UK. Br. J. Nutr. 2007, 98, 667–675. [Google Scholar] [CrossRef] [PubMed]
- Obeid, R.; Herrmann, W. The Emerging Role of Unmetabolized Folic Acid in Human Diseases: Myth or Reality? Curr. Drug Metab. 2012, 13, 1184–1195. [Google Scholar] [CrossRef] [PubMed]
- Rong, N.; Selhub, J.; Goldin, B.R.; Rosenberg, I.H. Bacterially Synthesized Folate in Rat Large Intestine is Incorporated into Host Tissue Folyl Polyglutamates. J. Nutr. 1991, 121, 1955–1959. [Google Scholar] [CrossRef]
- Beydoun, S.; Fardous, A.M.; Saruna, M.M.; Beydoun, A.G.; Sorge, J.A.; Ma, H.; Aoun, G.; Unnikrishnan, A.; Cabelof, D.C.; Heydari, A.R. Succinylsulfathiazole modulates the mTOR signaling pathway in the liver of c57BL/6 mice via a folate independent mechanism. Exp. Gerontol. 2021, 150, 111387. [Google Scholar] [CrossRef]
- Ventrella-Lucente, L.F.; Unnikrishnan, A.; Pilling, A.B.; Patel, H.V.; Kushwaha, D.; Dombkowski, A.; Schmelz, E.M.; Cabelof, D.C.; Heydari, A.R. Folate Deficiency Provides Protection against Colon Carcinogenesis in DNA Polymerase β Haploinsufficient Mice. J. Biol. Chem. 2010, 285, 19246–19258. [Google Scholar] [CrossRef] [Green Version]
- Hanley, M.P.; Aladelokun, O.; Kadaveru, K.; Rosenberg, D.W. Methyl Donor Deficiency Blocks Colorectal Cancer Development by Affecting Key Metabolic Pathways. Cancer Prev. Res. 2019, 13, 1–14. [Google Scholar] [CrossRef]
- Macfarlane, A.J.; McEntee, M.; Stover, P.J. Azoxymethane-Induced Colon Carcinogenesis in Mice Occurs Independently of De Novo Thymidylate Synthesis Capacity. J. Nutr. 2014, 144, 419–424. [Google Scholar] [CrossRef] [PubMed]
- Taylor, S.C.; Posch, A. The Design of a Quantitative Western Blot Experiment. BioMed Res. Int. 2014, 2014, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Cabelof, D.C.; Raffoul, J.J.; Nakamura, J.; Kapoor, D.; Abdalla, H.; Heydari, A.R. Imbalanced Base Excision Repair in Response to Folate Deficiency Is Accelerated by Polymerase β Haploinsufficiency. J. Biol. Chem. 2004, 279, 36504–36513. [Google Scholar] [CrossRef] [Green Version]
- Fazili, Z.; Pfeiffer, C.M.; Zhang, M. Comparison of Serum Folate Species Analyzed by LC-MS/MS with Total Folate Measured by Microbiologic Assay and Bio-Rad Radioassay. Clin. Chem. 2007, 53, 781–784. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Golja, M.V.; Šmid, A.; Kuželički, N.K.; Trontelj, J.; Geršak, K.; Mlinarič-Raščan, I. Folate Insufficiency Due to MTHFR Deficiency Is Bypassed by 5-Methyltetrahydrofolate. J. Clin. Med. 2020, 9, 2836. [Google Scholar] [CrossRef] [PubMed]
- Blount, B.C.; Ames, B.N. 2 DNA damage in folate deficiency. Baillière’s Clin. Haematol. 1995, 8, 461–478. [Google Scholar] [CrossRef]
- Kim, Y.-I. Folate and colorectal cancer: An evidence-based critical review. Mol. Nutr. Food Res. 2007, 51, 267–292. [Google Scholar] [CrossRef] [PubMed]
- Gylling, B.; Van Guelpen, B.; Schneede, J.; Hultdin, J.; Ueland, P.M.; Hallmans, G.; Johansson, I.; Palmqvist, R. Low Folate Levels Are Associated with Reduced Risk of Colorectal Cancer in a Population with Low Folate Status. Cancer Epidemiol. Biomark. Prev. 2014, 23, 2136–2144. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mason, J.B.; Dickstein, A.; Jacques, P.F.; Haggarty, P.; Selhub, J.; Dallal, G.; Rosenberg, I.H. A Temporal Association between Folic Acid Fortification and an Increase in Colorectal Cancer Rates May Be Illuminating Important Biological Principles: A Hypothesis. Cancer Epidemiol. Biomark. Prev. 2007, 16, 1325–1329. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Williams, E. Folate, colorectal cancer and the involvement of DNA methylation. Proc. Nutr. Soc. 2012, 71, 592–597. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cheng, Z.; Shao, X.; Xu, M.; Wang, J.; Kuai, X.; Zhang, L.; Wu, J.; Zhou, C.; Mao, J. Rab1A promotes proliferation and migration abilities via regulation of the HER2/AKT-independent mTOR/S6K1 pathway in colorectal cancer. Oncol. Rep. 2019, 41, 2717–2728. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zeng, J.-D.; Wu, W.K.; Wang, H.-Y.; Li, X.-X. Serine and one-carbon metabolism, a bridge that links mTOR signaling and DNA methylation in cancer. Pharmacol. Res. 2019, 149, 104352. [Google Scholar] [CrossRef]
- Silva, E.; Rosario, F.J.; Powell, T.L.; Jansson, T. Mechanistic Target of Rapamycin Is a Novel Molecular Mechanism Linking Folate Availability and Cell Function. J. Nutr. 2017, 147, 1237–1242. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hwang, S.Y.; Kang, Y.J.; Sung, B.; Kim, M.; Kim, D.H.; Lee, Y.; Yoo, M.-A.; Kim, C.M.; Chung, H.Y.; Kim, N.D. Folic acid promotes the myogenic differentiation of C2C12 murine myoblasts through the Akt signaling pathway. Int. J. Mol. Med. 2015, 36, 1073–1080. [Google Scholar] [CrossRef] [Green Version]
- Ducker, G.S.; Rabinowitz, J.D. One-Carbon Metabolism in Health and Disease. Cell Metab. 2016, 25, 27–42. [Google Scholar] [CrossRef] [Green Version]
- Cetrullo, S.; D’Adamo, S.; Tantini, B.; Borzì, R.M.; Flamigni, F. mTOR, AMPK, and Sirt1: Key Players in Metabolic Stress Management. Crit. Rev. Eukaryot. Gene Expr. 2015, 25, 59–75. [Google Scholar] [CrossRef]
- Rathor, L.; Akhoon, B.A.; Pandey, S.; Srivastava, S.; Pandey, R. Folic acid supplementation at lower doses increases oxidative stress resistance and longevity in Caenorhabditis elegans. AGE 2015, 37, 1–15. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mariana, R.; Isadora, C.B.; Camila, L.; Augusto, D.; Fernando, M. The S6K protein family in health and disease. Life Sci. 2015, 131, 1–10. [Google Scholar] [CrossRef]
- Roux, P.P.; Shahbazian, D.; Vu, H.; Holz, M.K.; Cohen, M.S.; Taunton, J.; Sonenberg, N.; Blenis, J. RAS/ERK Signaling Promotes Site-specific Ribosomal Protein S6 Phosphorylation via RSK and Stimulates Cap-dependent Translation. J. Biol. Chem. 2007, 282, 14056–14064. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jennings, B.A.; Willis, G. How folate metabolism affects colorectal cancer development and treatment; a story of heterogeneity and pleiotropy. Cancer Lett. 2015, 356, 224–230. [Google Scholar] [CrossRef] [PubMed]
- Ryan, B.M.; Weir, D.G. Relevance of folate metabolism in the pathogenesis of colorectal cancer. J. Lab. Clin. Med. 2001, 138, 164–176. [Google Scholar] [CrossRef] [PubMed]
- Caudill, M.A.; Wang, J.C.; Melnyk, S.; Pogribny, I.P.; Jernigan, S.; Collins, M.D.; Santos-Guzman, J.; Swendseid, M.E.; Cogger, E.A.; James, S.J. Intracellular S-Adenosylhomocysteine Concentrations Predict Global DNA Hypomethylation in Tissues of Methyl-Deficient Cystathionine β-Synthase Heterozygous Mice. J. Nutr. 2001, 131, 2811–2818. [Google Scholar] [CrossRef] [PubMed]
- Oliva, A.; Galletti, P.; Zappia, V.; Paik, W.K.; Kim, S. Studies on Substrate Specificity of S-Adenosylmethionine: Protein-Carboxyl Methyltransferase from Calf Brain. JBIC J. Biol. Inorg. Chem. 1980, 104, 595–602. [Google Scholar] [CrossRef] [PubMed]
- Ding, Y.B.; He, J.L.; Liu, X.Q.; Chen, X.M.; Long, C.L.; Wang, Y.X. Expression of DNA methyltransferases in the mouse uterus during early pregnancy and susceptibility to dietary folate deficiency. Reproduction 2012, 144, 91–100. [Google Scholar] [CrossRef]
- Kim, Y.-I. Folate, colorectal carcinogenesis, and DNA methylation: Lessons from animal studies. Environ. Mol. Mutagen. 2004, 44, 10–25. [Google Scholar] [CrossRef]
- Eads, C.A.; Nickel, A.E.; Laird, P.W. Complete genetic suppression of polyp formation and reduction of CPG-island hypermethylation in Apc (Min/+) Dnmt1-hypomorphic mice. Cancer Res. 2002, 62, 1296–1299. [Google Scholar]
- Weis, B.; Schmidt, J.; Maamar, H.; Raj, A.; Lin, H.; Tóth, C.; Riedmann, K.; Raddatz, G.; Seitz, H.-K.; Ho, A.D.; et al. Inhibition of intestinal tumor formation by deletion of the DNA methyltransferase 3a. Oncogene 2014, 34, 1822–1830. [Google Scholar] [CrossRef] [PubMed]
- Unnikrishnan, A.; Raffoul, J.J.; Patel, H.V.; Prychitko, T.M.; Anyangwe, N.; Meira, L.B.; Friedberg, E.C.; Cabelof, D.C.; Heydari, A.R. Oxidative stress alters base excision repair pathway and increases apoptotic response in apurinic/apyrimidinic endonuclease 1/redox factor-1 haploinsufficient mice. Free Radic. Biol. Med. 2009, 46, 1488–1499. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Unnikrishnan, A.; Prychitko, T.M.; Patel, H.V.; Chowdhury, M.E.; Pilling, A.B.; Ventrella-Lucente, L.F.; Papakonstantinou, E.V.; Cabelof, D.C.; Heydari, A.R. Folate deficiency regulates expression of DNA polymerase β in response to oxidative stress. Free Radic. Biol. Med. 2011, 50, 270–280. [Google Scholar] [CrossRef] [PubMed] [Green Version]


Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Fardous, A.M.; Beydoun, S.; James, A.A.; Ma, H.; Cabelof, D.C.; Unnikrishnan, A.; Heydari, A.R. The Timing and Duration of Folate Restriction Differentially Impacts Colon Carcinogenesis. Nutrients 2022, 14, 16. https://doi.org/10.3390/nu14010016
Fardous AM, Beydoun S, James AA, Ma H, Cabelof DC, Unnikrishnan A, Heydari AR. The Timing and Duration of Folate Restriction Differentially Impacts Colon Carcinogenesis. Nutrients. 2022; 14(1):16. https://doi.org/10.3390/nu14010016
Chicago/Turabian StyleFardous, Ali M., Safa Beydoun, Andrew A. James, Hongzhi Ma, Diane C. Cabelof, Archana Unnikrishnan, and Ahmad R. Heydari. 2022. "The Timing and Duration of Folate Restriction Differentially Impacts Colon Carcinogenesis" Nutrients 14, no. 1: 16. https://doi.org/10.3390/nu14010016