
Vasorin Deletion in C57BL/6J Mice Induces Hepatocyte Autophagy through Glycogen-Mediated mTOR Regulation
 ,
, {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Animals
2.2. Liver Function Tests (LFTs)
2.3. miRNA Sequencing
2.4. Transmission Electron Microscopy (TEM)
2.5. Histopathology
2.6. Periodic Acid Schiff (PAS) Glycogen Staining
2.7. Quantitative Analysis and Detection of Glycogen
(Cstandard × V1) × (A3 − A1) ÷ (A2 − A1) ÷ (V1 × Cpr) ÷ 1.11 = 0.09 × (A3 − A1) ÷ (A2 − A1) ÷ Cpr
2.8. Quantitative Real-Time Reverse-Transcription PCR (qRT-PCR)
2.9. Western Blot Assay
2.10. Statistical Analyses
3. Results
3.1. Vasn Deficiency Causes Liver/Body Weight Loss
3.2. Vasn Deficiency Leads to Pathological Damage in the Liver
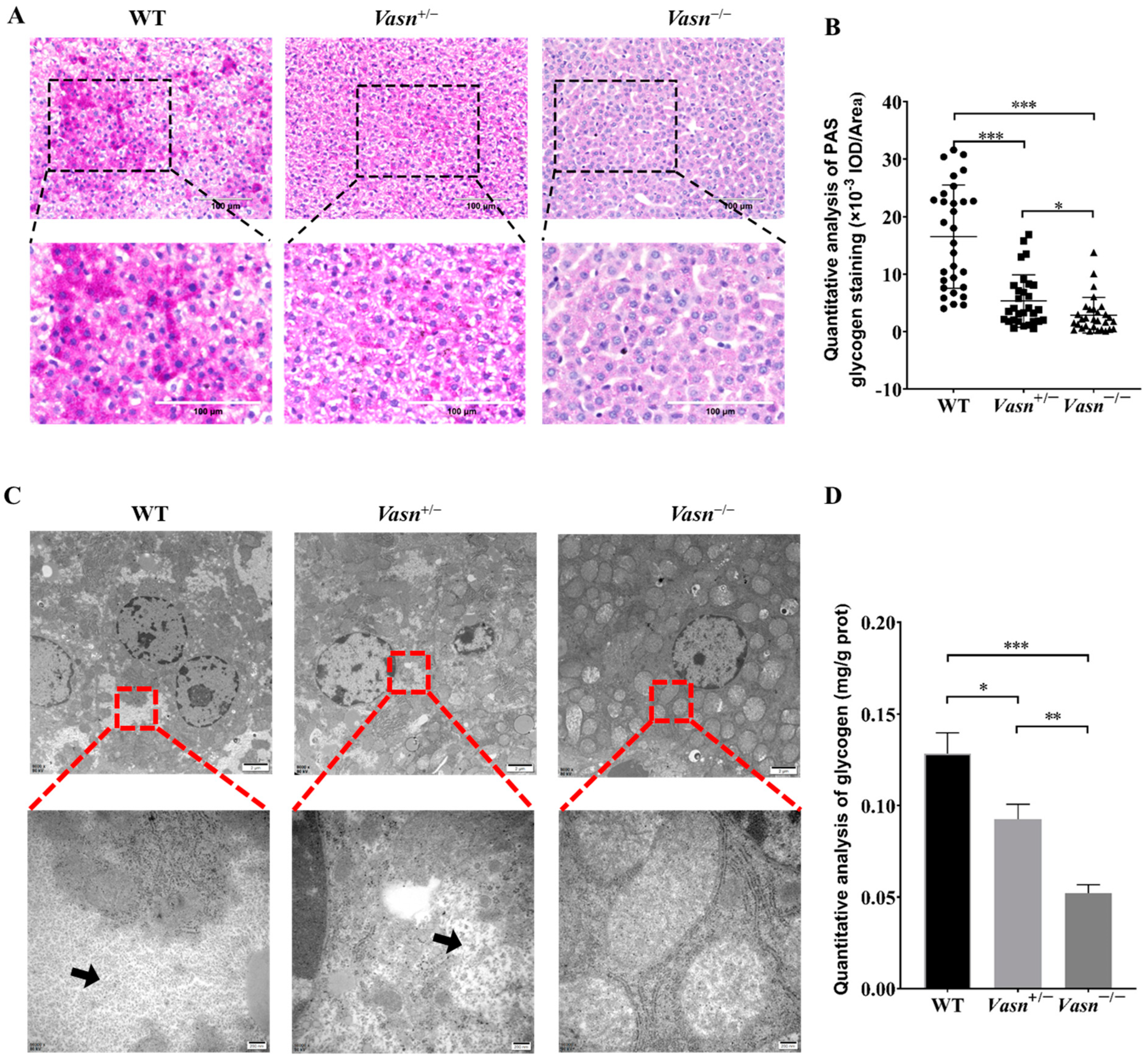
3.3. Vasn Deficiency Leads to Glycogen Depletion in the Liver
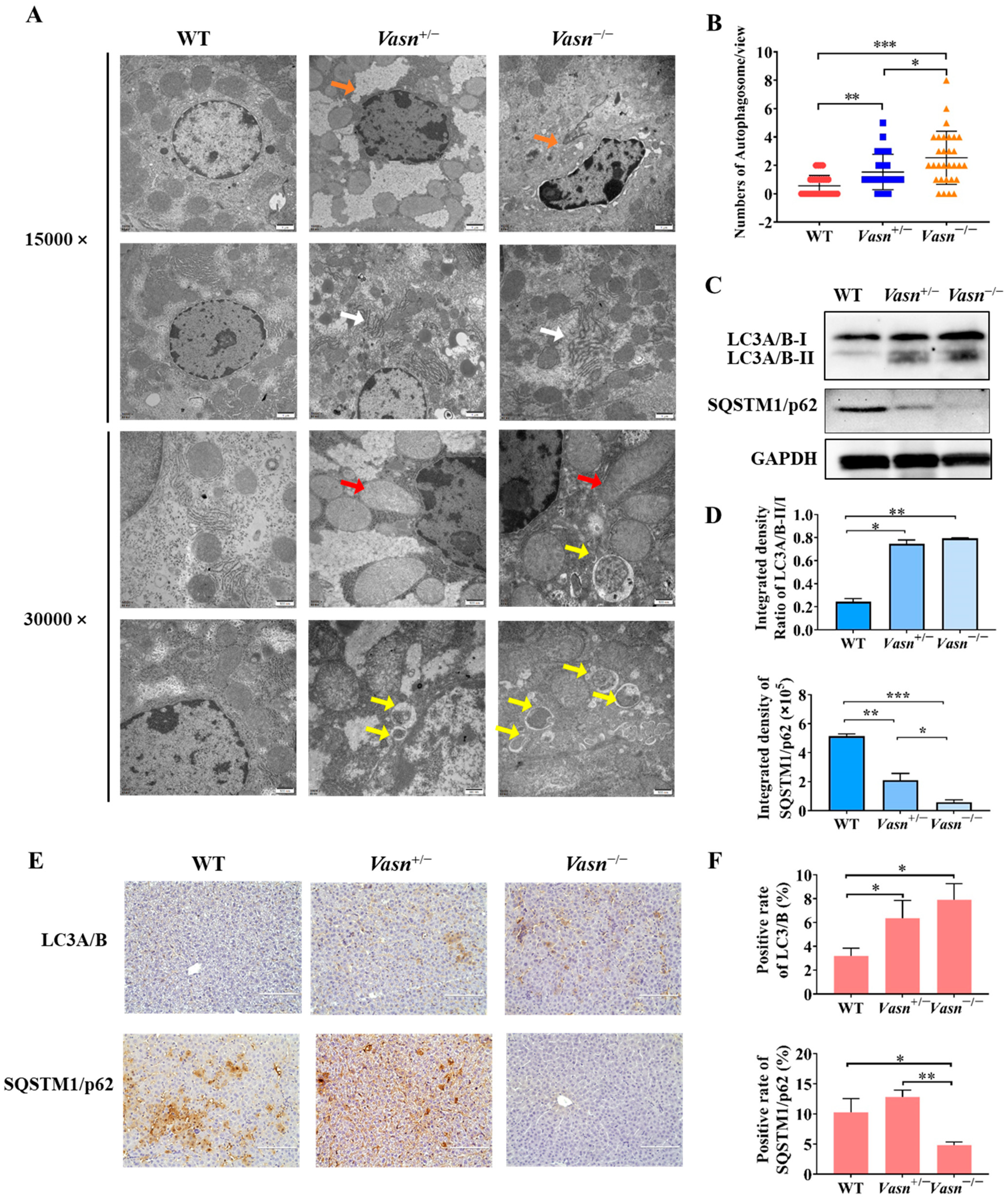
3.4. Vasn Deficiency Induces Hepatocyte Autophagy
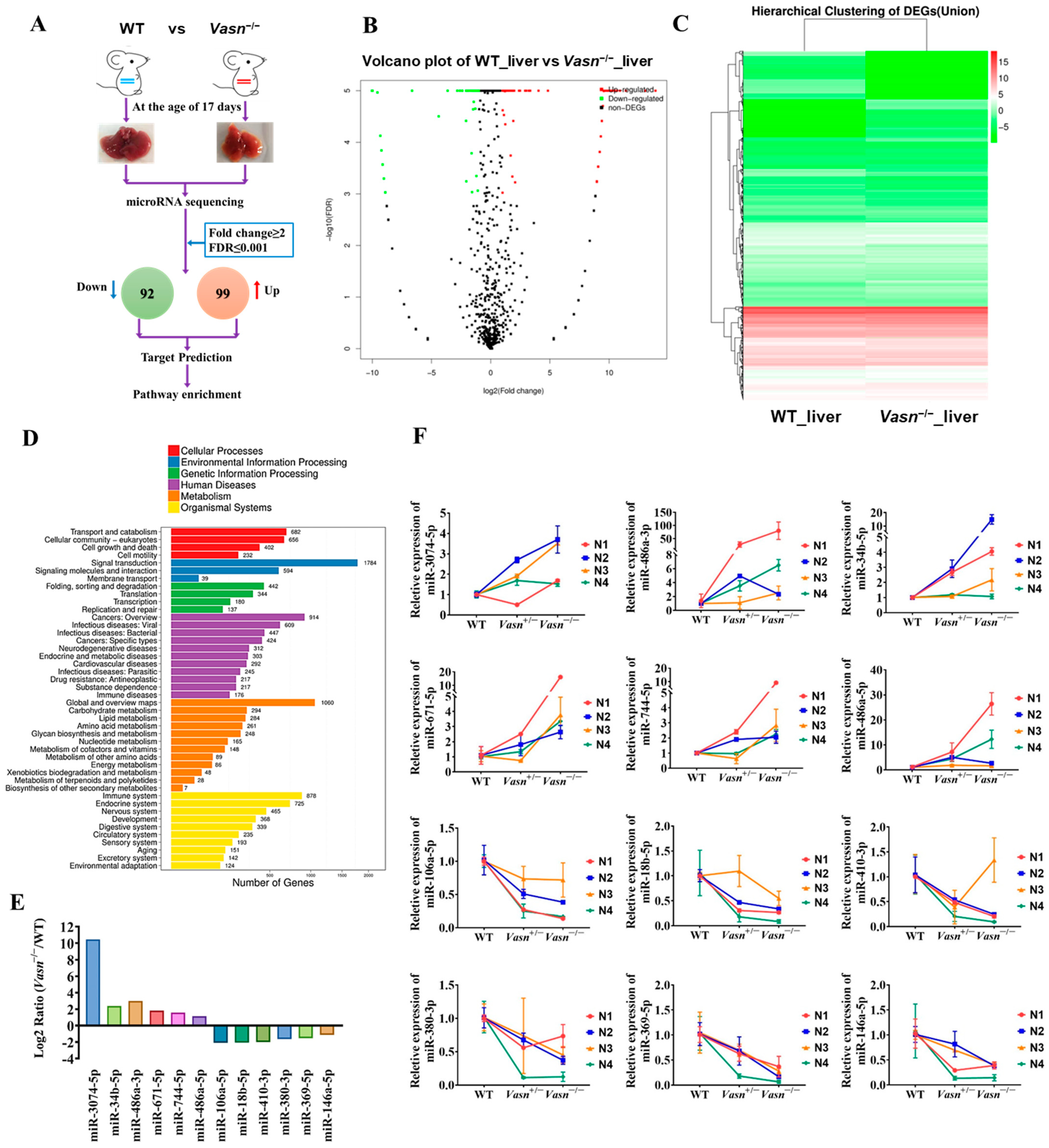
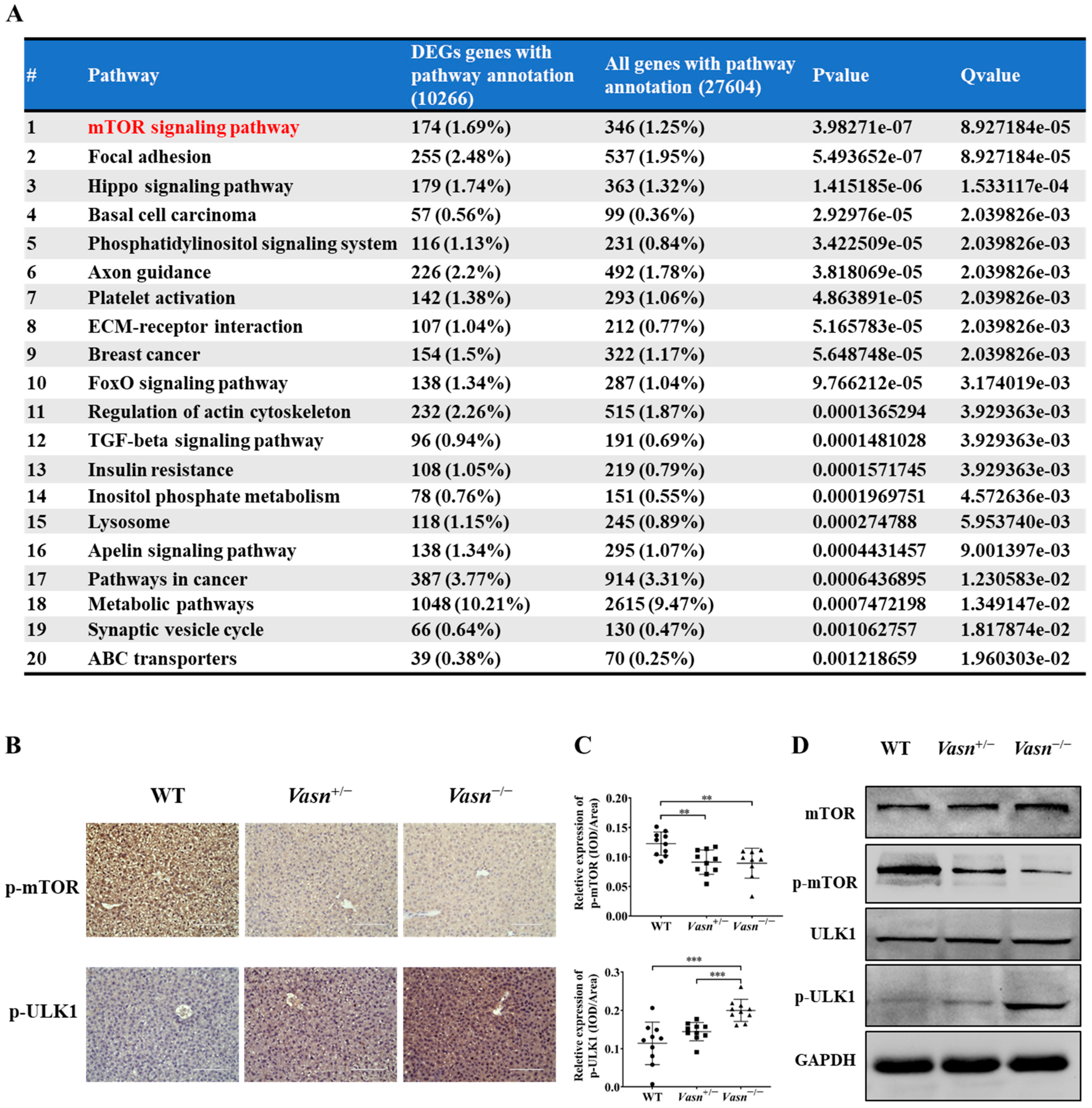
3.5. Hepatic Differential Expressed microRNA Profile Shows That Vasn Deficiency Regulates the mTOR-ULK1 Pathway
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Krautzberger, A.M.; Kosiol, B.; Scholze, M.; Schrewe, H. Expression of vasorin (Vasn) during embryonic development of the mouse. Gene Expr. Patterns 2012, 12, 167–171. [Google Scholar] [CrossRef] [PubMed]
- Bonnet, A.L.; Chaussain, C.; Broutin, I.; Rochefort, G.Y.; Schrewe, H.; Gaucher, C. From vascular smooth muscle cells to folliculogenesis: What about Vasorin? Front. Med. 2018, 5, 335. [Google Scholar] [CrossRef] [PubMed]
- Choksi, S.; Lin, Y.; Pobezinskaya, Y.; Chen, L.; Park, C.; Morgan, M.; Li, T.; Jitkaew, S.; Cao, X.; Kim, Y.S.; et al. A HIF-1 target, ATIA, protects cells from apoptosis by modulating the mitochondrial thioredoxin, TRX2. Mol. Cell 2011, 42, 597–609. [Google Scholar] [CrossRef]
- Rimon-Dahari, N.; Heinemann-Yerushalmi, L.; Hadas, R.; Kalich-Philosoph, L.; Ketter, D.; Nevo, N.; Galiani, D.; Dekel, N. Vasorin: A newly identified regulator of ovarian folliculogenesis. FASEB J. 2018, 32, 2124–2136. [Google Scholar] [CrossRef] [PubMed]
- Ikeda, Y.; Imai, Y.; Kumagai, H.; Nosaka, T.; Morikawa, Y.; Hisaoka, T.; Manabe, I.; Maemura, K.; Nakaoka, T.; Imamura, T.; et al. Vasorin, a transforming growth factor beta-binding protein expressed in vascular smooth muscle cells, modulates the arterial response to injury in vivo. Proc. Natl. Acad. Sci. USA 2004, 101, 10732–10737. [Google Scholar] [CrossRef] [PubMed]
- Li, S.; Li, H.; Yang, X.; Wang, W.; Huang, A.; Li, J.; Qin, X.; Li, F.; Lu, G.; Ding, H.; et al. Vasorin is a potential serum biomarker and drug target of hepatocarcinoma screened by subtractive-EMSA-SELEX to clinic patient serum. Oncotarget 2015, 6, 10045–10059. [Google Scholar] [CrossRef]
- Malapeira, J.; Esselens, C.; Bech-Serra, J.J.; Canals, F.; Arribas, J. ADAM17 (TACE) regulates TGFbeta signaling through the cleavage of vasorin. Oncogene 2011, 30, 1912–1922. [Google Scholar] [CrossRef]
- Man, J.; Yu, X.; Huang, H.; Zhou, W.; Xiang, C.; Huang, H.; Miele, L.; Liu, Z.; Bebek, G.; Bao, S.; et al. Hypoxic induction of Vasorin regulates Notch1 turnover to maintain glioma stem-like cells. Cell Stem Cell 2018, 22, 104–118.e6. [Google Scholar] [CrossRef]
- Liang, W.; Guo, B.; Ye, J.; Liu, H.; Deng, W.; Lin, C.; Zhong, X.; Wang, L. Vasorin stimulates malignant progression and angiogenesis in glioma. Cancer Sci. 2019, 110, 2558–2572. [Google Scholar] [CrossRef]
- Bhandari, A.; Guan, Y.; Xia, E.; Huang, Q.; Chen, Y. VASN promotes YAP/TAZ and EMT pathway in thyroid carcinogenesis in vitro. Am. J. Transl. Res. 2019, 11, 3589–3599. [Google Scholar]
- Huang, A.; Dong, J.; Li, S.; Wang, C.; Ding, H.; Li, H.; Su, X.; Ge, X.; Sun, L.; Bai, C.; et al. Exosomal transfer of vasorin expressed in hepatocellular carcinoma cells promotes migration of human umbilical vein endothelial cells. Int. J. Biol. Sci. 2015, 11, 961–969. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, D.; Zhang, T.; Yang, X.; Geng, J.; Li, S.; Ding, H.; Li, H.; Huang, A.; Wang, C.; Sun, L.; et al. Identification of Functional mimotopes of human Vasorin Ectodomain by Biopanning. Int. J. Biol. Sci. 2018, 14, 461–470. [Google Scholar] [CrossRef] [PubMed]
- Ke, P.Y. Diverse Functions of Autophagy in Liver Physiology and Liver Diseases. Int. J. Mol. Sci. 2019, 20, 300. [Google Scholar] [CrossRef]
- Sun, J.; Guo, X.; Yu, P.; Liang, J.; Mo, Z.; Zhang, M.; Yang, L.; Huang, X.; Hu, B.; Liu, J.; et al. Vasorin deficiency leads to cardiac hypertrophy by targeting MYL7 in young mice. J. Cell. Mol. Med. 2022, 26, 88–98. [Google Scholar] [CrossRef]
- Trefts, E.; Gannon, M.; Wasserman, D.H. The liver. Curr. Biol. 2017, 27, R1147–R1151. [Google Scholar] [CrossRef] [PubMed]
- Jackson, A.A. Nutrition and Liver Health. Dig. Dis. 2017, 35, 411–417. [Google Scholar] [CrossRef]
- de Toro-Martin, J.; Fernandez-Marcelo, T.; Gonzalez-Rodriguez, A.; Escriva, F.; Valverde, A.M.; Alvarez, C.; Fernandez-Millan, E. Defective liver glycogen autophagy related to hyperinsulinemia in intrauterine growth-restricted newborn wistar rats. Sci. Rep. 2020, 10, 17651. [Google Scholar] [CrossRef]
- Nagarajan, S.R.; Paul-Heng, M.; Krycer, J.R.; Fazakerley, D.J.; Sharland, A.F.; Hoy, A.J. Lipid and glucose metabolism in hepatocyte cell lines and primary mouse hepatocytes: A comprehensive resource for in vitro studies of hepatic metabolism. Am. J. Physiol. Endocrinol. Metab. 2019, 316, E578–E589. [Google Scholar] [CrossRef]
- Pursell, N.; Gierut, J.; Zhou, W.; Dills, M.; Diwanji, R.; Gjorgjieva, M.; Saxena, U.; Yang, J.S.; Shah, A.; Venkat, N.; et al. Inhibition of Glycogen Synthase II with RNAi Prevents Liver Injury in Mouse Models of Glycogen Storage Diseases. Mol. Ther. 2018, 26, 1771–1782. [Google Scholar] [CrossRef]
- Anno, T.; Kaneto, H.; Shigemoto, R.; Kawasaki, F.; Kawai, Y.; Urata, N.; Kawamoto, H.; Kaku, K.; Okimoto, N. Hypoinsulinemic hypoglycemia triggered by liver injury in elderly subjects with low body weight: Case reports. Endocrinol. Diabetes Metab. Case Rep. 2018, 2018, 17-0155. [Google Scholar] [CrossRef]
- Jung, C.H.; Ro, S.H.; Cao, J.; Otto, N.M.; Kim, D.H. mTOR regulation of autophagy. FEBS Lett. 2010, 584, 1287–1295. [Google Scholar] [CrossRef] [Green Version]
- Schneider, J.L.; Cuervo, A.M. Liver autophagy: Much more than just taking out the trash. Nat. Rev. Gastroenterol. Hepatol. 2014, 11, 187–200. [Google Scholar] [CrossRef]
- Langmead, B.; Trapnell, C.; Pop, M.; Salzberg, S.L. Ultrafast and memory-efficient alignment of short DNA sequences to the human genome. Genome Biol. 2009, 10, R25. [Google Scholar] [CrossRef] [PubMed]
- ’t Hoen, P.A.; Ariyurek, Y.; Thygesen, H.H.; Vreugdenhil, E.; Vossen, R.H.; de Menezes, R.X.; Boer, J.M.; van Ommen, G.J.; den Dunnen, J.T. Deep sequencing-based expression analysis shows major advances in robustness, resolution and inter-lab portability over five microarray platforms. Nucleic Acids Res. 2008, 36, e141. [Google Scholar] [CrossRef] [PubMed]
- Benjamini, Y.; Yekutieli, D. The Control of the False Discovery Rate in Multiple Testing. Ann. Stat. 2001, 29, 1165–1188. [Google Scholar] [CrossRef]
- Kruger, J.; Rehmsmeier, M. RNAhybrid: microRNA target prediction easy, fast and flexible. Nucleic Acids Res. 2006, 34, W451–W454. [Google Scholar] [CrossRef]
- John, B.; Enright, A.J.; Aravin, A.; Tuschl, T.; Sander, C.; Marks, D.S. Human MicroRNA targets. PLoS Biol. 2004, 2, e363. [Google Scholar] [CrossRef]
- Agarwal, V.; Bell, G.W.; Nam, J.W.; Bartel, D.P. Predicting effective microRNA target sites in mammalian mRNAs. eLife 2015, 4, e05005. [Google Scholar] [CrossRef] [PubMed]
- Kanehisa, M.; Araki, M.; Goto, S.; Hattori, M.; Hirakawa, M.; Itoh, M.; Katayama, T.; Kawashima, S.; Okuda, S.; Tokimatsu, T.; et al. KEGG for linking genomes to life and the environment. Nucleic Acids Res. 2008, 36, D480–D484. [Google Scholar] [CrossRef]
- Zubrzycki, A.; Wronska, A.; Zauszkiewicz-Pawlak, A.; Kmiec, Z. Short-term fenofibrate treatment improves ultrastructure of hepatocytes of old rats. Folia Histochem. Cytobiol. 2021, 59, 167–177. [Google Scholar] [CrossRef]
- Wang, X.; Song, X.; Si, Y.; Xia, J.; Wang, B.; Wang, P. Effect of autophagy-associated proteins on the arecoline-induced liver injury in mice. Exp. Ther. Med. 2018, 16, 3041–3049. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yoshii, S.R.; Mizushima, N. Monitoring and Measuring Autophagy. Int. J. Mol. Sci. 2017, 18, 1865. [Google Scholar] [CrossRef] [PubMed]
- Kabeya, Y.; Mizushima, N.; Ueno, T.; Yamamoto, A.; Kirisako, T.; Noda, T.; Kominami, E.; Ohsumi, Y.; Yoshimori, T. LC3, a mammalian homologue of yeast Apg8p, is localized in autophagosome membranes after processing. EMBO J. 2000, 19, 5720–5728. [Google Scholar] [CrossRef]
- Zhao, Y.; Wang, Z.; Zhang, W.; Zhang, L. MicroRNAs play an essential role in autophagy regulation in various disease phenotypes. Biofactors 2019, 45, 844–856. [Google Scholar] [CrossRef]
- Resaz, R.; Cangelosi, D.; Segalerba, D.; Morini, M.; Uva, P.; Bosco, M.C.; Banderali, G.; Estrella, A.; Wanner, C.; Weinstein, D.A.; et al. Exosomal MicroRNAs as Potential Biomarkers of Hepatic Injury and Kidney Disease in Glycogen Storage Disease Type Ia Patients. Int. J. Mol. Sci. 2021, 23, 328. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.; Liu, Y.; Wang, D.; Xu, Y.; Dong, R.; Yang, Y.; Lv, Q.; Chen, X.; Zhang, Z. The Upstream Pathway of mTOR-Mediated Autophagy in Liver Diseases. Cells 2019, 8, 1597. [Google Scholar] [CrossRef] [PubMed]
- Egan, D.; Kim, J.; Shaw, R.J.; Guan, K.L. The autophagy initiating kinase ULK1 is regulated via opposing phosphorylation by AMPK and mTOR. Autophagy 2011, 7, 643–644. [Google Scholar] [CrossRef]
- Laplante, M.; Sabatini, D.M. mTOR signaling in growth control and disease. Cell 2012, 149, 274–293. [Google Scholar] [CrossRef]
- Saxton, R.A.; Sabatini, D.M. mTOR Signaling in Growth, Metabolism, and Disease. Cell 2017, 169, 361–371. [Google Scholar] [CrossRef]
- Cui, F.L.; Mahmud, A.N.; Xu, Z.P.; Wang, Z.Y.; Hu, J.P. VASN promotes proliferation of prostate cancer through the YAP/TAZ axis. Eur. Rev. Med. Pharmacol. Sci. 2020, 24, 6589–6596. [Google Scholar] [CrossRef]
- Liu, H.; Kong, W.; Wen, S.; Wu, J.; Yin, X.; Liu, Y. VASN promotes proliferation of laryngeal cancer cells via YAP/TAZ. J. BUON 2021, 26, 1563–1570. [Google Scholar] [PubMed]
- Yeo, H.L.; Fan, T.C.; Lin, R.J.; Yu, J.C.; Liao, G.S.; Chen, E.S.; Ho, M.Y.; Lin, W.D.; Chen, K.; Chen, C.H.; et al. Sialylation of vasorin by ST3Gal1 facilitates TGF-beta1-mediated tumor angiogenesis and progression. Int. J. Cancer 2019, 144, 1996–2007. [Google Scholar] [CrossRef] [PubMed]
- Aydin, M.; Kiziltan, R.; Algul, S.; Kemik, O. The utility of serum vasorin levels as a novel potential biomarker for early detection of colon cancer. Cureus 2022, 14, e21653. [Google Scholar] [CrossRef]
- Ding, W.X.; Manley, S.; Ni, H.M. The emerging role of autophagy in alcoholic liver disease. Exp. Biol. Med. 2011, 236, 546–556. [Google Scholar] [CrossRef]
- Chao, X.; Wang, S.; Zhao, K.; Li, Y.; Williams, J.A.; Li, T.; Chavan, H.; Krishnamurthy, P.; He, X.C.; Li, L.; et al. Impaired TFEB-Mediated Lysosome Biogenesis and Autophagy Promote Chronic Ethanol-Induced Liver Injury and Steatosis in Mice. Gastroenterology 2018, 155, 865–879.e12. [Google Scholar] [CrossRef] [PubMed]
- Tanaka, S.; Hikita, H.; Tatsumi, T.; Sakamori, R.; Nozaki, Y.; Sakane, S.; Shiode, Y.; Nakabori, T.; Saito, Y.; Hiramatsu, N.; et al. Rubicon inhibits autophagy and accelerates hepatocyte apoptosis and lipid accumulation in nonalcoholic fatty liver disease in mice. Hepatology 2016, 64, 1994–2014. [Google Scholar] [CrossRef] [PubMed]
- Mizushima, N.; Yoshimori, T.; Levine, B. Methods in mammalian autophagy research. Cell 2010, 140, 313–326. [Google Scholar] [CrossRef]
- Klionsky, D.J.; Abdel-Aziz, A.K.; Abdelfatah, S.; Abdellatif, M.; Abdoli, A.; Abel, S.; Abeliovich, H.; Abildgaard, M.H.; Abudu, Y.P.; Acevedo-Arozena, A.; et al. Guidelines for the use and interpretation of assays for monitoring autophagy (4th edition) (1). Autophagy 2021, 17, 1–382. [Google Scholar] [CrossRef]
- Levine, B.; Packer, M.; Codogno, P. Development of autophagy inducers in clinical medicine. J. Clin. Investig. 2015, 125, 14–24. [Google Scholar] [CrossRef]
- Li, Q.; Ni, Y.; Zhang, L.; Jiang, R.; Xu, J.; Yang, H.; Hu, Y.; Qiu, J.; Pu, L.; Tang, J.; et al. HIF-1alpha-induced expression of m6A reader YTHDF1 drives hypoxia-induced autophagy and malignancy of hepatocellular carcinoma by promoting ATG2A and ATG14 translation. Signal Transduct. Target. Ther. 2021, 6, 76. [Google Scholar] [CrossRef]
- Zhang, M.; Liu, S.; Chua, M.S.; Li, H.; Luo, D.; Wang, S.; Zhang, S.; Han, B.; Sun, C. SOCS5 inhibition induces autophagy to impair metastasis in hepatocellular carcinoma cells via the PI3K/Akt/mTOR pathway. Cell Death Dis. 2019, 10, 612. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xu, J.; Wang, Y.; Tan, X.; Jing, H. MicroRNAs in autophagy and their emerging roles in crosstalk with apoptosis. Autophagy 2012, 8, 873–882. [Google Scholar] [CrossRef] [PubMed]
- Zhao, X.; Su, L.; He, X.; Zhao, B.; Miao, J. Long noncoding RNA CA7-4 promotes autophagy and apoptosis via sponging MIR877-3P and MIR5680 in high glucose-induced vascular endothelial cells. Autophagy 2020, 16, 70–85. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.; Kundu, M.; Viollet, B.; Guan, K.L. AMPK and mTOR regulate autophagy through direct phosphorylation of Ulk1. Nat. Cell Biol. 2011, 13, 132–141. [Google Scholar] [CrossRef]
- Gambardella, G.; Staiano, L.; Moretti, M.N.; De Cegli, R.; Fagnocchi, L.; Di Tullio, G.; Polletti, S.; Braccia, C.; Armirotti, A.; Zippo, A.; et al. GADD34 is a modulator of autophagy during starvation. Sci. Adv. 2020, 6, eabb0205. [Google Scholar] [CrossRef]
- Roach, P.J. Glycogen and its metabolism. Curr. Mol. Med. 2002, 2, 101–120. [Google Scholar] [CrossRef]
- Choi, J.A.; Maddala, R.; Karnam, S.; Skiba, N.P.; Vann, R.; Challa, P.; Rao, P.V. Role of vasorin, an anti-apoptotic, anti-TGF-beta and hypoxia-induced glycoprotein in the trabecular meshwork cells and glaucoma. J. Cell. Mol. Med. 2022, 26, 2063–2075. [Google Scholar] [CrossRef]
- Nur, T.; Sela, I.; Webster, N.J.; Madar, Z. Starvation and.d refeeding regulate glycogen synthase gene expression in rat liver at the posttranscriptional level. J. Nutr. 1995, 125, 2457–2462. [Google Scholar] [CrossRef]
- Deter, R.L.; Baudhuin, P.; De Duve, C. Participation of lysosomes in cellular autophagy induced in rat liver by glucagon. J. Cell Biol. 1967, 35, C11–C16. [Google Scholar] [CrossRef]
- Karabiyik, C.; Vicinanza, M.; Son, S.M.; Rubinsztein, D.C. Glucose starvation induces autophagy via ULK1-mediated activation of PIKfyve in an AMPK-dependent manner. Dev. Cell 2021, 56, 1961–1975.e5. [Google Scholar] [CrossRef]
- Leprivier, G.; Rotblat, B. How does mTOR sense glucose starvation? AMPK is the usual suspect. Cell Death Discov. 2020, 6, 27. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hopkins, B.D.; Pauli, C.; Du, X.; Wang, D.G.; Li, X.; Wu, D.; Amadiume, S.C.; Goncalves, M.D.; Hodakoski, C.; Lundquist, M.R.; et al. Suppression of insulin feedback enhances the efficacy of PI3K inhibitors. Nature 2018, 560, 499–503. [Google Scholar] [CrossRef] [PubMed]
- Singh, B.K.; Tripathi, M.; Sandireddy, R.; Tikno, K.; Zhou, J.; Yen, P.M. Decreased autophagy and fuel switching occur in a senescent hepatic cell model system. Aging 2020, 12, 13958–13978. [Google Scholar] [CrossRef] [PubMed]
- Zhou, J.; Peng, X.; Ru, Y.; Xu, J. Circ_0060077 knockdown alleviates high-glucose-induced cell apoptosis, oxidative stress, inflammation and fibrosis in HK-2 Cells via miR-145-5p/VASN pathway. Inflammation, 2022; epub ahead of print. [Google Scholar] [CrossRef]



Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Yang, L.; Cheng, X.; Shi, W.; Li, H.; Zhang, Q.; Huang, S.; Huang, X.; Wen, S.; Gan, J.; Liao, Z.; et al. Vasorin Deletion in C57BL/6J Mice Induces Hepatocyte Autophagy through Glycogen-Mediated mTOR Regulation. Nutrients 2022, 14, 3600. https://doi.org/10.3390/nu14173600
Yang L, Cheng X, Shi W, Li H, Zhang Q, Huang S, Huang X, Wen S, Gan J, Liao Z, et al. Vasorin Deletion in C57BL/6J Mice Induces Hepatocyte Autophagy through Glycogen-Mediated mTOR Regulation. Nutrients. 2022; 14(17):3600. https://doi.org/10.3390/nu14173600
Chicago/Turabian StyleYang, Lichao, Xiaojing Cheng, Wei Shi, Hui Li, Qi Zhang, Shiping Huang, Xuejing Huang, Sha Wen, Ji Gan, Zhouxiang Liao, and et al. 2022. "Vasorin Deletion in C57BL/6J Mice Induces Hepatocyte Autophagy through Glycogen-Mediated mTOR Regulation" Nutrients 14, no. 17: 3600. https://doi.org/10.3390/nu14173600