

Reduction in Insulin Mediated ERK Phosphorylation by Palmitate in Liver Cells Is Independent of Fatty Acid Induced ER Stress

 ,
,  and
and
Abstract
:1. Introduction
2. Materials and Methods
2.1. Materials
2.2. Cell Culture and Treatment
2.3. Western Blotting
2.4. mRNA Analysis via Quantitative Real Time PCR
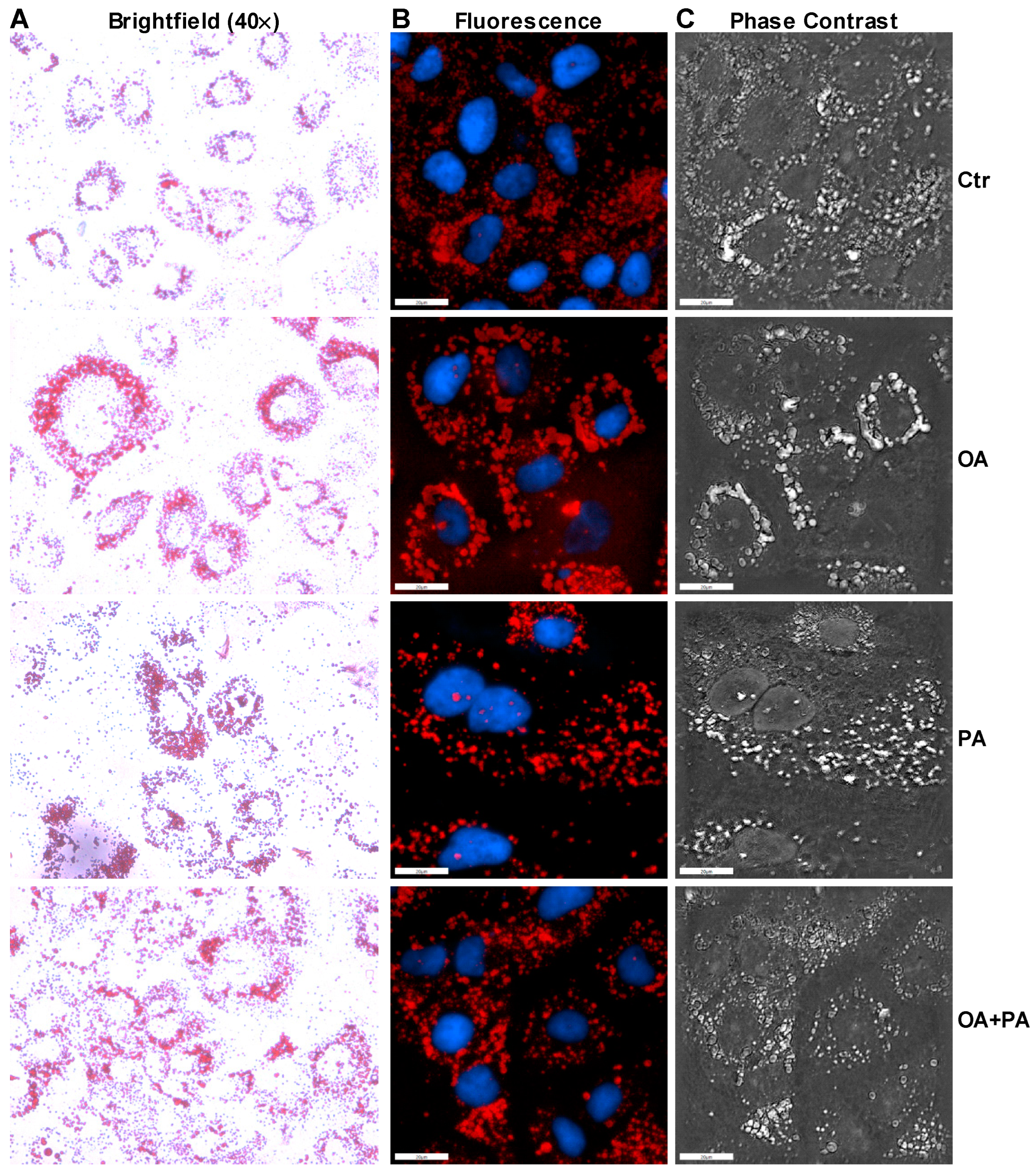
2.5. Oil Red O Staining
2.6. Statistical Analysis
3. Results
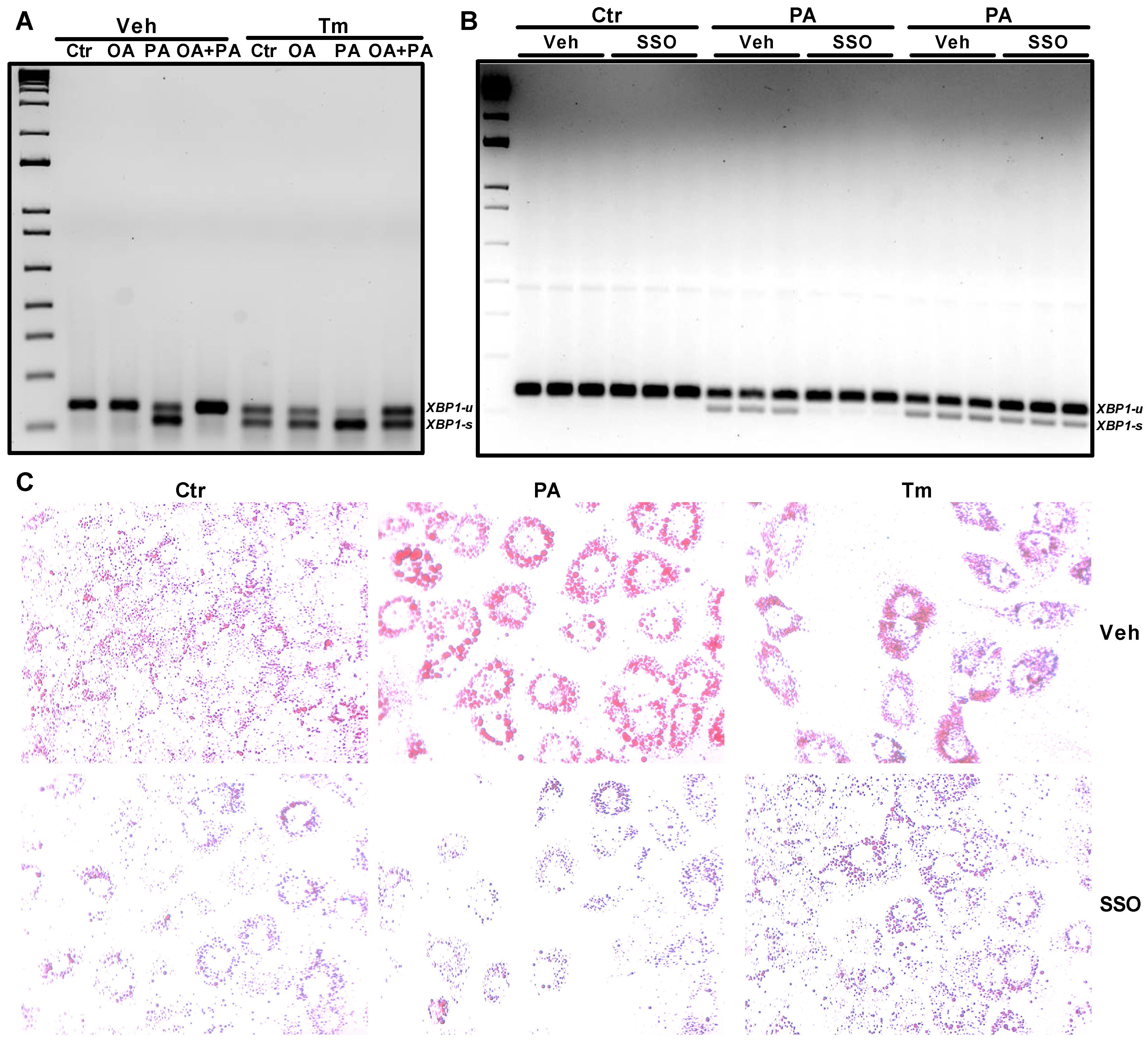
3.1. Induction of ER Stress by Tunicamycin in Huh-7 Cells Impairs Insulin Signaling through a Reduction in ERK Phosphorylation
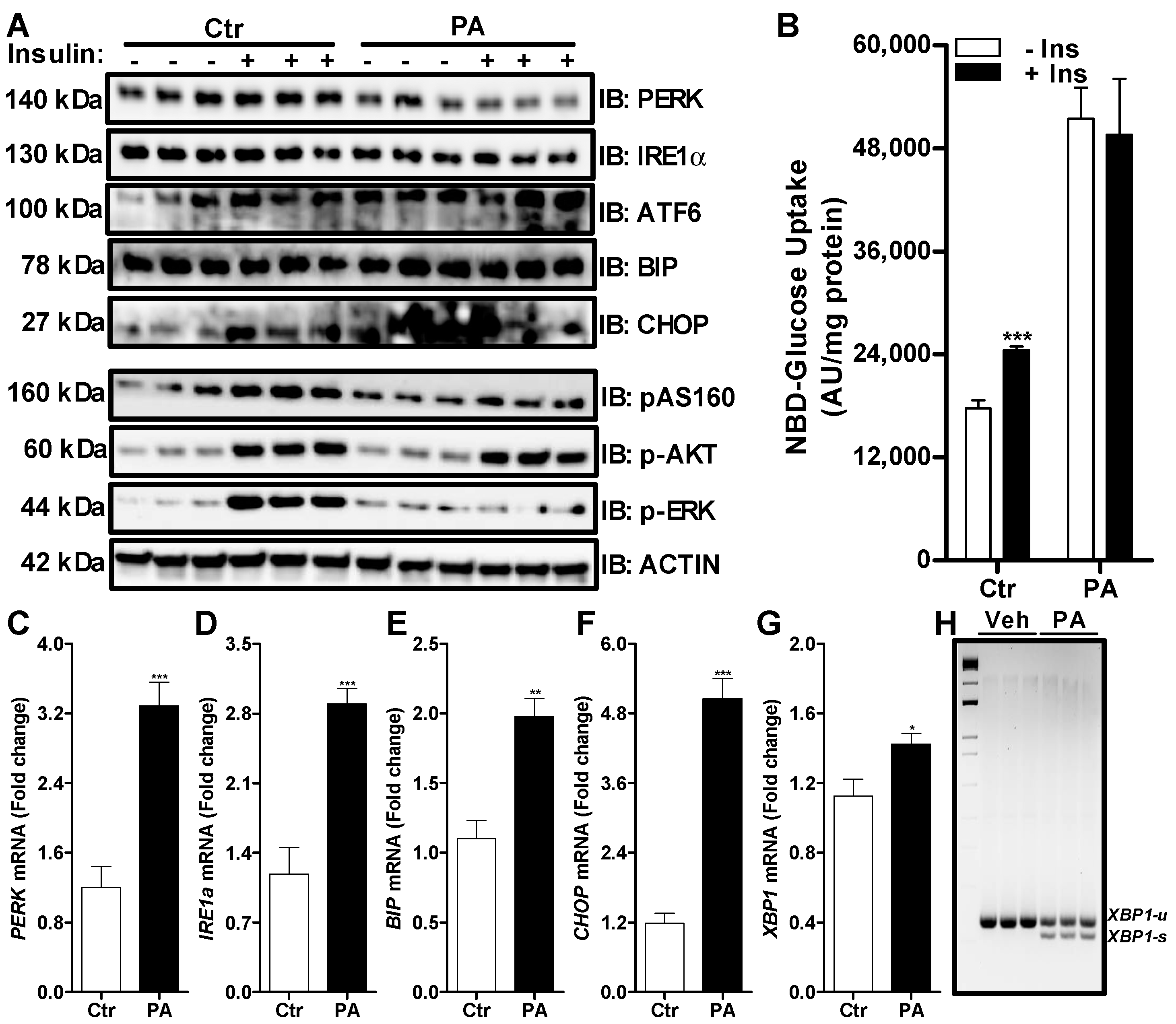
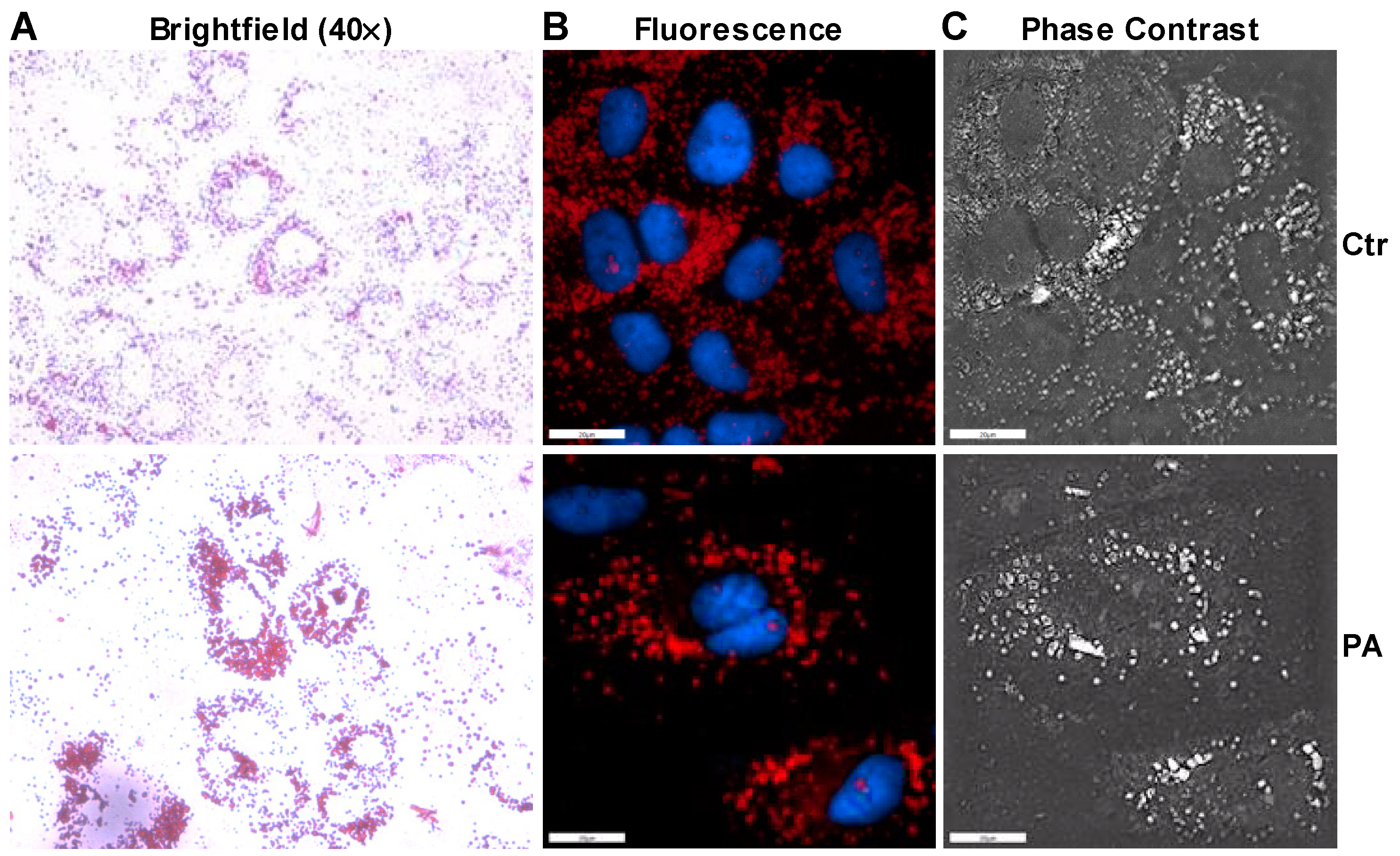
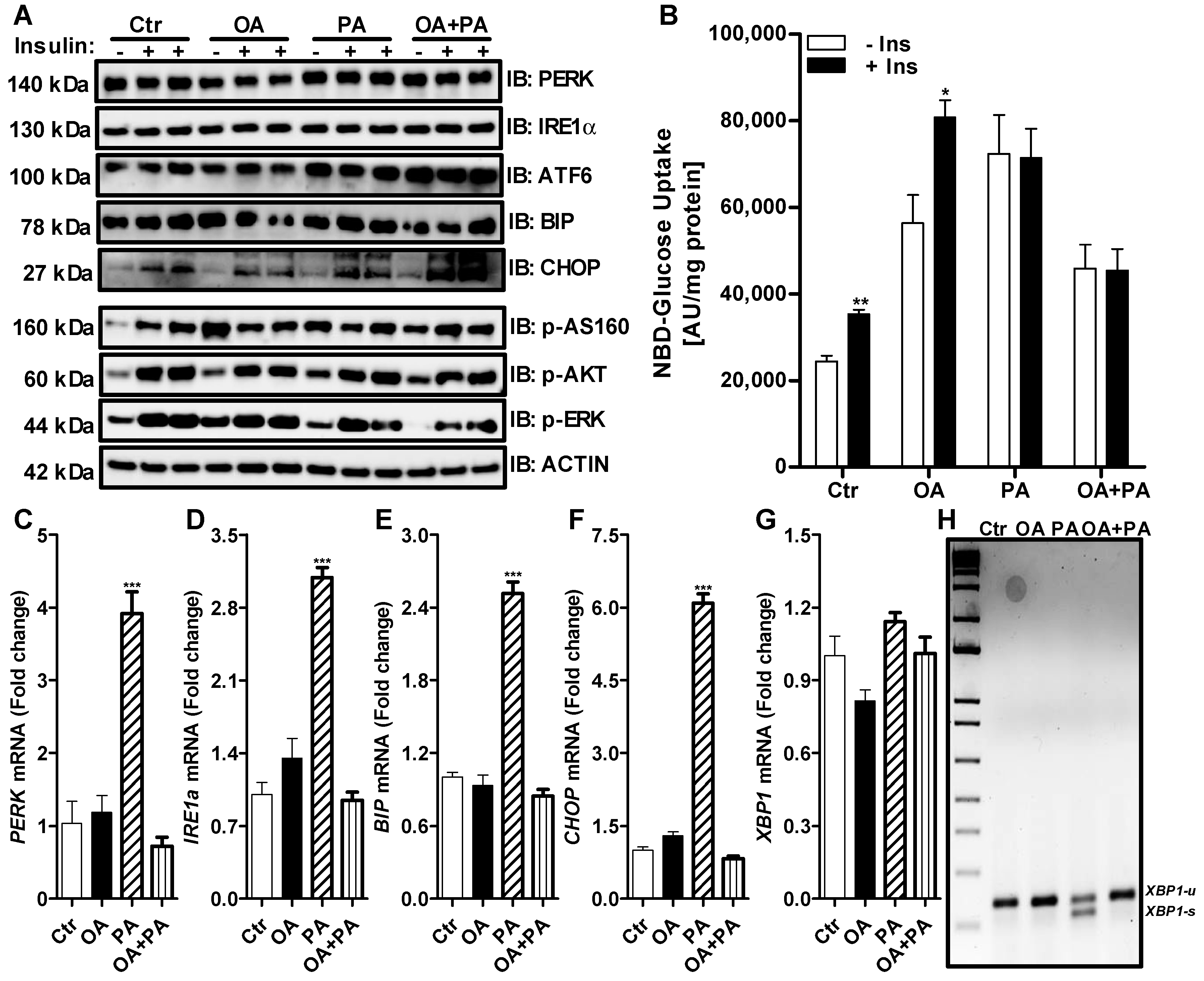
3.2. Palmitate Induces ER Stress and Reduces ERK Phosphorylation That Affects Insulin Dependent Glucose Uptake in Huh-7 Cells
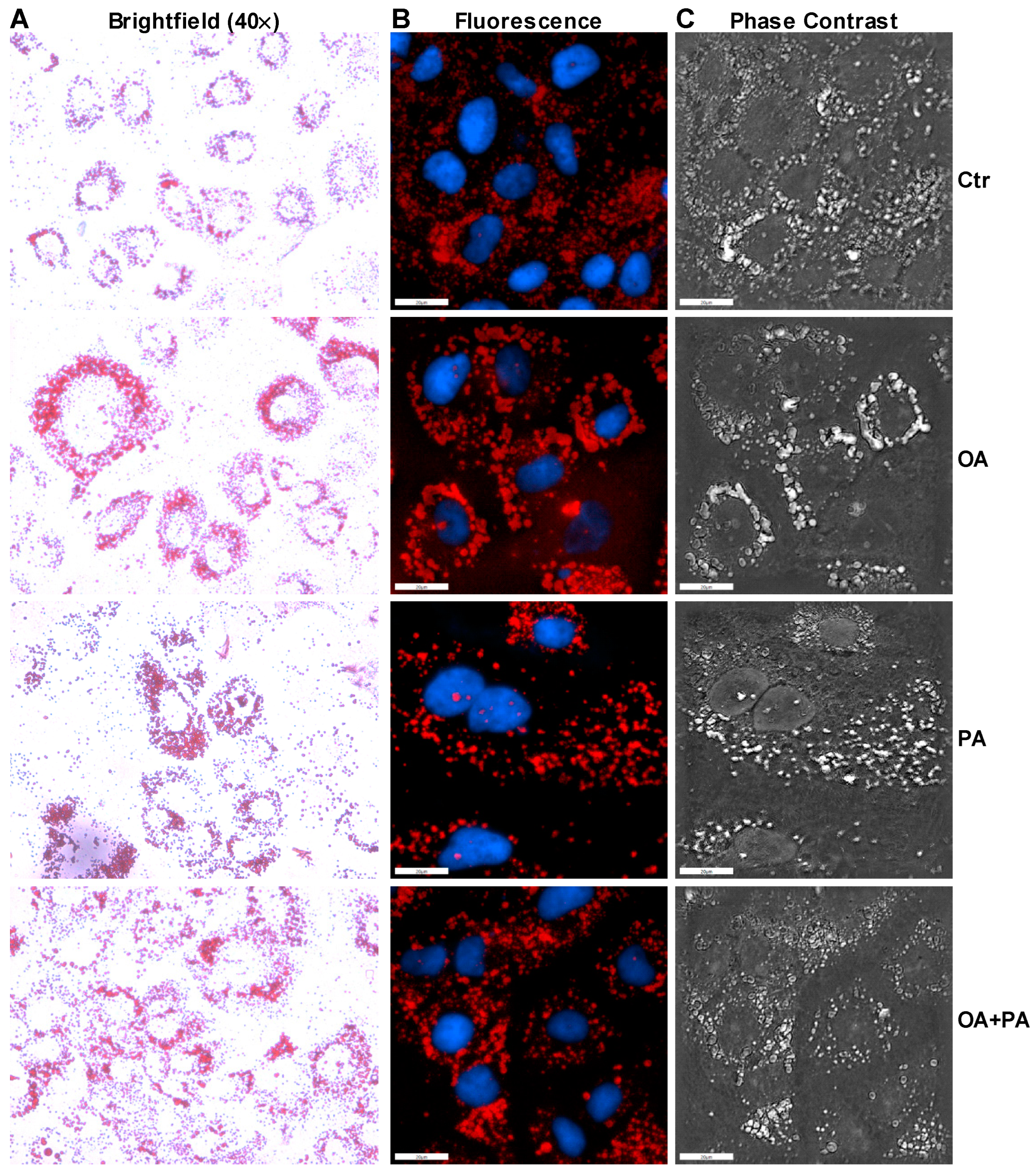
3.3. Oleate Prevents Palmitate-Induced ER Stress without Affecting Insulin Signaling in Huh-7 Cells
3.4. CD36 Inhibitor Prevents Palmitate-Induced XBP1 Splicing in Huh-7 Cells
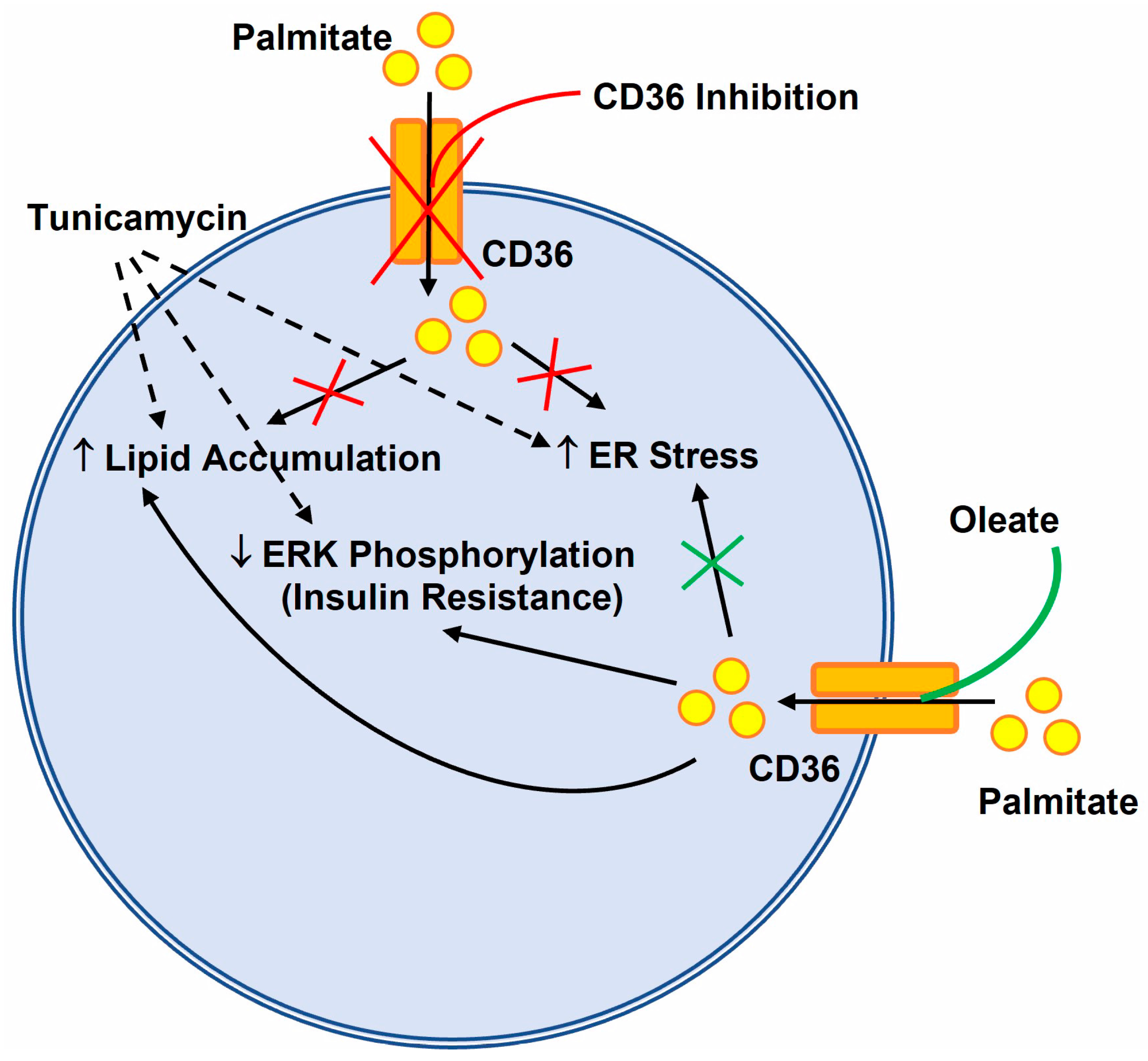
4. Discussion
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Boden, G. Role of fatty acids in the pathogenesis of insulin resistance and NIDDM. Diabetes 1997, 46, 3–10. [Google Scholar] [CrossRef] [PubMed]
- Ertunc, M.E.; Hotamisligil, G.S. Lipid signaling and lipotoxicity in metaflammation: Indications for metabolic disease pathogenesis and treatment. J. Lipid Res. 2016, 57, 2099–2114. [Google Scholar] [CrossRef] [PubMed]
- Kien, C.L.; Bunn, J.Y.; Stevens, R.; Bain, J.; Ikayeva, O.; Crain, K.; Koves, T.R.; Muoio, D.M. Dietary intake of palmitate and oleate has broad impact on systemic and tissue lipid profiles in humans. Am. J. Clin. Nutr. 2014, 99, 436–445. [Google Scholar] [CrossRef] [PubMed]
- Ricchi, M.; Odoardi, M.R.; Carulli, L.; Anzivino, C.; Ballestri, S.; Pinetti, A.; Fantoni, L.I.; Marra, F.; Bertolotti, M.; Banni, S.; et al. Differential effect of oleic and palmitic acid on lipid accumulation and apoptosis in cultured hepatocytes. J. Gastroenterol. Hepatol. 2009, 24, 830–840. [Google Scholar] [CrossRef] [PubMed]
- Staiger, K.; Staiger, H.; Weigert, C.; Haas, C.; Haring, H.U.; Kellerer, M. Saturated, but not unsaturated, fatty acids induce apoptosis of human coronary artery endothelial cells via nuclear factor-kappaB activation. Diabetes 2006, 55, 3121–3126. [Google Scholar] [CrossRef]
- El-Assaad, W.; Buteau, J.; Peyot, M.L.; Nolan, C.; Roduit, R.; Hardy, S.; Joly, E.; Dbaibo, G.; Rosenberg, L.; Prentki, M. Saturated fatty acids synergize with elevated glucose to cause pancreatic beta-cell death. Endocrinology 2003, 144, 4154–4163. [Google Scholar] [CrossRef]
- Karaskov, E.; Scott, C.; Zhang, L.; Teodoro, T.; Ravazzola, M.; Volchuk, A. Chronic palmitate but not oleate exposure induces endoplasmic reticulum stress, which may contribute to INS-1 pancreatic beta-cell apoptosis. Endocrinology 2006, 147, 3398–3407. [Google Scholar] [CrossRef]
- Miller, T.A.; LeBrasseur, N.K.; Cote, G.M.; Trucillo, M.P.; Pimentel, D.R.; Ido, Y.; Ruderman, N.B.; Sawyer, D.B. Oleate prevents palmitate-induced cytotoxic stress in cardiac myocytes. Biochem. Biophys. Res. Commun. 2005, 336, 309–315. [Google Scholar] [CrossRef]
- Wei, Y.; Wang, D.; Topczewski, F.; Pagliassotti, M.J. Saturated fatty acids induce endoplasmic reticulum stress and apoptosis independently of ceramide in liver cells. Am. J. Physiol Endocrinol. Metab. 2006, 291, E275–E281. [Google Scholar] [CrossRef]
- Listenberger, L.L.; Han, X.; Lewis, S.E.; Cases, S.; Farese, R.V., Jr.; Ory, D.S.; Schaffer, J.E. Triglyceride accumulation protects against fatty acid-induced lipotoxicity. Proc. Natl. Acad. Sci. USA 2003, 100, 3077–3082. [Google Scholar] [CrossRef] [Green Version]
- Soumura, M.; Kume, S.; Isshiki, K.; Takeda, N.; Araki, S.; Tanaka, Y.; Sugimoto, T.; Chin-Kanasaki, M.; Nishio, Y.; Haneda, M.; et al. Oleate and eicosapentaenoic acid attenuate palmitate-induced inflammation and apoptosis in renal proximal tubular cell. Biochem. Biophys. Res. Commun. 2010, 402, 265–271. [Google Scholar] [CrossRef] [PubMed]
- Thorn, K.; Bergsten, P. Fatty acid-induced oxidation and triglyceride formation is higher in insulin-producing MIN6 cells exposed to oleate compared to palmitate. J. Cell Biochem. 2010, 111, 497–507. [Google Scholar] [CrossRef] [PubMed]
- Peng, G.; Li, L.; Liu, Y.; Pu, J.; Zhang, S.; Yu, J.; Zhao, J.; Liu, P. Oleate blocks palmitate-induced abnormal lipid distribution, endoplasmic reticulum expansion and stress, and insulin resistance in skeletal muscle. Endocrinology 2011, 152, 2206–2218. [Google Scholar] [CrossRef]
- Kim, D.H.; Cho, Y.M.; Lee, K.H.; Jeong, S.W.; Kwon, O.J. Oleate protects macrophages from palmitate-induced apoptosis through the downregulation of CD36 expression. Biochem. Biophys. Res. Commun. 2017, 488, 477–482. [Google Scholar] [CrossRef]
- Colvin, B.N.; Longtine, M.S.; Chen, B.; Costa, M.L.; Nelson, D.M. Oleate attenuates palmitate-induced endoplasmic reticulum stress and apoptosis in placental trophoblasts. Reproduction 2017, 153, 369–380. [Google Scholar] [CrossRef] [PubMed]
- Salvado, L.; Coll, T.; Gomez-Foix, A.M.; Salmeron, E.; Barroso, E.; Palomer, X.; Vazquez-Carrera, M. Oleate prevents saturated-fatty-acid-induced ER stress, inflammation and insulin resistance in skeletal muscle cells through an AMPK-dependent mechanism. Diabetologia 2013, 56, 1372–1382. [Google Scholar] [CrossRef] [PubMed]
- Flamment, M.; Kammoun, H.L.; Hainault, I.; Ferre, P.; Foufelle, F. Endoplasmic reticulum stress: A new actor in the development of hepatic steatosis. Curr. Opin. Lipidol. 2010, 21, 239–246. [Google Scholar] [CrossRef]
- Nakamura, S.; Takamura, T.; Matsuzawa-Nagata, N.; Takayama, H.; Misu, H.; Noda, H.; Nabemoto, S.; Kurita, S.; Ota, T.; Ando, H.; et al. Palmitate induces insulin resistance in H4IIEC3 hepatocytes through reactive oxygen species produced by mitochondria. J. Biol. Chem. 2009, 284, 14809–14818. [Google Scholar] [CrossRef]
- Ruddock, M.W.; Stein, A.; Landaker, E.; Park, J.; Cooksey, R.C.; McClain, D.; Patti, M.E. Saturated fatty acids inhibit hepatic insulin action by modulating insulin receptor expression and post-receptor signalling. J. Biochem. 2008, 144, 599–607. [Google Scholar] [CrossRef]
- Bjornholm, M.; Kawano, Y.; Lehtihet, M.; Zierath, J.R. Insulin receptor substrate-1 phosphorylation and phosphatidylinositol 3-kinase activity in skeletal muscle from NIDDM subjects after in vivo insulin stimulation. Diabetes 1997, 46, 524–527. [Google Scholar] [CrossRef] [Green Version]
- Krook, A.; Bjornholm, M.; Galuska, D.; Jiang, X.J.; Fahlman, R.; Myers, M.G., Jr.; Wallberg-Henriksson, H.; Zierath, J.R. Characterization of signal transduction and glucose transport in skeletal muscle from type 2 diabetic patients. Diabetes 2000, 49, 284–292. [Google Scholar] [CrossRef] [PubMed]
- Andreasson, K.; Galuska, D.; Thorne, A.; Sonnenfeld, T.; Wallberg-Henriksson, H. Decreased insulin-stimulated 3-0-methylglucose transport in in vitro incubated muscle strips from type II diabetic subjects. Acta Physiol. Scand. 1991, 142, 255–260. [Google Scholar] [CrossRef] [PubMed]
- Jiang, Z.Y.; Zhou, Q.L.; Coleman, K.A.; Chouinard, M.; Boese, Q.; Czech, M.P. Insulin signaling through Akt/protein kinase B analyzed by small interfering RNA-mediated gene silencing. Proc. Natl. Acad. Sci. USA 2003, 100, 7569–7574. [Google Scholar] [CrossRef]
- Ozaki, K.I.; Awazu, M.; Tamiya, M.; Iwasaki, Y.; Harada, A.; Kugisaki, S.; Tanimura, S.; Kohno, M. Targeting the ERK signaling pathway as a potential treatment for insulin resistance and type 2 diabetes. Am. J. Physiol. Endocrinol. Metab. 2016, 310, E643–E651. [Google Scholar] [CrossRef] [PubMed]
- Tang, X.; Shen, H.; Chen, J.; Wang, X.; Zhang, Y.; Chen, L.L.; Rukachaisirikul, V.; Jiang, H.L.; Shen, X. Activating transcription factor 6 protects insulin receptor from ER stress-stimulated desensitization via p42/44 ERK pathway. Acta Pharmacol. Sin. 2011, 32, 1138–1147. [Google Scholar] [CrossRef]
- Hwang, S.L.; Jeong, Y.T.; Li, X.; Kim, Y.D.; Lu, Y.; Chang, Y.C.; Lee, I.K.; Chang, H.W. Inhibitory cross-talk between the AMPK and ERK pathways mediates endoplasmic reticulum stress-induced insulin resistance in skeletal muscle. Br. J. Pharmacol. 2013, 169, 69–81. [Google Scholar] [CrossRef]
- Powell, D.J.; Turban, S.; Gray, A.; Hajduch, E.; Hundal, H.S. Intracellular ceramide synthesis and protein kinase Czeta activation play an essential role in palmitate-induced insulin resistance in rat L6 skeletal muscle cells. Biochem. J. 2004, 382, 619–629. [Google Scholar] [CrossRef]
- Yoshioka, K.; Oh, K.B.; Saito, M.; Nemoto, Y.; Matsuoka, H. Evaluation of 2-[N-(7-nitrobenz-2-oxa-1,3-diazol-4-yl)amino]-2-deoxy-D-glucose, a new fluorescent derivative of glucose, for viability assessment of yeast Candida albicans. Appl. Microbiol. Biotechnol. 1996, 46, 400–404. [Google Scholar] [CrossRef]
- Kuda, O.; Pietka, T.A.; Demianova, Z.; Kudova, E.; Cvacka, J.; Kopecky, J.; Abumrad, N.A. Sulfo-N-succinimidyl oleate (SSO) inhibits fatty acid uptake and signaling for intracellular calcium via binding CD36 lysine 164: SSO also inhibits oxidized low density lipoprotein uptake by macrophages. J. Biol. Chem. 2013, 288, 15547–15555. [Google Scholar] [CrossRef]
- Nath, A.; Li, I.; Roberts, L.R.; Chan, C. Elevated free fatty acid uptake via CD36 promotes epithelial-mesenchymal transition in hepatocellular carcinoma. Sci. Rep. 2015, 5, 14752. [Google Scholar] [CrossRef] [Green Version]
- Drury, J.; Rychahou, P.G.; He, D.; Jafari, N.; Wang, C.; Lee, E.Y.; Weiss, H.L.; Evers, B.M.; Zaytseva, Y.Y. Inhibition of Fatty Acid Synthase Upregulates Expression of CD36 to Sustain Proliferation of Colorectal Cancer Cells. Front. Oncol. 2020, 10, 1185. [Google Scholar] [CrossRef]
- Huang, X.; Liu, G.; Guo, J.; Su, Z. The PI3K/AKT pathway in obesity and type 2 diabetes. Int. J. Biol. Sci. 2018, 14, 1483–1496. [Google Scholar] [CrossRef] [PubMed]
- Ijuin, T.; Hosooka, T.; Takenawa, T. Phosphatidylinositol 3,4,5-Trisphosphate Phosphatase SKIP Links Endoplasmic Reticulum Stress in Skeletal Muscle to Insulin Resistance. Mol. Cell Biol. 2016, 36, 108–118. [Google Scholar] [CrossRef] [PubMed]
- Hage Hassan, R.; Hainault, I.; Vilquin, J.T.; Samama, C.; Lasnier, F.; Ferre, P.; Foufelle, F.; Hajduch, E. Endoplasmic reticulum stress does not mediate palmitate-induced insulin resistance in mouse and human muscle cells. Diabetologia 2012, 55, 204–214. [Google Scholar] [CrossRef] [PubMed]
- Schmitz-Peiffer, C.; Craig, D.L.; Biden, T.J. Ceramide generation is sufficient to account for the inhibition of the insulin-stimulated PKB pathway in C2C12 skeletal muscle cells pretreated with palmitate. J. Biol. Chem. 1999, 274, 24202–24210. [Google Scholar] [CrossRef]
- Achard, C.S.; Laybutt, D.R. Lipid-induced endoplasmic reticulum stress in liver cells results in two distinct outcomes: Adaptation with enhanced insulin signaling or insulin resistance. Endocrinology 2012, 153, 2164–2177. [Google Scholar] [CrossRef]
- Pardo, V.; Gonzalez-Rodriguez, A.; Muntane, J.; Kozma, S.C.; Valverde, A.M. Role of hepatocyte S6K1 in palmitic acid-induced endoplasmic reticulum stress, lipotoxicity, insulin resistance and in oleic acid-induced protection. Food Chem. Toxicol. 2015, 80, 298–309. [Google Scholar] [CrossRef]
- Murer, E.; Boden, G.; Gyda, M.; Deluca, F. Effects of oleate and insulin on glucose uptake, oxidation, and glucose transporter proteins in rat adipocytes. Diabetes 1992, 41, 1063–1068. [Google Scholar] [CrossRef]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Gene | Forward Primer | Reverse Primer |
|---|---|---|
| PERK | 5′-CAGAGATTGAGACTGCGTGGC-3′ | 5′-AAGGAACCGGATCCCACATC-3′ |
| IRE1a | 5′-TTGGGCGAACAGAGATGTCC-3′ | 5′-CACAGGGGAGGCGTAGTTTT-3′ |
| BIP | 5′-CGGGCAAAGATGTCAGGAAAG-3′ | 5′-CAGATGATACTTCGGGCAGGTC-3′ |
| CHOP | 5′-ACCAAGGGAGAACCAGGAAACG-3′ | 5′-CGAGACTAACTGGCTTACCACT-3′ |
| XBP1 | 5′-TTACGAGAGAAAACTCATGGC-3′ | 5′-CGTAAGACCTGTTGAACCTGGG-3′ |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Mubarak, S.A.; Otaibi, A.A.; Qarni, A.A.; Bakillah, A.; Iqbal, J. Reduction in Insulin Mediated ERK Phosphorylation by Palmitate in Liver Cells Is Independent of Fatty Acid Induced ER Stress. Nutrients 2022, 14, 3641. https://doi.org/10.3390/nu14173641
Mubarak SA, Otaibi AA, Qarni AA, Bakillah A, Iqbal J. Reduction in Insulin Mediated ERK Phosphorylation by Palmitate in Liver Cells Is Independent of Fatty Acid Induced ER Stress. Nutrients. 2022; 14(17):3641. https://doi.org/10.3390/nu14173641
Chicago/Turabian StyleMubarak, Sindiyan Alshaikh, Abeer Al Otaibi, Ali Al Qarni, Ahmed Bakillah, and Jahangir Iqbal. 2022. "Reduction in Insulin Mediated ERK Phosphorylation by Palmitate in Liver Cells Is Independent of Fatty Acid Induced ER Stress" Nutrients 14, no. 17: 3641. https://doi.org/10.3390/nu14173641
APA StyleMubarak, S. A., Otaibi, A. A., Qarni, A. A., Bakillah, A., & Iqbal, J. (2022). Reduction in Insulin Mediated ERK Phosphorylation by Palmitate in Liver Cells Is Independent of Fatty Acid Induced ER Stress. Nutrients, 14(17), 3641. https://doi.org/10.3390/nu14173641