
Wentilactone A Reverses the NF-κB/ECM1 Signaling-Induced Cisplatin Resistance through Inhibition of IKK/IκB in Ovarian Cancer Cells

 and
and
Abstract
:1. Introduction
2. Materials and Methods
2.1. Chemical Compound and Preparation
2.2. Cell Culture
2.3. Establishment of Cell Lines by Gene Silencing or Over-Expression
2.4. Cell Counting Kit-8 (CCK-8) Assay
2.5. Western Blotting Analysis
2.6. Mouse Xenograft Model
2.7. ECM1 Promoter Cloning
2.8. Luciferase Assay
2.9. Immunofluorescence Staining
2.10. Cell Culture Conditioned Medium (CM) and Detection of Secreted ECM1
2.11. 3D Spheroid Formation Assay
2.12. Immunohistochemistry Staining
2.13. Statistical Analysis
3. Results
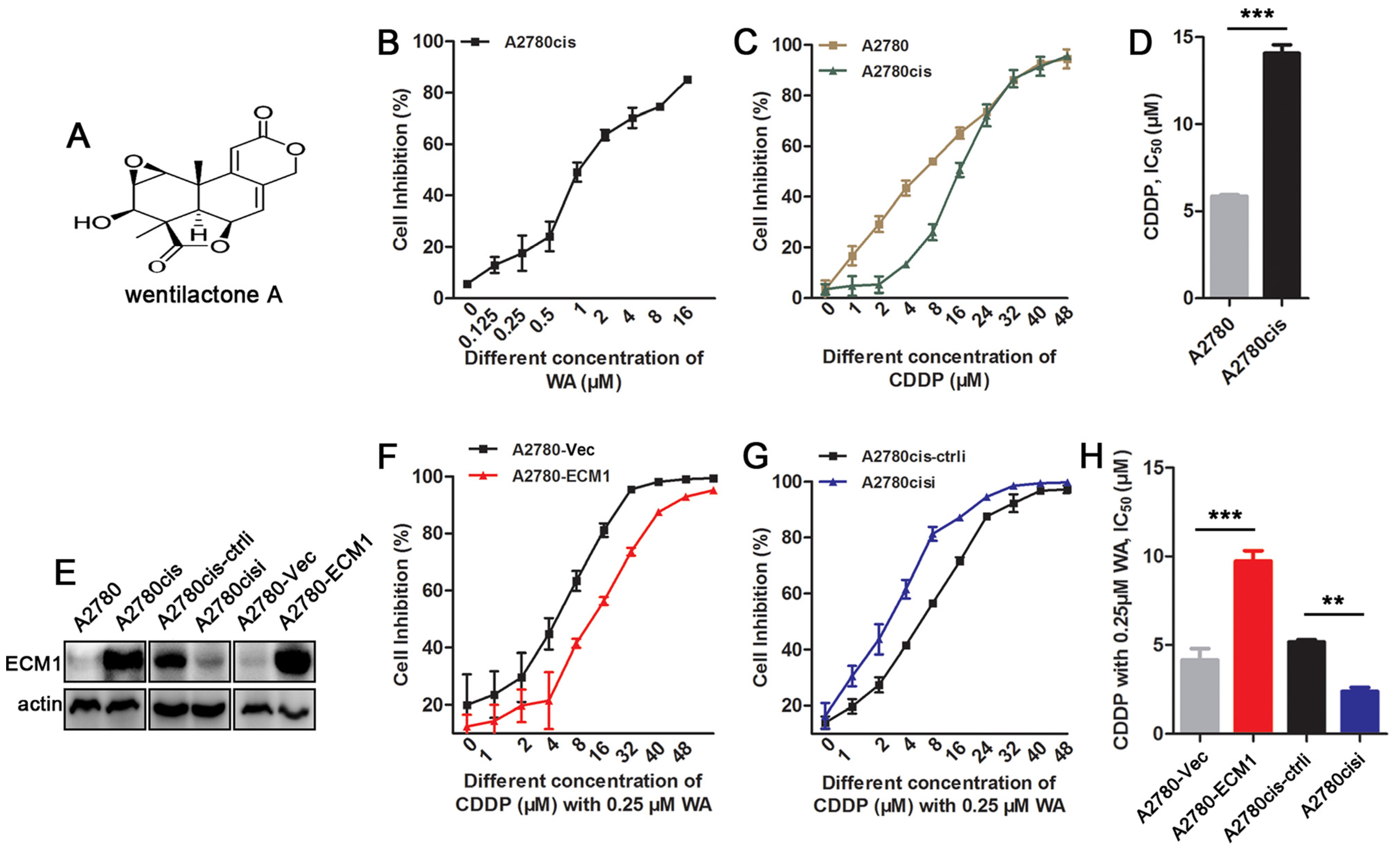
3.1. WA Reverses Cisplatin Resistance in ECM1 Highly-Expressed Ovarian Cancer Cells
3.2. Resistent Reversal Efficacy of WA + Cisplatin Is Associated with ECM1 Expression
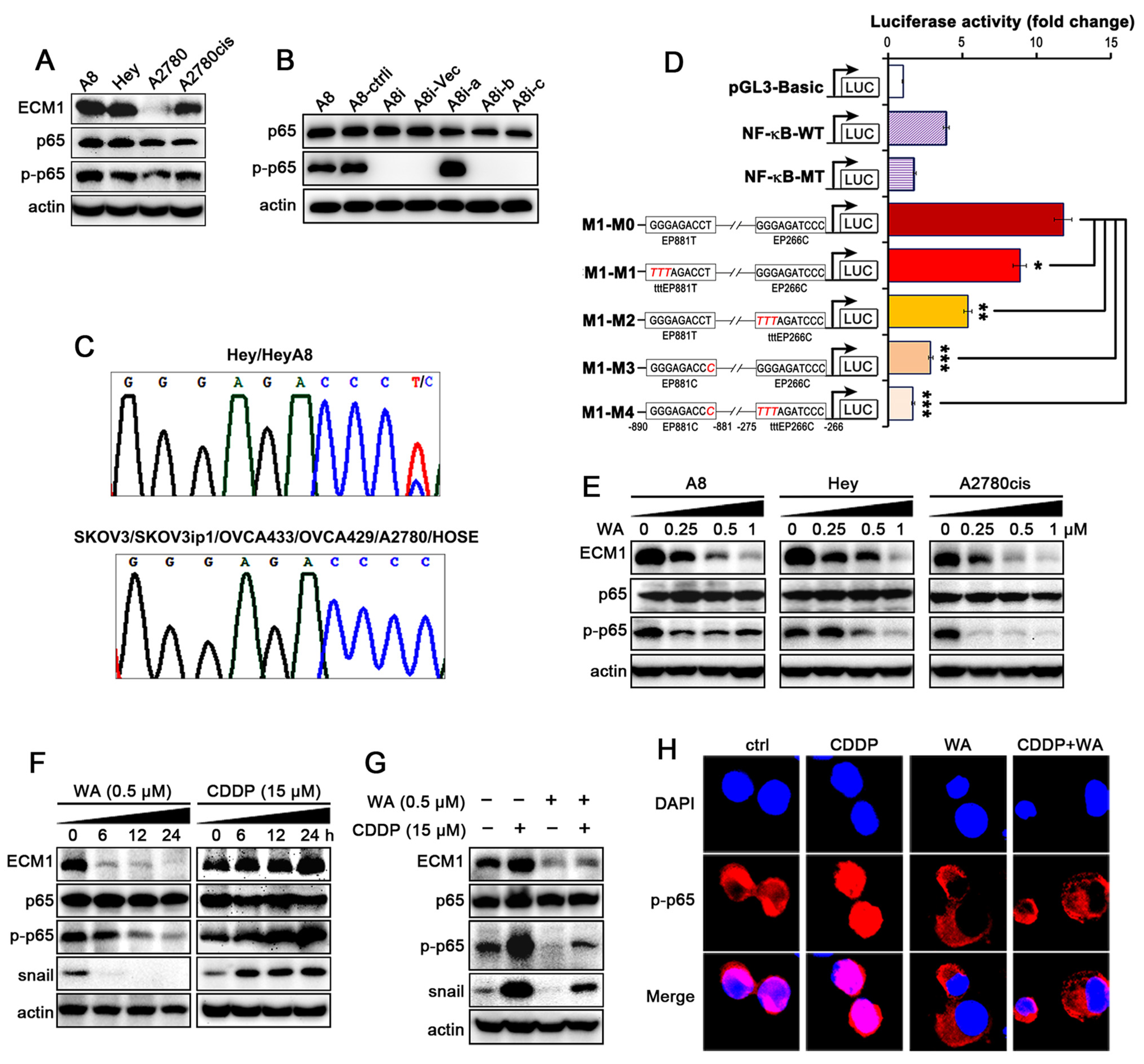
3.3. WA Inhibits the Cisplatin-Induced Activation of NF-κB/ECM1
3.4. The Secreted ECM1 Induces Malignant Transformation of NFs
3.5. WA Inhibits IKKs/IκBα Phosphorylation
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Ferlay, J.; Soerjomataram, I.; Dikshit, R.; Eser, S.; Mathers, C.; Rebelo, M.; Parkin, D.M.; Forman, D.; Bray, F. Cancer incidence and mortality worldwide: Sources, methods and major patterns in GLOBOCAN 2012. Int. J. Cancer 2015, 136, 359–386. [Google Scholar] [CrossRef]
- Ahmed, S.; Khan, H.; Fakhri, S.; Aschner, M.; Cheang, W.S. Therapeutic potential of marine peptides in cervical and ovarian cancers. Mol. Cell. Biochem. 2022, 477, 605–619. [Google Scholar] [CrossRef]
- Eisenhauer, E.A. Real-world evidence in the treatment of ovarian cancer. Ann. Oncol. 2017, 28, viii61–viii65. [Google Scholar] [CrossRef]
- Park, K.J.; Krishnan, V.; O’malley, B.W.; Yamamoto, Y.; Gaynor, R.B. Formation of an IKKalpha-dependent transcription complex is required for estrogen receptor-mediated gene activation. Mol. Cell 2005, 18, 71–82. [Google Scholar] [CrossRef]
- Nakanishi, C.; Toi, M. Nuclear factor-kappaB inhibitors as sensitizers to anticancer drugs. Nat. Rev. Cancer 2005, 5, 297–309. [Google Scholar] [CrossRef]
- Yeh, P.Y.; Yeh, K.H.; Chuang, S.E.; Song, Y.C.; Cheng, A.L. Suppression of MEK/ERK signaling pathway enhances cisplatin-induced NF-kappaB activation by protein phosphatase 4-mediated NF-kappaB p65 Thr dephosphorylation. J. Biol. Chem. 2004, 279, 26143–26148. [Google Scholar] [CrossRef]
- Von Ahrens, D.; Bhagat, T.D.; Nagrath, D.; Maitra, A.; Verma, A. The role of stromal cancer-associated fibroblasts in pancreatic cancer. J. Hematol. Oncol. 2017, 10, 76. [Google Scholar] [CrossRef]
- Wu, T.; Dai, Y. Tumor microenvironment and therapeutic response. Cancer Lett. 2017, 387, 61–68. [Google Scholar] [CrossRef]
- Hui, L.; Chen, Y. Tumor microenvironment: Sanctuary of the devil. Cancer Lett. 2015, 368, 7–13. [Google Scholar] [CrossRef]
- Johnson, M.R.; Wilkin, D.J.; Vos, H.L.; Ortiz De Luna, R.I.; Dehejia, A.M.; Polymeropoulos, M.H.; Francomano, C.A. Characterization of the human extracellular matrix protein 1 gene on chromosome 1q21. Matrix Biol. 1997, 16, 289–292. [Google Scholar] [CrossRef] [Green Version]
- Smits, P.; Ni, J.; Feng, P.; Wauters, J.; Van Hul, W.; Boutaibi, M.E.; Dillon, P.J.; Merregaert, J. The human extracellular matrix gene 1 (ECM1): Genomic structure, cDNA cloning, expression pattern, and chromosomal localization. Genomics 1997, 45, 487–495. [Google Scholar] [CrossRef]
- Han, Z.; Ni, J.; Smits, P.; Underhill, C.B.; Xie, B.; Chen, Y.; Liu, N.; Tylzanowski, P.; Parmelee, D.; Feng, P.; et al. Extracellular matrix protein 1 (ECM1) has angiogenic properties and is expressed by breast tumor cells. FASEB J. 2001, 15, 988–994. [Google Scholar] [CrossRef]
- Deckers, M.M.; Smits, P.; Karperien, M.; Ni, J.; Tylzanowski, P.; Feng, P.; Parmelee, D.; Zhang, J.; Bouffard, E.; Gentz, R.; et al. Recombinant human extracellular matrix protein 1 inhibits alkaline phosphatase activity and mineralization of mouse embryonic metatarsals in vitro. Bone 2001, 28, 14–20. [Google Scholar] [CrossRef]
- Wang, L.; Yu, J.; Ni, J.; Xu, X.M.; Wang, J.; Ning, H.; Pei, X.F.; Chen, J.; Yang, S.; Underhill, C.B.; et al. Extracellular matrix protein 1 (ECM1) is over-expressed in malignant epithelial tumors. Cancer Lett. 2003, 200, 57–67. [Google Scholar] [CrossRef]
- Gómez-Contreras, P.; Ramiro-Díaz, J.M.; Sierra, A.; Stipp, C.; Domann, F.E.; Weigel, R.J.; Lal, G. Extracellular matrix 1 (ECM1) regulates the actin cytoskeletal architecture of aggressive breast cancer cells in part via S100A4 and Rho-family GTPases. Clin. Exp. Metastasis 2017, 34, 37–49. [Google Scholar] [CrossRef]
- Chen, H.; Jia, W.; Li, J. ECM1 promotes migration and invasion of hepatocellular carcinoma by inducing epithelial-mesenchymal transition. World J. Surg. Oncol. 2016, 14, 195. [Google Scholar] [CrossRef]
- Lee, K.M.; Nam, K.; Oh, S.; Lim, J.; Kim, Y.P.; Lee, J.W.; Yu, J.H.; Ahn, S.H.; Kim, S.B.; Noh, D.Y.; et al. Extracellular matrix protein 1 regulates cell proliferation and trastuzumab resistance through activation of epidermal growth factor signaling. Breast Cancer Res. 2014, 16, 479. [Google Scholar] [CrossRef]
- Lal, G.; Hashimi, S.; Smith, B.J.; Lynch, C.F.; Zhang, L.; Robinson, R.A.; Weigel, R.J. Extracellular matrix 1 (ECM1) expression is a novel prognostic marker for poor long-term survival in breast cancer: A Hospital-based Cohort Study in Iowa. Ann. Surg. Oncol. 2009, 16, 2280–2287. [Google Scholar] [CrossRef]
- Sun, H.F.; Li, X.M.; Meng, L.; Cui, C.M.; Gao, S.S.; Li, C.S.; Huang, C.G.; Wang, B.G. Asperolides A-C, tetranorlabdane diterpenoids from the marine alga-derived endophytic fungus Aspergillus wentii EN-48. J. Nat. Prod. 2012, 75, 148–152. [Google Scholar] [CrossRef]
- Lv, C.; Hong, Y.; Miao, L.; Li, C.; Xu, G.; Wei, S.; Wang, B.; Huang, C.; Jiao, B. Wentilactone A as a novel potential antitumor agent induces apoptosis and G2/M arrest of human lung carcinoma cells, and is mediated by HRas-GTP accumulation to excessively activate the Ras/Raf/ERK/p53-p21 pathway. Cell Death Dis. 2013, 4, e952. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Z.; Miao, L.; Lv, C.; Sun, H.; Wei, S.; Wang, B.; Huang, C.; Jiao, B. Wentilactone B induces G2/M phase arrest and apoptosis via the Ras/Raf/MAPK signaling pathway in human hepatoma SMMC-7721 cells. Cell Death Dis. 2013, 4, e657. [Google Scholar] [CrossRef]
- Norouzi, O.; Jafarian, S.; Safari, F.; Tavasoli, A.; Nejati, B. Promotion of hydrogen-rich gas and phenolic-rich bio-oil production from green macroalgae Cladophora glomerata via pyrolysis over its bio-char. Bioresour. Technol. 2016, 219, 643–651. [Google Scholar] [CrossRef]
- Liu, J.; Luthuli, S.; Wu, Q.; Wu, M.; Choi, J.I.; Tong, H. Pharmaceutical and Nutraceutical Potential Applications of Sargassum fulvellum. Biomed. Res. Int. 2020, 2020, 2417410. [Google Scholar] [CrossRef] [PubMed]
- Abdul, Q.A.; Choi, R.J.; Jung, H.A.; Choi, J.S. Health benefit of fucosterol from marine algae: A review. J. Sci. Food Agric. 2016, 96, 1856–1866. [Google Scholar] [CrossRef]
- Amador-Castro, F.; García-Cayuela, T.; Alper, H.S.; Rodriguez-Martinez, V.; Carrillo-Nieves, D. Valorization of pelagic sargassum biomass into sustainable applications: Current trends and challenges. J. Environ. Manag. 2021, 283, 112013. [Google Scholar] [CrossRef]
- Huang, C.Y.; Wu, S.J.; Yang, W.N.; Kuan, A.W.; Chen, C.Y. Antioxidant activities of crude extracts of fucoidan extracted from Sargassum glaucescens by a compressional-puffing-hydrothermal extraction process. Food Chem. 2016, 197 Pt B, 1121–1129. [Google Scholar] [CrossRef]
- Yin, H.; Wang, J.; Li, H.; Yu, Y.; Wang, X.; Lu, L.; Lv, C.; Chang, B.; Jin, W.; Guo, W.; et al. Extracellular matrix protein-1 secretory isoform promotes ovarian cancer through increasing alternative mRNA splicing and stemness. Nat. Commun. 2021, 12, 4230. [Google Scholar] [CrossRef]
- Li, S.; Yang, L.; Wang, J.; Liang, F.; Chang, B.; Gu, H.; Wang, H.; Yang, G.; Chen, Y. Analysis of the chemotherapeutic effects of a propadiene compound on malignant ovarian cancer cells. Oncotarget 2016, 7, 57145–57159. [Google Scholar] [CrossRef]
- Shao, Y.; Liu, X.; Meng, J.; Zhang, X.; Ma, Z.; Yang, G. MicroRNA-1251-5p Promotes Carcinogenesis and Autophagy via Targeting the Tumor Suppressor TBCC in Ovarian Cancer Cells. Mol. Ther. 2019, 27, 1653–1664. [Google Scholar] [CrossRef]
- Yang, G.; Rosen, D.G.; Liu, G.; Yang, F.; Guo, X.; Xiao, X.; Xue, F.; Mercado-Uribe, I.; Huang, J.; Lin, S.H.; et al. CXCR2 promotes ovarian cancer growth through dysregulated cell cycle, diminished apoptosis, and enhanced angiogenesis. Clin. Cancer Res. 2010, 16, 3875–3886. [Google Scholar] [CrossRef] [Green Version]
- Zhang, X.; Qi, Z.; Yin, H.; Yang, G. Interaction between p53 and Ras signaling controls cisplatin resistance via HDAC4- and HIF-1α-mediated regulation of apoptosis and autophagy. Theranostics 2019, 9, 1096–1114. [Google Scholar] [CrossRef]
- Martone, R.; Euskirchen, G.; Bertone, P.; Hartman, S.; Royce, T.E.; Luscombe, N.M.; Rinn, J.L.; Nelson, F.K.; Miller, P.; Gerstein, M.; et al. Distribution of NF-kappaB-binding sites across human chromosome 22. Proc. Natl. Acad. Sci. USA 2003, 100, 12247–12252. [Google Scholar] [CrossRef]
- Yang, G.; Xiao, X.; Rosen, D.G.; Cheng, X.; Wu, X.; Chang, B.; Liu, G.; Xue, F.; Mercado-Uribe, I.; Chiao, P.; et al. The biphasic role of NF-kappaB in progression and chemoresistance of ovarian cancer. Clin. Cancer Res. 2011, 17, 2181–2194. [Google Scholar] [CrossRef]
- Yang, G.; Chang, B.; Yang, F.; Guo, X.; Cai, K.Q.; Xiao, X.S.; Wang, H.; Sen, S.; Hung, M.C.; Mills, G.B.; et al. Aurora kinase A promotes ovarian tumorigenesis through dysregulation of the cell cycle and suppression of BRCA2. Clin. Cancer Res. 2010, 16, 3171–3181. [Google Scholar] [CrossRef]
- Xiong, G.P.; Zhang, J.X.; Gu, S.P.; Wu, Y.B.; Liu, J.F. Overexpression of ECM1 contributes to migration and invasion in cholangiocarcinoma cell. Neoplasma 2012, 59, 409–415. [Google Scholar] [CrossRef]
- Lu, P.; Weaver, V.M.; Werb, Z. The extracellular matrix: A dynamic niche in cancer progression. J. Cell Biol. 2012, 196, 395–406. [Google Scholar] [CrossRef]
- Reuter, J.A.; Ortiz-Urda, S.; Kretz, M.; Garcia, J.; Scholl, F.A.; Pasmooij, A.M.; Cassarino, D.; Chang, H.Y.; Khavari, P.A. Modeling inducible human tissue neoplasia identifies an extracellular matrix interaction network involved in cancer progression. Cancer Cell 2009, 15, 477–488. [Google Scholar] [CrossRef]
- Perkins, N.D. The diverse and complex roles of NF-κB subunits in cancer. Nat. Rev. Cancer 2012, 12, 121–132. [Google Scholar] [CrossRef]
- Tao, Y.; Liu, Z.; Hou, Y.; Wang, S.; Liu, S.; Jiang, Y.; Tan, D.; Ge, Q.; Li, C.; Hu, Y.; et al. Alternative NF-κB signaling promotes colorectal tumorigenesis through transcriptionally upregulating Bcl-3. Oncogene 2018, 37, 5887–5900. [Google Scholar] [CrossRef]
- Park, J.U.; Kang, B.Y.; Lee, H.J.; Kim, S.; Bae, D.; Park, J.H.; Kim, Y.R. Tetradecanol reduces EL-4 T cell growth by the down regulation of NF-κB mediated IL-2 secretion. Eur. J. Pharmacol. 2017, 799, 135–142. [Google Scholar] [CrossRef]
- Hoesel, B.; Schmid, J.A. The complexity of NF-κB signaling in inflammation and cancer. Mol. Cancer 2013, 12, 86. [Google Scholar] [CrossRef] [PubMed]
- Xin, L.; Yang, W.F.; Zhang, H.T.; Li, Y.F.; Liu, C. The mechanism study of lentiviral vector carrying methioninase enhances the sensitivity of drug-resistant gastric cancer cells to Cisplatin. Br. J. Cancer 2018, 118, 1189–1199. [Google Scholar] [CrossRef] [PubMed]
- Kim, H.J.; Hawke, N.; Baldwin, A.S. NF-kappaB and IKK as therapeutic targets in cancer. Cell Death Differ. 2006, 13, 738–747. [Google Scholar] [CrossRef] [PubMed]
- Orlowski, R.Z.; Baldwin, A.S., Jr. NF-kappaB as a therapeutic target in cancer. Trends Mol. Med. 2002, 8, 385–389. [Google Scholar] [CrossRef]
- Fang, S.; Yu, L.; Mei, H.; Yang, J.; Gao, T.; Cheng, A.; Guo, W.; Xia, K.; Liu, G. Cisplatin promotes mesenchymal-like characteristics in osteosarcoma through Snail. Oncol. Lett. 2016, 12, 5007–5014. [Google Scholar] [CrossRef]
- Pistollato, F.; Calderón Iglesias, R.; Ruiz, R.; Aparicio, S.; Crespo, J.; Dzul Lopez, L.; Giampieri, F.; Battino, M. The use of natural compounds for the targeting and chemoprevention of ovarian cancer. Cancer Lett. 2017, 411, 191–200. [Google Scholar] [CrossRef]
- Baghbani, F.; Moztarzadeh, F. Bypassing multidrug resistant ovarian cancer using ultrasound responsive doxorubicin/curcumin co-deliver alginate nanodroplets. Colloids Surf. B Biointerfaces 2017, 153, 132–140. [Google Scholar] [CrossRef]
- Zhang, J.; Liu, J.; Xu, X.; Li, L. Curcumin suppresses cisplatin resistance development partly via modulating extracellular vesicle-mediated transfer of MEG3 and miR-214 in ovarian cancer. Cancer Chemother. Pharmacol. 2017, 79, 479–487. [Google Scholar] [CrossRef]
- Haslehurst, A.M.; Koti, M.; Dharsee, M.; Nuin, P.; Evans, K.; Geraci, J.; Childs, T.; Chen, J.; Li, J.; Weberpals, J.; et al. EMT transcription factors snail and slug directly contribute to cisplatin resistance in ovarian cancer. BMC Cancer 2012, 12, 91. [Google Scholar] [CrossRef]
- Perkins, N.D. Post-translational modifications regulating the activity and function of the nuclear factor kappa B pathway. Oncogene 2006, 25, 6717–6730. [Google Scholar] [CrossRef] [Green Version]
- Lee, K.M.; Nam, K.; Oh, S.; Lim, J.; Lee, T.; Shin, I. ECM1 promotes the Warburg effect through EGF-mediated activation of PKM2. Cell. Signal. 2015, 27, 228–235. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Tang, H.; Cai, J.; Zhang, T.; Guo, J.; Feng, D.; Wang, Z. Ovarian cancer-associated fibroblasts contribute to epithelial ovarian carcinoma metastasis by promoting angiogenesis, lymphangiogenesis and tumor cell invasion. Cancer Lett. 2011, 303, 47–55. [Google Scholar] [CrossRef] [PubMed]
- Augsten, M. Cancer-associated fibroblasts as another polarized cell type of the tumor microenvironment. Front. Oncol. 2014, 4, 62. [Google Scholar] [CrossRef]
- Fang, H.; Declerck, Y.A. Targeting the tumor microenvironment: From understanding pathways to effective clinical trials. Cancer Res. 2013, 73, 4965–4977. [Google Scholar] [CrossRef] [PubMed]
- Deying, W.; Feng, G.; Shumei, L.; Hui, Z.; Ming, L.; Hongqing, W. CAF-derived HGF promotes cell proliferation and drug resistance by up-regulating the c-Met/PI3K/Akt and GRP78 signalling in ovarian cancer cells. Biosci. Rep. 2017, 37, BSR20160470. [Google Scholar] [CrossRef]
- Mathieu, E.; Meheus, L.; Raymackers, J.; Merregaert, J. Characterization of the osteogenic stromal cell line MN7: Identification of secreted MN7 proteins using two-dimensional polyacrylamide gel electrophoresis, western blotting, and microsequencing. J. Bone Miner. Res. 1994, 9, 903–913. [Google Scholar] [CrossRef]
- Wang, X.; Peng, Y.; Xie, M.; Gao, Z.; Yin, L.; Pu, Y.; Liu, R. Identification of extracellular matrix protein 1 as a potential plasma biomarker of ESCC by proteomic analysis using iTRAQ and 2D-LC-MS/MS. Proteom. Clin. Appl. 2017, 11, 201600163. [Google Scholar] [CrossRef]
- Santasusagna, S.; Moreno, I.; Navarro, A.; Castellano, J.J.; Martinez, F.; Hernández, R.; Muñoz, C.; Monzo, M. Proteomic Analysis of Liquid Biopsy from Tumor-Draining Vein Indicates that High Expression of Exosomal ECM1 Is Associated with Relapse in Stage I–III Colon Cancer. Transl. Oncol. 2018, 11, 715–721. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| N | Names | Sequences and Notes | Purposes |
|---|---|---|---|
| 1 | ECM1-P-FW | 5′-GTGGAGCTACAGAACACGAGGGTC-3′ | ECM1 promoter cloning |
| 2 | ECM1-P-RV | 5′-CACATCCAAACAGCTACAGCTTCCC-3′ | ECM1 promoter cloning |
| 3 | tttEP881T-FW | 5′-cctccctcacatTTTagaccctaacccagctgac-3′ | ECM1 promoter mutation |
| 4 | tttEP881T-RV | 5′-gtcagctgggttagggtctAAAatgtgagggagg-3′ | ECM1 promoter mutation |
| 5 | tttEP266C-FW | 5′-cacactggtagTTTagatcccttggataggtt-3′ | ECM1 promoter mutation |
| 6 | tttEP266C-RV | 5′-aacctatccaagggatctAAActaccagtgtg-3′ | ECM1 promoter mutation |
| 7 | EP881C-FW | 5′-cctccctcacatgggagacccCaacccagctgac-3′ | ECM1 promoter mutation |
| 8 | EP881C-RV | 5′-gtcagctgggttGgggtctcccatgtgagggagg-3′ | ECM1 promoter mutation |
| Population | Chrom.SampleCnt. | Source | C | T |
|---|---|---|---|---|
| EAS * | 1008 | AF | 1 | 0 |
| EUR | 1006 | AF | 0.9811 | 0.0189 |
| AFR | 1322 | AF | 0.9985 | 0.0015 |
| AMR | 694 | AF | 0.9914 | 0.0086 |
| SAS | 978 | AF | 1 | 0 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lv, C.; Ren, C.; Yu, Y.; Yin, H.; Huang, C.; Yang, G.; Hong, Y. Wentilactone A Reverses the NF-κB/ECM1 Signaling-Induced Cisplatin Resistance through Inhibition of IKK/IκB in Ovarian Cancer Cells. Nutrients 2022, 14, 3790. https://doi.org/10.3390/nu14183790
Lv C, Ren C, Yu Y, Yin H, Huang C, Yang G, Hong Y. Wentilactone A Reverses the NF-κB/ECM1 Signaling-Induced Cisplatin Resistance through Inhibition of IKK/IκB in Ovarian Cancer Cells. Nutrients. 2022; 14(18):3790. https://doi.org/10.3390/nu14183790
Chicago/Turabian StyleLv, Cuiting, Chunxia Ren, Yinjue Yu, Huijing Yin, Caiguo Huang, Gong Yang, and Yang Hong. 2022. "Wentilactone A Reverses the NF-κB/ECM1 Signaling-Induced Cisplatin Resistance through Inhibition of IKK/IκB in Ovarian Cancer Cells" Nutrients 14, no. 18: 3790. https://doi.org/10.3390/nu14183790
APA StyleLv, C., Ren, C., Yu, Y., Yin, H., Huang, C., Yang, G., & Hong, Y. (2022). Wentilactone A Reverses the NF-κB/ECM1 Signaling-Induced Cisplatin Resistance through Inhibition of IKK/IκB in Ovarian Cancer Cells. Nutrients, 14(18), 3790. https://doi.org/10.3390/nu14183790