Hibiscus sabdariffa Extract Protects HaCaT Cells against Phenanthrene-Induced Toxicity through the Regulation of Constitutive Androstane Receptor/Pregnane X Receptor Pathway

 ,
,  and
and 
Abstract
:
1. Introduction
2. Materials and Methods
2.1. Chemicals
2.2. Plant Collection and Extraction
2.3. Cell Viability Assay
2.4. Measurement of Reactive Oxygen Species (ROS) Level and Mitochondrial Membrane Potential (ΔΨm)
2.5. Real-Time PCR Analysis
2.6. Western Blot Analysis
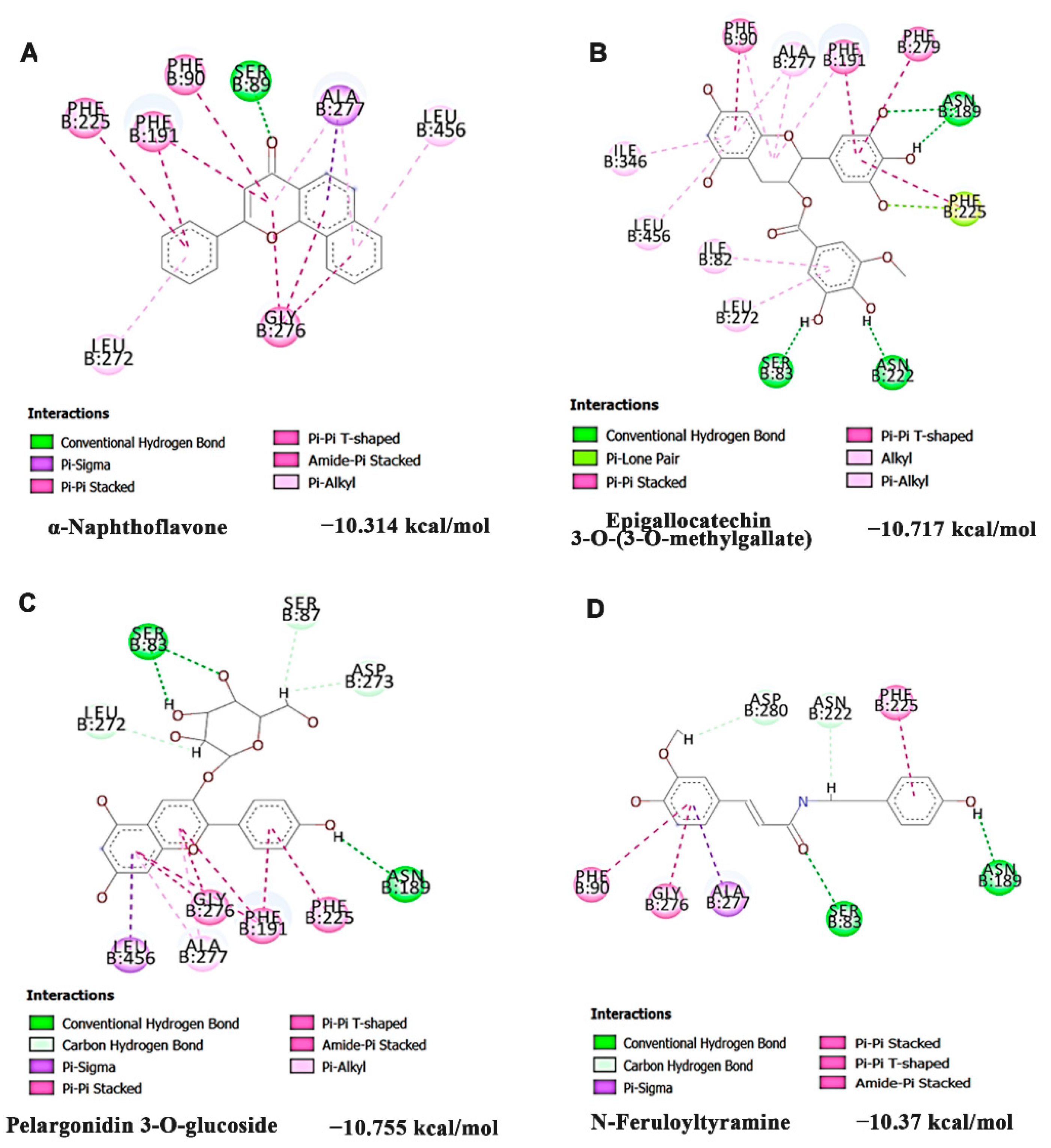
2.7. Molecular Docking Analysis
2.8. Statistical Analysis
3. Results
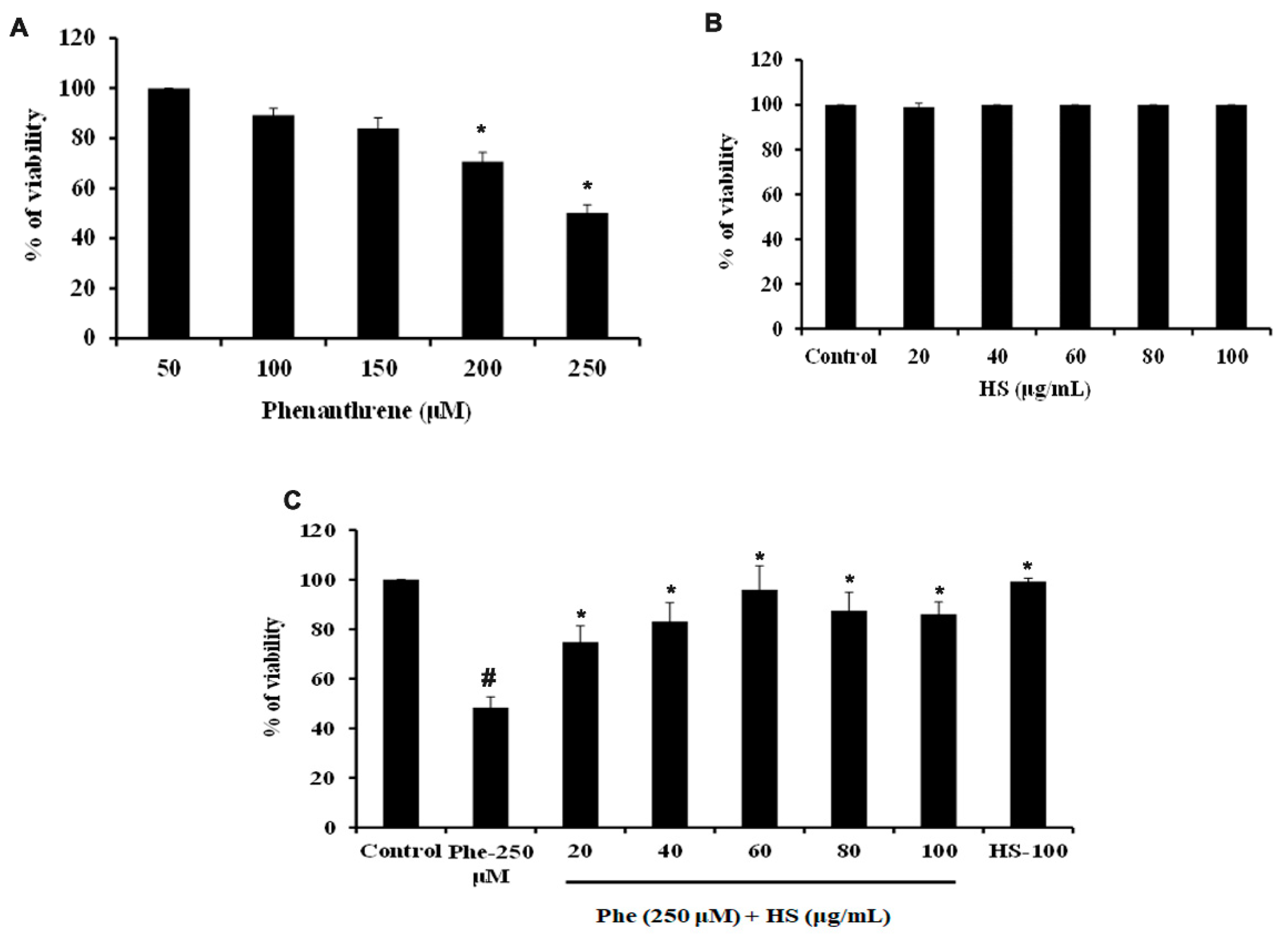
3.1. HS Extract Pre-Treatment Protects HaCaT Cells from Phe Induced Cell Death
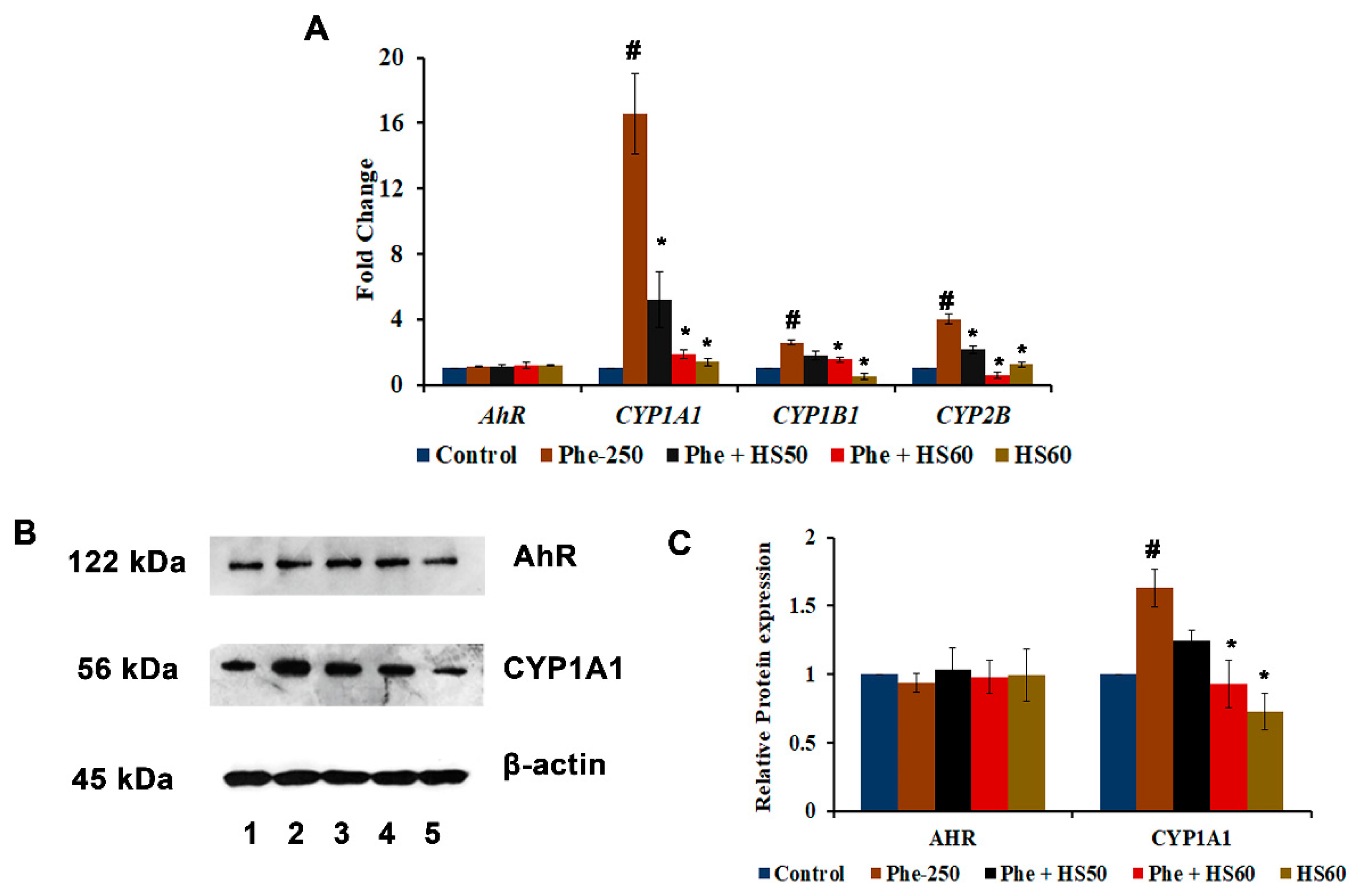
3.2. Phe Induced Toxicity through the AhR-Independent Activation of CYP1A1
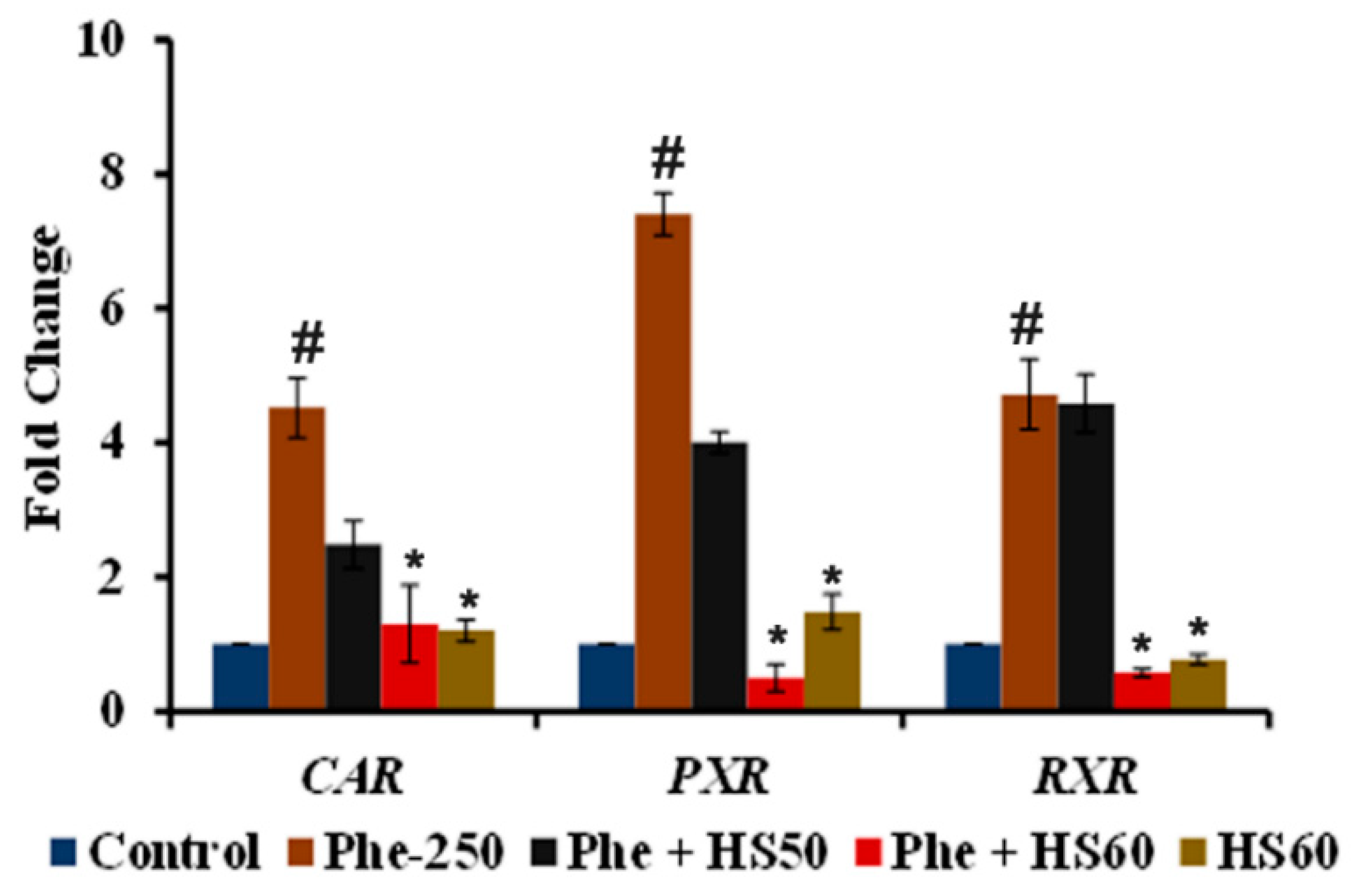
3.3. Phe Induced CYP1A1 Activity Was Mediated through the Activation of CAR/PXR/RXR Nuclear Receptors
3.4. HS Extract Restored the Expression of CAR/PXR Target Genes Encoding Phase-I and II Drug Metabolizing Enzymes to Protect against Phe Toxicity
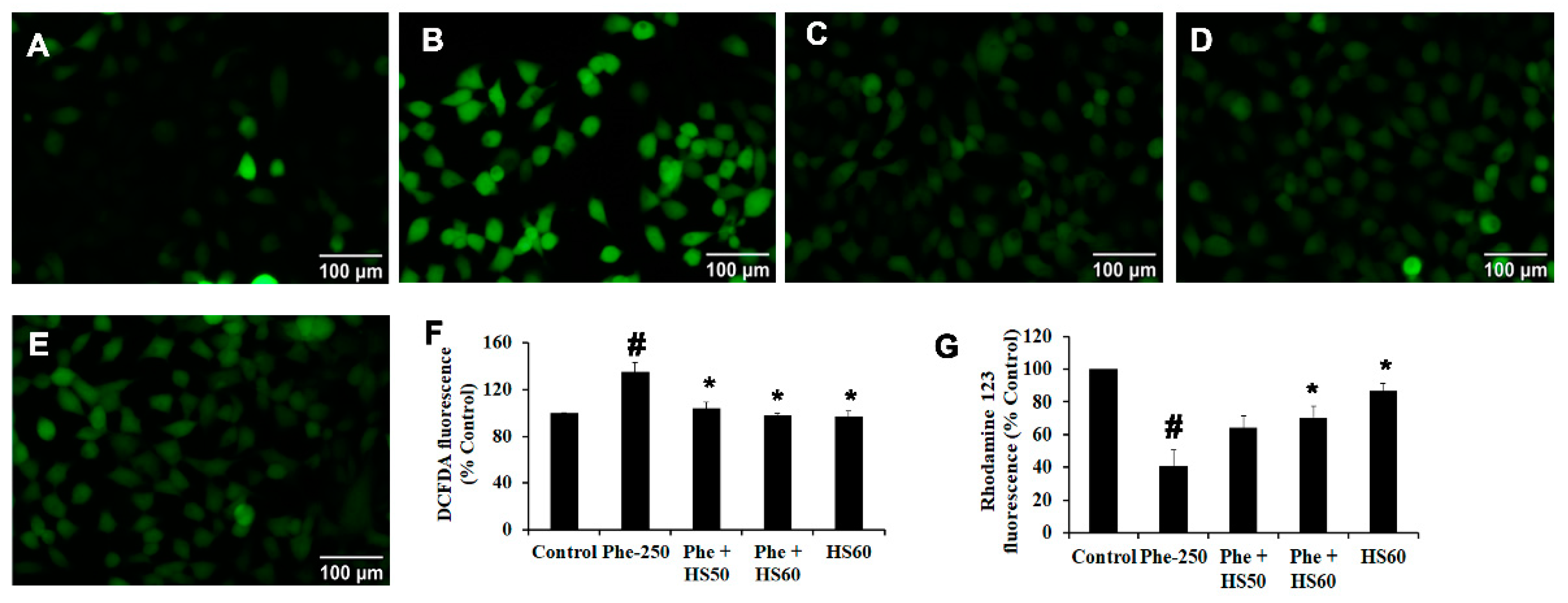
3.5. HS Extract Inhibited CYP1A1-Induced ROS Generation upon Phe Toxicity and Restored Mitochondrial Membrane Potential (ΔΨm) in HaCaT Cells
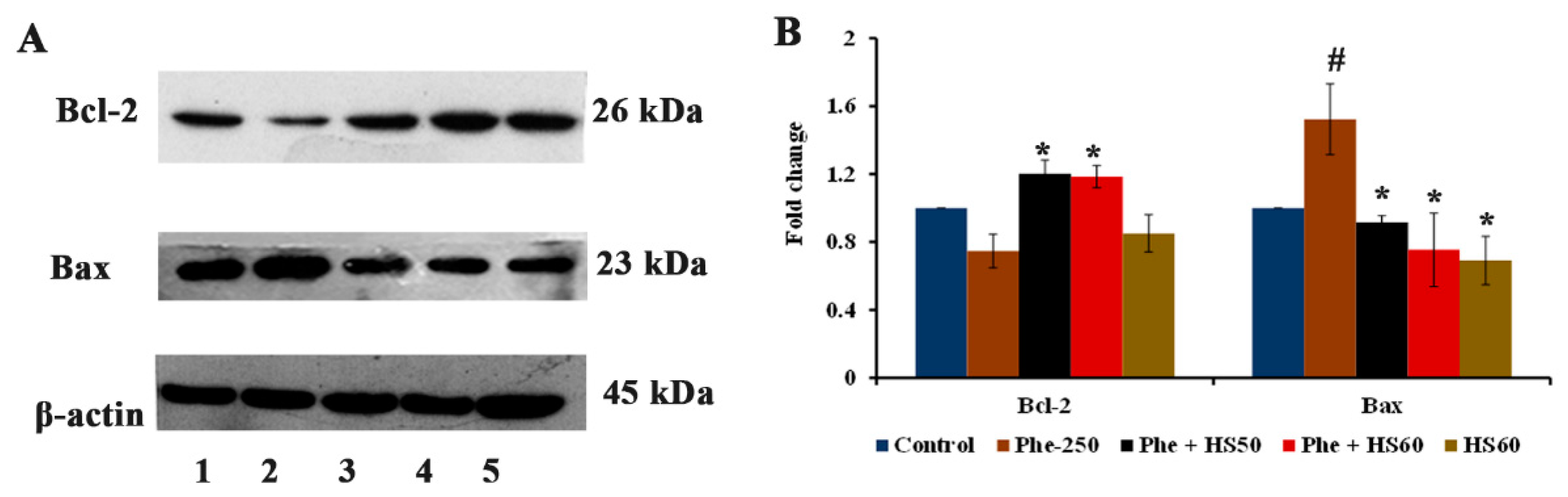
3.6. HS Extract Protected HaCat Cells from Phe-Induced Apoptosis
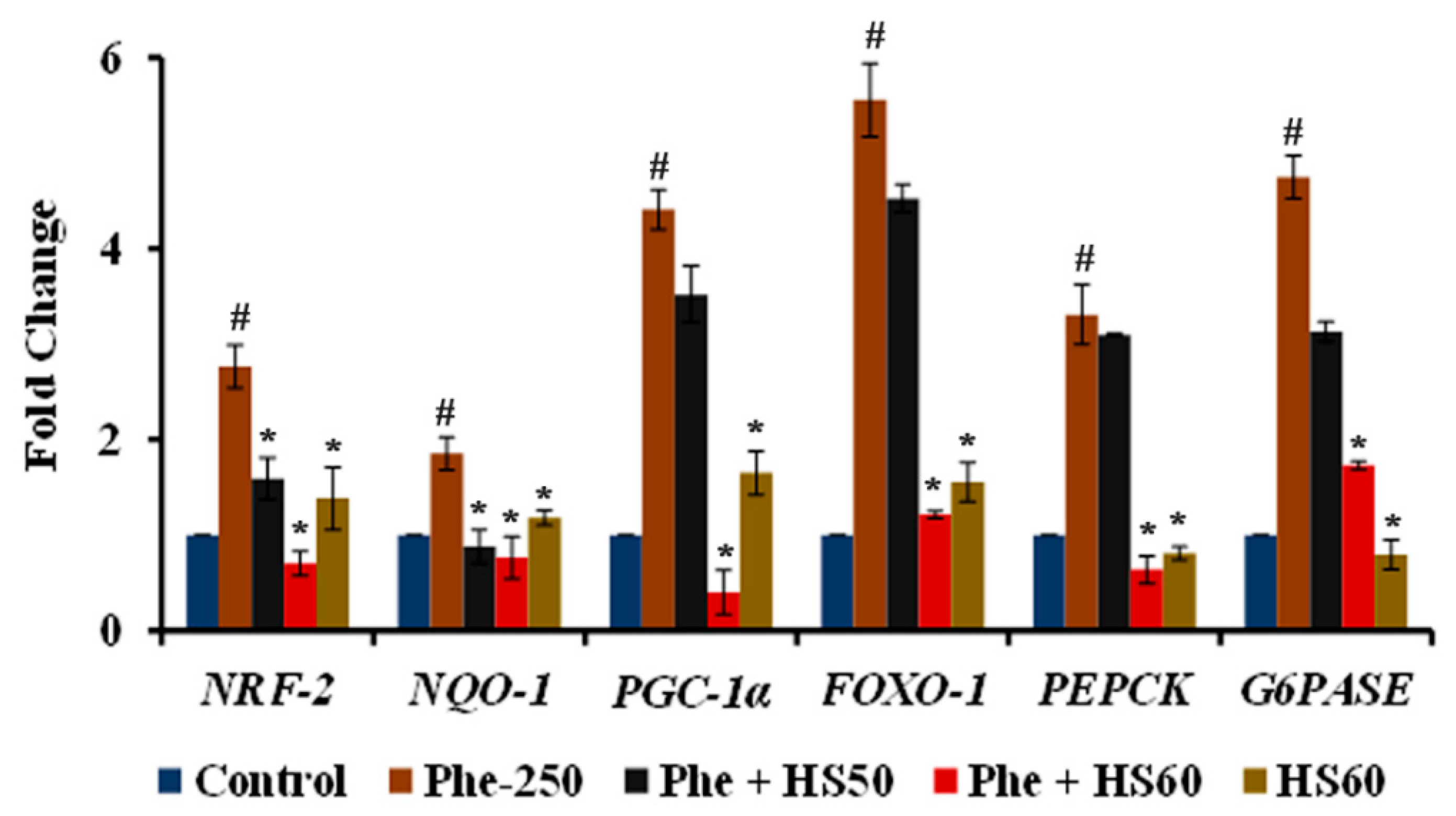
3.7. HS Extract Regulated the Expression of Stress Response and Gluconeogenesis Related Genes upon Phe Toxicity
4. Discussion
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- WHO. Ambient Air Pollution: A Global Assessment of Exposure and Burden of Disease; World Health Organization: Geneva, Switzerland, 2016. [Google Scholar]
- Xing, Y.F.; Xu, Y.H.; Shi, M.H.; Lian, Y.X. The impact of PM2.5 on the human respiratory system. J. Thorac. Dis. 2016, 8, E69–E74. [Google Scholar] [CrossRef] [PubMed]
- Javed, W.; Iakovides, M.; Stephanou, E.G.; Wolfson, J.M.; Koutrakis, P.; Guo, B. Concentrations of aliphatic and polycyclic aromatic hydrocarbons in ambient PM2.5 and PM10 particulates in Doha, Qatar. J. Air Waste Manag. Assoc. 2019, 69, 162–177. [Google Scholar] [CrossRef] [PubMed]
- Patel, A.B.; Shaikh, S.; Jain, K.R.; Desai, C.; Madamwar, D. Polycyclic Aromatic Hydrocarbons: Sources, Toxicity, and Remediation Approaches. Front. Microbiol. 2020, 11, 562813. [Google Scholar] [CrossRef] [PubMed]
- Goedtke, L.; Sprenger, H.; Hofmann, U.; Schmidt, F.F.; Hammer, H.S.; Zanger, U.M.; Poetz, O.; Seidel, A.; Braeuning, A.; Hessel-Pras, S. Polycyclic Aromatic Hydrocarbons Activate the Aryl Hydrocarbon Receptor and the Constitutive Androstane Receptor to Regulate Xenobiotic Metabolism in Human Liver Cells. Int. J. Mol. Sci. 2020, 22, 372. [Google Scholar] [CrossRef]
- Baird, W.M.; Hooven, L.A.; Mahadevan, B. Carcinogenic polycyclic aromatic hydrocarbon-DNA adducts and mechanism of action. Environ. Mol. Mutagen. 2005, 45, 106–114. [Google Scholar] [CrossRef]
- Zhang, Y.; Dong, S.; Wang, H.; Tao, S.; Kiyama, R. Biological impact of environmental polycyclic aromatic hydrocarbons (ePAHs) as endocrine disruptors. Environ. Pollut. 2016, 213, 809–824. [Google Scholar] [CrossRef]
- Luckert, C.; Ehlers, A.; Buhrke, T.; Seidel, A.; Lampen, A.; Hessel, S. Polycyclic aromatic hydrocarbons stimulate human CYP3A4 promoter activity via PXR. Toxicol. Lett. 2013, 222, 180–188. [Google Scholar] [CrossRef]
- Šimečková, P.; Pěnčíková, K.; Kováč, O.; Slavík, J.; Pařenicová, M.; Vondráček, J.; Machala, M. In vitro profiling of toxic effects of environmental polycyclic aromatic hydrocarbons on nuclear receptor signaling, disruption of endogenous metabolism and induction of cellular stress. Sci. Total Environ. 2022, 815, 151967. [Google Scholar] [CrossRef]
- Rusni, S.; Sassa, M.; Takagi, T.; Kinoshita, M.; Takehana, Y.; Inoue, K. Establishment of cytochrome P450 1a gene-knockout Javanese medaka, Oryzias javanicus, which distinguishes toxicity modes of the polycyclic aromatic hydrocarbons, pyrene and phenanthrene. Mar. Pollut. Bull. 2022, 178, 113578. [Google Scholar] [CrossRef]
- Hussar, E.; Richards, S.; Lin, Z.Q.; Dixon, R.P.; Johnson, K.A. Human Health Risk Assessment of 16 Priority Polycyclic Aromatic Hydrocarbons in Soils of Chattanooga, Tennessee, USA. Water Air Soil Pollut. 2012, 223, 5535–5548. [Google Scholar] [CrossRef]
- Samanta, S.K.; Chakraborti, A.K.; Jain, R.K. Degradation of phenanthrene by different bacteria: Evidence for novel transformation sequences involving the formation of 1-naphthol. Appl. Microbiol. Biotechnol. 1999, 53, 98–107. [Google Scholar] [CrossRef] [PubMed]
- Wu, S.; Wu, E.; Qiu, L.; Zhong, W.; Chen, J. Effects of phenanthrene on the mortality, growth, and anti-oxidant system of earthworms (Eisenia fetida) under laboratory conditions. Chemosphere 2011, 83, 429–434. [Google Scholar] [CrossRef] [PubMed]
- Guo, Y.; Senthilkumar, K.; Alomirah, H.; Moon, H.B.; Minh, T.B.; Mohd, M.A.; Nakata, H.; Kannan, K. Concentrations and profiles of urinary polycyclic aromatic hydrocarbon metabolites (OH-PAHs) in several Asian countries. Environ. Sci. Technol. 2013, 47, 2932–2938. [Google Scholar] [CrossRef]
- Polachova, A.; Gramblicka, T.; Parizek, O.; Sram, R.J.; Stupak, M.; Hajslova, J.; Pulkrabova, J. Estimation of human exposure to polycyclic aromatic hydrocarbons (PAHs) based on the dietary and outdoor atmospheric monitoring in the Czech Republic. Environ. Res. 2020, 182, 108977. [Google Scholar] [CrossRef] [PubMed]
- Yu, Z.; Lin, Q.; Gu, Y.; Du, F.; Wang, X.; Shi, F.; Ke, C.; Xiang, M.; Yu, Y. Bioaccumulation of polycyclic aromatic hydrocarbons (PAHs) in wild marine fish from the coastal waters of the northern South China Sea: Risk assessment for human health. Ecotoxicol. Environ. Saf. 2019, 180, 742–748. [Google Scholar] [CrossRef]
- Acharya, N.; Gautam, B.; Subbiah, S.; Rogge, M.M.; Anderson, T.A.; Gao, W. Polycyclic aromatic hydrocarbons in breast milk of obese vs normal women: Infant exposure and risk assessment. Sci. Total Environ. 2019, 668, 658–667. [Google Scholar] [CrossRef]
- Bohrer, D.; Barichello, M.M.; Viana, C.; Nascimento, P.C.; Carvalho, L.M. Newborn Exposure to Polycyclic Aromatic Hydrocarbons through Parenteral Nutrition. J. Pediatr. Gastroenterol. Nutr. 2018, 67, 671–676. [Google Scholar] [CrossRef]
- He, F.; Wan, J.; Chu, S.; Li, X.; Zong, W.; Liu, R. Toxic mechanism on phenanthrene-triggered cell apoptosis, genotoxicity, immunotoxicity and activity changes of immunity protein in Eisenia fetida: Combined analysis at cellular and molecular levels. Sci. Total Environ. 2022, 819, 153167. [Google Scholar] [CrossRef]
- Nam, T.H.; Jeon, H.J.; Mo, H.H.; Cho, K.; Ok, Y.S.; Lee, S.E. Determination of biomarkers for polycyclic aromatic hydrocarbons (PAHs) toxicity to earthworm (Eisenia fetida). Environ. Geochem. Health 2015, 37, 943–951. [Google Scholar] [CrossRef]
- Huang, L.; Wang, C.; Zhang, Y.; Wu, M.; Zuo, Z. Phenanthrene causes ocular developmental toxicity in zebrafish embryos and the possible mechanisms involved. J. Hazard. Mater. 2013, 261, 172–180. [Google Scholar] [CrossRef]
- Huang, L.; Xi, Z.; Wang, C.; Zhang, Y.; Yang, Z.; Zhang, S.; Chen, Y.; Zuo, Z. Phenanthrene exposure induces cardiac hypertrophy via reducing miR-133a expression by DNA methylation. Sci. Rep. 2016, 6, 20105. [Google Scholar] [CrossRef] [PubMed]
- Huang, J.; Fang, L.; Zhang, S.; Zhang, Y.; Ou, K.; Wang, C. Long-term exposure to environmental levels of phenanthrene disrupts spermatogenesis in male mice. Environ. Pollut. 2021, 285, 117488. [Google Scholar] [CrossRef] [PubMed]
- Sun, L.; Zuo, Z.; Luo, H.; Chen, M.; Zhong, Y.; Chen, Y.; Wang, C. Chronic exposure to phenanthrene influences the spermatogenesis of male Sebastiscus marmoratus: U-shaped effects and the reason for them. Environ. Sci. Technol. 2011, 45, 10212–10218. [Google Scholar] [CrossRef] [PubMed]
- Yin, Y.; Jia, H.; Sun, Y.; Yu, H.; Wang, X.; Wu, J.; Xue, Y. Bioaccumulation and ROS generation in liver of Carassius auratus, exposed to phenanthrene. Comp. Biochem. Physiol. C Toxicol. Pharmacol. 2007, 145, 288–293. [Google Scholar] [CrossRef]
- Sarker, S.; Vashistha, D.; Saha Sarker, M.; Sarkar, A. DNA damage in marine rock oyster (Saccostrea Cucullata) exposed to environmentally available PAHs and heavy metals along the Arabian Sea coast. Ecotoxicol. Environ. Saf. 2018, 151, 132–143. [Google Scholar] [CrossRef]
- Puri, P.; Nandar, S.K.; Kathuria, S.; Ramesh, V. Effects of air pollution on the skin: A review. Indian J. Dermatol. Venereol. Leprol. 2017, 83, 415. [Google Scholar] [CrossRef]
- Eshak, G.A.; Malak, H.M.; Ahmed, M.I. Polycyclic aromatic hydrocarbons: Role of apoptosis in dermatotoxic and carcinogenic effect in asphalt road paving workers. J. Clin. Toxicol. 2012, 2, 137. [Google Scholar] [CrossRef]
- Luo, K.; Zeng, D.; Kang, Y.; Lin, X.; Sun, N.; Li, C.; Zhu, M.; Chen, Z.; Man, Y.B.; Li, H. Dermal bioaccessibility and absorption of polycyclic aromatic hydrocarbons (PAHs) in indoor dust and its implication in risk assessment. Environ. Pollut. 2020, 264, 114829. [Google Scholar] [CrossRef]
- Hecht, S.S.; Carmella, S.G.; Murphy, S.E. Effects of watercress consumption on urinary metabolites of nicotine in smokers. Cancer Epidemiol. Biomark. Prev. 1999, 8, 907–913. [Google Scholar]
- Bailey, D.G.; Dresser, G.K.; Kreeft, J.H.; Munoz, C.; Freeman, D.J.; Bend, J.R. Grapefruit-felodipine interaction: Effect of unprocessed fruit and probable active ingredients. Clin. Pharmacol. Ther. 2000, 68, 468–477. [Google Scholar] [CrossRef]
- Walters, D.G.; Young, P.J.; Agus, C.; Knize, M.G.; Boobis, A.R.; Gooderham, N.J.; Lake, B.G. Cruciferous vegetable consumption alters the metabolism of the dietary carcinogen 2-amino-1-methyl-6-phenylimidazo [4,5-b]pyridine (PhIP) in humans. Carcinogenesis 2004, 25, 1659–1669. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rocejanasaroj, A.; Tencomnao, T.; Sangkitikomol, W. Thunbergia laurifolia extract minimizes the adverse effects of toxicants by regulating P-glycoprotein activity, CYP450, and lipid metabolism gene expression in HepG2 cells. Genet. Mol. Res. 2014, 13, 205–219. [Google Scholar] [CrossRef] [PubMed]
- Lee, S.E.; Park, S.H.; Yoo, J.A.; Kwon, K.; Kim, J.W.; Oh, S.W.; Park, S.J.; Kim, J.; Yu, E.; Han, B.S.; et al. Antagonizing Effects of Clematis apiifolia DC. Extract against Benzo[a]pyrene-Induced Damage to Human Keratinocytes. Oxid. Med. Cell Longev. 2019, 2019, 2386163. [Google Scholar] [CrossRef] [PubMed]
- Kallimanis, P.; Chinou, I.; Panagiotopoulou, A.; Soshilov, A.A.; He, G.; Denison, M.S.; Magiatis, P. Rosmarinus officinalis L. Leaf Extracts and Their Metabolites Inhibit the Aryl Hydrocarbon Receptor (AhR) Activation In Vitro and in Human Keratinocytes: Potential Impact on Inflammatory Skin Diseases and Skin Cancer. Molecules 2022, 27, 2499. [Google Scholar] [CrossRef]
- Pattarachotanant, N.; Prasansuklab, A.; Tencomnao, T. Momordica charantia L. Extract Protects Hippocampal Neuronal Cells against PAHs-Induced Neurotoxicity: Possible Active Constituents Include Stigmasterol and Vitamin E. Nutrients 2021, 13, 2368. [Google Scholar] [CrossRef]
- Hoang, H.T.; Moon, J.Y.; Lee, Y.C. Natural Antioxidants from Plant Extracts in Skincare Cosmetics: Recent Applications, Challenges and Perspectives. Cosmetics 2021, 8, 106. [Google Scholar] [CrossRef]
- Nobile, V.; Schiano, I.; Peral, A.; Giardina, S.; Spartà, E.; Caturla, N. Antioxidant and reduced skin-ageing effects of a polyphenol-enriched dietary supplement in response to air pollution: A randomized, double-blind, placebo-controlled study. Food Nutr. Res. 2021, 65, 5619. [Google Scholar] [CrossRef]
- Khalil, R.; Yahya, G.; Abdo, W.S.; El-Tanbouly, G.S.; Johar, D.; Abdel-Halim, M.S.; Eissa, H.; Magheru, C.; Saber, S.; Cavalu, S. Emerging Approach for the Application of Hibiscus sabdariffa Extract Ointment in the Superficial Burn Care. Sci. Pharm. 2022, 90, 41. [Google Scholar] [CrossRef]
- Song, K.; An, S.M.; Kim, M.; Koh, J.S.; Boo, Y.C. Comparison of the antimelanogenic effects of p-coumaric acid and its methyl ester and their skin permeabilities. J. Dermatol. Sci. 2011, 63, 17–22. [Google Scholar] [CrossRef]
- Park, H.J.; Cho, J.H.; Hong, S.H.; Kim, D.H.; Jung, H.Y.; Kang, I.K.; Cho, Y.J. Whitening and anti-wrinkle activities of ferulic acid isolated from Tetragonia tetragonioides in B16F10 melanoma and CCD-986sk fibroblast cells. J. Nat. Med. 2018, 72, 127–135. [Google Scholar] [CrossRef]
- Rho, H.S.; Ghimeray, A.K.; Yoo, D.S.; Ahn, S.M.; Kwon, S.S.; Lee, K.H.; Cho, D.H.; Cho, J.Y. Kaempferol and kaempferol rhamnosides with depigmenting and anti-inflammatory properties. Molecules 2011, 16, 3338–3344. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Koch, K.; Weldle, N.; Baier, S.; Büchter, C.; Wätjen, W. Hibiscus sabdariffa L. extract prolongs lifespan and protects against amyloid-β toxicity in Caenorhabditis elegans: Involvement of the FoxO and Nrf2 orthologues DAF-16 and SKN-1. Eur. J. Nutr. 2020, 59, 137–150. [Google Scholar] [CrossRef] [PubMed]
- Malar, D.S.; Prasanth, M.I.; Brimson, J.M.; Verma, K.; Prasansuklab, A.; Tencomnao, T. Hibiscus sabdariffa Extract Protects HT-22 Cells from Glutamate-induced Neurodegeneration by Upregulating Glutamate Transporters and Exerts Lifespan Extension in C. Elegans via DAF-16 Mediated Pathway. Nutr. Healthy Aging 2021, 6, 229–247. [Google Scholar] [CrossRef]
- Malar, D.S.; Prasanth, M.I.; Brimson, J.M.; Verma, K.; Prasansuklab, A.; Tencomnao, T. Neuroprotective effect of Hibiscus sabdariffa extract against high glucose-induced toxicity in Neuro-2a cells and transgenic Caenorhabditis elegans strain of Alzheimer’s disease. Am. J. Chin. Med. 2022; submitted. [Google Scholar]
- Guex, N.; Peitsch, M.C. Swiss-PdbViewer: A fast and easy-to-use PDB viewer for Macintosh and PC. Protein Data Bank Q. Newsl. 1996, 77, 7. [Google Scholar]
- Kim, S.; Thiessen, P.A.; Bolton, E.E.; Chen, J.; Fu, G.; Gindulyte, A.; Han, L.; He, J.; He, S.; Shoemaker, B.A.; et al. PubChem Substance and Compound databases. Nucleic Acids Res. 2016, 44, D1202–D1213. [Google Scholar] [CrossRef]
- Verma, K.; Lahariya, A.K.; Dubey, S.; Verma, A.K.; Das, A.; Schneider, K.A.; Bharti, P.K. An integrated virtual screening and drug repurposing strategy for the discovery of new antimalarial drugs against Plasmodium falciparum phosphatidylinositol 3-kinase. J. Cell Biochem. 2021, 122, 1326–1336. [Google Scholar] [CrossRef]
- Alsulaimany, F.A.; Almukadi, H.; Zabermawi, N.M.O.; Aljuhani, T.A.; Rashidie, O.M.; Albaqami, W.F.; Alghamdi, A.A.; Ahmad, A.; Shaik, N.A.; Banaganapalli, B. Identification of novel mycobacterium tuberculosis leucyl-tRNA synthetase inhibitor using a knowledge-based computational screening approach. J. King Saud Univ. Sci. 2022, 34, 102032. [Google Scholar] [CrossRef]
- Jiang, G.; Kang, H.; Yu, Y. Cross-platform metabolomics investigating the intracellular metabolic alterations of HaCaT cells exposed to phenanthrene. J. Chromatogr. B Analyt. Technol. Biomed. Life Sci. 2017, 1060, 15–21. [Google Scholar] [CrossRef]
- Brette, F.; Shiels, H.; Galli, G.; Cros, C.; Incardona, J.P.; Scholz, N.L.; Block, B.A. A Novel Cardiotoxic Mechanism for a Pervasive Global Pollutant. Sci. Rep. 2017, 7, 41476. [Google Scholar] [CrossRef]
- Sun, Y.; Miller, C.A.; Wiese, T.E.; Blake, D.A. Methylated phenanthrenes are more potent than phenanthrene in a bioassay of human aryl hydrocarbon receptor (AhR) signaling. Environ. Toxicol. Chem. 2014, 33, 2363–2367. [Google Scholar] [CrossRef] [PubMed]
- Ito, S.; Chen, C.; Satoh, J.; Yim, S.; Gonzalez, F.J. Dietary phytochemicals regulate whole-body CYP1A1 expression through an arylhydrocarbon receptor nuclear translocator-dependent system in gut. J. Clin. Investig. 2007, 117, 1940–1950. [Google Scholar] [CrossRef] [PubMed]
- Taura, K.; Naito, E.; Ishii, Y.; Mori, M.A.; Oguri, K.; Yamada, H. Cytochrome P450 1A1 (CYP1A1) inhibitor alpha-naphthoflavone interferes with UDP-glucuronosyltransferase (UGT) activity in intact but not in permeabilized hepatic microsomes from 3-methylcholanthrene-treated rats: Possible involvement of UGT-P450 interactions. Biol. Pharm. Bull. 2004, 27, 56–60. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yan, J.; Xie, W. A brief history of the discovery of PXR and CAR as xenobiotic receptors. Acta Pharm. Sin. B 2016, 6, 450–452. [Google Scholar] [CrossRef]
- Yoshinari, K.; Yoda, N.; Toriyabe, T.; Yamazoe, Y. Constitutive androstane receptor transcriptionally activates human CYP1A1 and CYP1A2 genes through a common regulatory element in the 5′-flanking region. Biochem. Pharmacol. 2010, 79, 261–269. [Google Scholar] [CrossRef]
- Yang, H.; Shi, Z.; Wang, X.X.; Cheng, R.; Lu, M.; Zhu, J.; Deng, W.; Zeng, Y.; Zhao, L.Y.; Zhang, S.Y. Phenanthrene, but not its isomer anthracene, effectively activates both human and mouse nuclear receptor constitutive androstane receptor (CAR) and induces hepatotoxicity in mice. Toxicol. Appl. Pharmacol. 2019, 378, 114618. [Google Scholar] [CrossRef]
- Lin, W.; Bwayi, M.; Wu, J.; Li, Y.; Chai, S.C.; Huber, A.D.; Chen, T. CITCO Directly Binds to and Activates Human Pregnane X Receptor. Mol. Pharmacol. 2020, 97, 180–190. [Google Scholar] [CrossRef]
- Maglich, J.M.; Parks, D.J.; Moore, L.B.; Collins, J.L.; Goodwin, B.; Billin, A.N.; Stoltz, C.A.; Kliewer, S.A.; Lambert, M.H.; Willson, T.M.; et al. Identification of a novel human constitutive androstane receptor (CAR) agonist and its use in the identification of CAR target genes. J. Biol. Chem. 2003, 278, 17277–17283. [Google Scholar] [CrossRef]
- Cherian, M.T.; Lin, W.; Wu, J.; Chen, T. CINPA1 is an inhibitor of constitutive androstane receptor that does not activate pregnane X receptor. Mol. Pharmacol. 2015, 87, 878–889. [Google Scholar] [CrossRef]
- Lim, Y.P.; Kuo, S.C.; Lai, M.L.; Huang, J.D. Inhibition of CYP3A4 expression by ketoconazole is mediated by the disruption of pregnane X receptor, steroid receptor coactivator-1, and hepatocyte nuclear factor 4α interaction. Pharm. Genom. 2009, 19, 11–24. [Google Scholar] [CrossRef]
- Sevrioukova, I.F.; Poulos, T.L. Structure and mechanism of the complex between cytochrome P4503A4 and ritonavir. Proc. Natl. Acad. Sci. USA 2010, 107, 18422–18427. [Google Scholar] [CrossRef] [PubMed]
- Satoh, T.; Fujisawa, H.; Nakamura, A.; Takahashi, N.; Watanabe, K. Inhibitory Effects of Eight Green Tea Catechins on Cytochrome P450 1A2, 2C9, 2D6, and 3A4 Activities. J. Pharm. Pharm. Sci. 2016, 19, 188–197. [Google Scholar] [CrossRef] [PubMed]
- Srovnalova, A.; Svecarova, M.; Zapletalova, M.K.; Anzenbacher, P.; Bachleda, P.; Anzenbacherova, E.; Dvorak, Z. Effects of anthocyanidins and anthocyanins on the expression and catalytic activities of CYP2A6, CYP2B6, CYP2C9, and CYP3A4 in primary human hepatocytes and human liver microsomes. J. Agric. Food Chem. 2014, 62, 789–797. [Google Scholar] [CrossRef]
- Sridar, C.; Kent, U.M.; Noon, K.; McCall, A.; Alworth, B.; Foroozesh, M.; Hollenberg, P.F. Differential inhibition of cytochromes P450 3A4 and 3A5 by the newly synthesized coumarin derivatives 7-coumarin propargyl ether and 7-(4-trifluoromethyl)coumarin propargyl ether. Drug Metab. Dispos. 2008, 36, 2234–2243. [Google Scholar] [CrossRef] [PubMed]
- Seidel, A.; Spickenheuer, A.; Straif, K.; Rihs, H.P.; Marczynski, B.; Scherenberg, M.; Dettbarn, G.; Angerer, J.; Wilhelm, M.; Brüning, T.; et al. New biomarkers of occupational exposure to polycyclic aromatic hydrocarbons. J. Toxicol. Environ. Health A 2008, 71, 734–745. [Google Scholar] [CrossRef]
- Schrlau, J.E.; Kramer, A.L.; Chlebowski, A.; Truong, L.; Tanguay, R.L.; Simonich, S.L.M.; Semprini, L. Formation of Developmentally Toxic Phenanthrene Metabolite Mixtures by Mycobacterium sp. ELW1. Environ. Sci. Technol. 2017, 51, 8569–8578. [Google Scholar] [CrossRef]
- Dasari, S.; Ganjayi, M.S.; Yellanurkonda, P.; Basha, S.; Meriga, B. Role of glutathione S-transferases in detoxification of a polycyclic aromatic hydrocarbon, methylcholanthrene. Chem. Biol. Interact. 2018, 294, 81–90. [Google Scholar] [CrossRef]
- Wang., J.; Zhong, Y.; Carmella, S.G.; Hochalter, J.B.; Rauch, D.; Oliver, A.; Jensen, J.; Hatsukami, D.K.; Upadhyaya, P.; Hecht, S.S.; et al. Phenanthrene metabolism in smokers: Use of a two-step diagnostic plot approach to identify subjects with extensive metabolic activation. J. Pharmacol. Exp. Ther. 2012, 342, 750–760. [Google Scholar] [CrossRef]
- Rojas, M.; Cascorbi, I.; Alexandrov, K.; Kriek, E.; Auburtin, G.; Mayer, L.; Kopp-Schneider, A.; Roots, I.; Bartsch, H. Modulation of benzo[a]pyrene diolepoxide-DNA adduct levels in human white blood cells by CYP1A1, GSTM1 and GSTT1 polymorphism. Carcinogenesis 2000, 21, 35–41. [Google Scholar] [CrossRef]
- Baglia, R.A.; Mills, K.R.; Mitra, K.; Tutol, J.N.; Ball, D.; Page, K.M.; Kallu, J.; Gottipolu, S.; D’Arcy, S.; Nielsen, S.O.; et al. An activity-based fluorescent sensor for the detection of the phenol sulfotransferase SULT1A1 in living cells. RSC Chem. Biol. 2021, 2, 830–834. [Google Scholar] [CrossRef]
- Piazza, R.S.; Trevisan, R.; Flores-Nunes, F.; Toledo-Silva, G.; Wendt, N.; Mattos, J.J.; Lima, D.; Taniguchi, S.; Sasaki, S.T.; Mello, Á.C.; et al. Exposure to phenanthrene and depuration: Changes on gene transcription, enzymatic activity and lipid peroxidation in gill of scallops Nodipecten nodosus. Aquat. Toxicol. 2016, 177, 146–155. [Google Scholar] [CrossRef] [PubMed]
- Klotz, L.O.; Steinbrenner, H. Cellular adaptation to xenobiotics: Interplay between xenosensors, reactive oxygen species and FOXO transcription factors. Redox. Biol. 2017, 13, 646–654. [Google Scholar] [CrossRef] [PubMed]
- Zhou, B.; Wang, X.; Li, F.; Wang, Y.; Yang, L.; Zhen, X.; Tan, W. Mitochondrial activity and oxidative stress functions are influenced by the activation of AhR-induced CYP1A1 overexpression in cardiomyocytes. Mol. Med. Rep. 2017, 16, 174–180. [Google Scholar] [CrossRef] [Green Version]
- Guo, H.; Zhang, Z.; Wang, H.; Ma, H.; Hu, F.; Zhang, W.; Liu, Y.; Huang, Y.; Zeng, Y.; Li, C.; et al. Oxidative stress and inflammatory effects in human lung epithelial A549 cells induced by phenanthrene, fluorene, and their binary mixture. Environ. Toxicol. 2021, 36, 95–104. [Google Scholar] [CrossRef] [PubMed]
- He, F.; Liu, Q.; Jing, M.; Wan, J.; Huo, C.; Zong, W.; Tang, J.; Liu, R. Toxic mechanism on phenanthrene-induced cytotoxicity, oxidative stress and activity changes of superoxide dismutase and catalase in earthworm (Eisenia foetida): A combined molecular and cellular study. J. Hazard. Mater. 2021, 418, 126302. [Google Scholar] [CrossRef] [PubMed]
- Hong, X.; Qin, J.; Chen, R.; Yuan, L.; Zha, J.; Huang, C.; Li, N.; Ji, X.; Wang, Z. Phenanthrene-Induced Apoptosis and Its Underlying Mechanism. Environ. Sci. Technol. 2017, 51, 14397–14405. [Google Scholar] [CrossRef]
- Kodama, S.; Moore, R.; Yamamoto, Y.; Negishi, M. Human nuclear pregnane X receptor cross-talk with CREB to repress cAMP activation of the glucose-6-phosphatase gene. Biochem. J. 2007, 407, 373–381. [Google Scholar] [CrossRef]
- Matsumoto, M.; Pocai, A.; Rossetti, L.; Depinho, R.A.; Accili, D. Impaired regulation of hepatic glucose production in mice lacking the forkhead transcription factor foxo1 in liver. Cell Metabol. 2007, 6, 208–216. [Google Scholar] [CrossRef]
- Puigserver, P.; Rhee, J.; Donovan, J.; Walkey, C.J.; Yoon, J.C.; Oriente, F.; Kitamura, Y.; Altomonte, J.; Dong, H.; Accili, D.; et al. Insulin-regulated hepatic gluconeogenesis through FOXO1-PGC-1alpha interaction. Nature 2003, 423, 550–555. [Google Scholar] [CrossRef]
- Lacroix, C.; Richard, G.; Seguineau, C.; Guyomarch, J.; Moraga, D.; Auffret, M. Active and passive biomonitoring suggest metabolic adaptation in blue mussels (Mytilus spp.) chronically exposed to a moderate contamination in Brest harbor (France). Aquat. Toxicol. 2015, 162, 126–137. [Google Scholar] [CrossRef]
- Lou, M.F. Redox regulation in the lens. Prog. Retin. Eye Res. 2003, 22, 657–682. [Google Scholar] [CrossRef]
- Housley, M.P.; Udeshi, N.D.; Rodgers, J.T.; Shabanowitz, J.; Puigserver, P.; Hunt, D.F.; Hart, G.W. A PGC-1α-O-GlcNAc transferase complex regulates FoxO transcription factor activity in response to glucose. J. Biol. Chem. 2009, 284, 5148–5157. [Google Scholar] [CrossRef] [PubMed]
- Kaarniranta, K.; Kajdanek, J.; Morawiec, J.; Pawlowska, E.; Blasiak, J. PGC-1α Protects RPE Cells of the Aging Retina against Oxidative Stress-Induced Degeneration through the Regulation of Senescence and Mitochondrial Quality Control. The Significance for AMD Pathogenesis. Int. J. Mol. Sci. 2018, 19, 2317. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yusni, Y.; Meutia, F. Action Mechanism of Rosella (Hibiscus sabdariffa L.) Used to Treat Metabolic Syndrome in Elderly Women. Evid. Based Complement. Alternat. Med. 2020, 2020, 5351318. [Google Scholar] [CrossRef]
- Yabaluri, N.; Bashyam, M.D. Hormonal regulation of gluconeogenic gene transcription in the liver. J. Biosci. 2010, 35, 473–484. [Google Scholar] [CrossRef]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Sl. No. | Gene Name | Forward Primer | Reverse Primer |
|---|---|---|---|
| 1 | AhR | GTCGTCTAAGGTGTCTGCTGGA | CGCAAACAAAGCCAACTGAGGTG |
| 2 | CYP1A1 | GATTGAGCACTGTCAGGAGAAGC | ATGAGGCTCCAGGAGATAGCAG |
| 3 | CYP1B1 | GCCACTATCACTGACATCTTCGG | CACGACCTGATCCAATTCTGCC |
| 4 | CYP2B | ACAGTGTGGAGAAGCACCGTGA | GGTTGAGGTTCTGGTGGCTGAA |
| 5 | CAR | GCAGAAGTGCTTAGATGCTGGC | GCTCCTTACTCAGTTGCACAGG |
| 6 | PXR | GCTGTCCTACTGCTTGGAAGAC | CTGCATCAGCACATACTCCTCC |
| 7 | RXR | TTCTCCACCCAGGTGAACTC | GAGCTGATGACCGAGAAAGG |
| 8 | CYP3A4 | CAAGACCCCTTTGTGGAAAA | CGAGGCGACTTTCTTTCATC |
| 9 | UGT1A1 | GCAAAGCGCATGGAGACTAAGG | GGTCCTTGTGAAGGCTGGAGAG |
| 10 | SULT1 | GCAACGCAAAGGATGTGGCA | TCCGTAGGACACTTCTCCGA |
| 11 | GSTP1 | TGGACATGGTGAATGACGGCGT | GGTCTCAAAAGGCTTCAGTTGCC |
| 12 | GSTM1 | TGATGTCCTTGACCTCCACCGT | GCTGGACTTCATGTAGGCAGAG |
| 13 | EPHX1 | GTTTTCCACCTGGACCAATACGG | TGGTGCCTGTTGTCCAGTAGAG |
| 14 | PEPCK | GTGGGGGATGATATTGCTTG | TGGTCTCAGCCACATTGGTA |
| 15 | G6PASE | GGGAAAGATAAAGCCGACCTAC | CAGCAAGGTAGATTCGTGACAG |
| 16 | NRF-2 | CACATCCAGTCAGAAACCAGTGG | GGAATGTCTGCGCCAAAAGCTG |
| 17 | NQO-1 | CCTGCCATTCTGAAAGGCTGGT | GTGGTGATGGAAAGCACTGCCT |
| 18 | PGC-1α | CCAAAGGATGCGCTCTCGTTCA | CGGTGTCTGTAGTGGCTTGACT |
| 19 | FOXO-1 | CTACGAGTGGATGGTCAAGAGC | CCAGTTCCTTCATTCTGCACACG |
| 20 | GAPDH | GTCTCCTCTGACTTCAACAGCG | ACCACCCTGTTGCTGTAGCCAA |
| Grid Box Center | Grid Box Size | CYP1A1 1 | CYP3A4 2 | CAR 3 | PXR 4 |
|---|---|---|---|---|---|
| X | 20 | −29.0937 | 22.5109 | 25.6018 | 12.2592 |
| Y | 20 | 83.4721 | 33.3657 | 54.1912 | 31.7877 |
| Z | 20 | 2.7617 | 138.7003 | 29.5870 | 24.5412 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Malar, D.S.; Prasanth, M.I.; Verma, K.; Prasansuklab, A.; Tencomnao, T. Hibiscus sabdariffa Extract Protects HaCaT Cells against Phenanthrene-Induced Toxicity through the Regulation of Constitutive Androstane Receptor/Pregnane X Receptor Pathway. Nutrients 2022, 14, 3829. https://doi.org/10.3390/nu14183829
Malar DS, Prasanth MI, Verma K, Prasansuklab A, Tencomnao T. Hibiscus sabdariffa Extract Protects HaCaT Cells against Phenanthrene-Induced Toxicity through the Regulation of Constitutive Androstane Receptor/Pregnane X Receptor Pathway. Nutrients. 2022; 14(18):3829. https://doi.org/10.3390/nu14183829
Chicago/Turabian StyleMalar, Dicson Sheeja, Mani Iyer Prasanth, Kanika Verma, Anchalee Prasansuklab, and Tewin Tencomnao. 2022. "Hibiscus sabdariffa Extract Protects HaCaT Cells against Phenanthrene-Induced Toxicity through the Regulation of Constitutive Androstane Receptor/Pregnane X Receptor Pathway" Nutrients 14, no. 18: 3829. https://doi.org/10.3390/nu14183829
APA StyleMalar, D. S., Prasanth, M. I., Verma, K., Prasansuklab, A., & Tencomnao, T. (2022). Hibiscus sabdariffa Extract Protects HaCaT Cells against Phenanthrene-Induced Toxicity through the Regulation of Constitutive Androstane Receptor/Pregnane X Receptor Pathway. Nutrients, 14(18), 3829. https://doi.org/10.3390/nu14183829