
Mulberry Leaf (Morus alba L.) Extracts and Its Chlorogenic Acid Isomer Component Improve Glucolipotoxicity-Induced Hepatic Lipid Accumulation via Downregulating miR-34a and Decreased Inflammation
 ,
, {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
2.1. Effect of Viability on HepG2 Cells Treated with MLE and CGA Isomer
2.2. Effect of Oleic Acid and High Glucose-Induced Lipid Accumulation
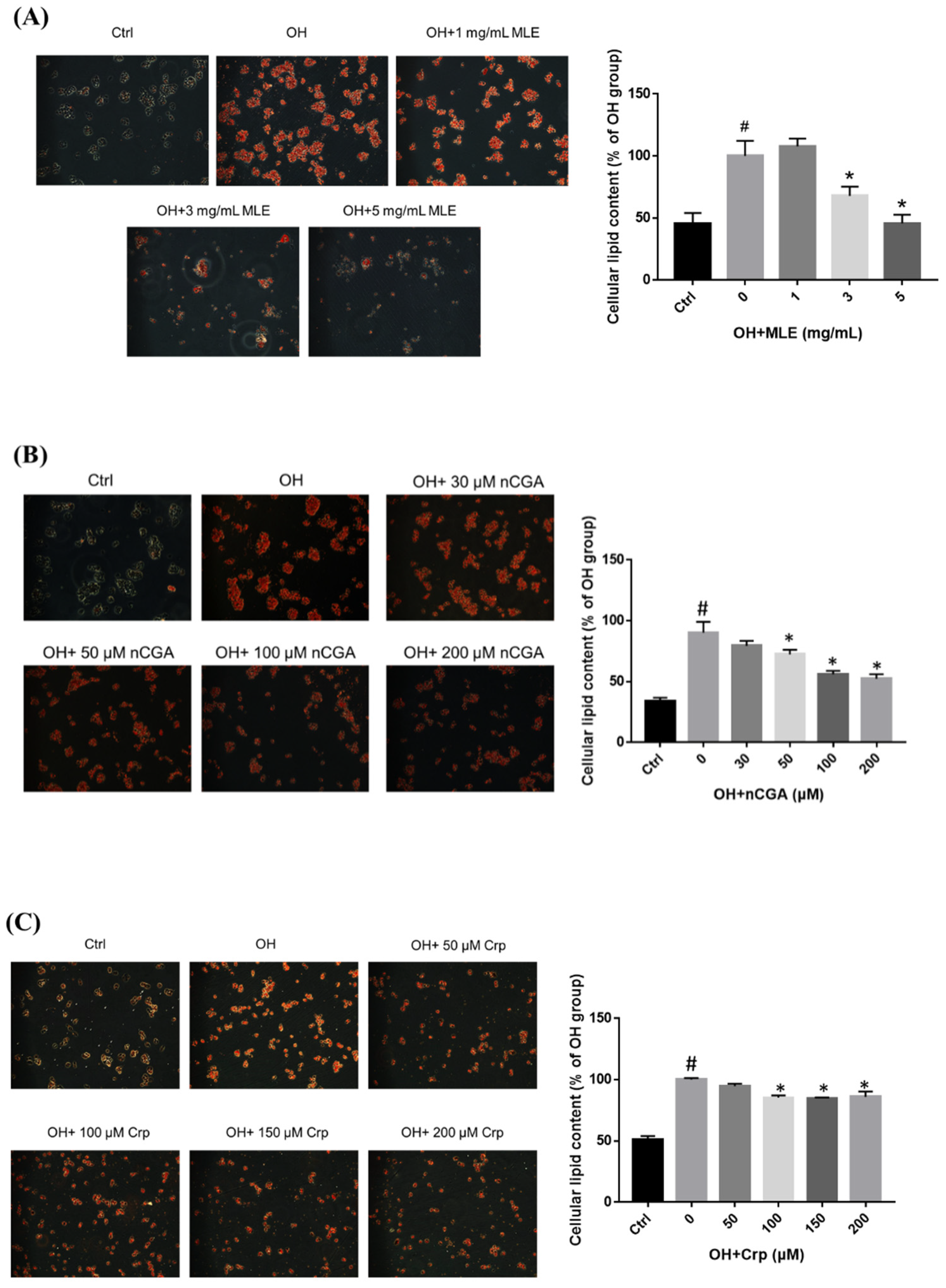
2.3. Inhibitory Effect of MLE and CGA Isomers on Lipid Accumulation
2.4. MLE and nCGA Activated the AMPK Signaling Pathway and β-Oxidative Signal Pathway in OH-Cultured HepG2 Cells
2.5. MLE and nCGA Reduced Lipogenesis-Related Protein Expression in OH-Cultured HepG2 Cells
2.6. Effects of MLE and nCGA on Fatty Acid Oxidation-Related and Inflammatory Protein Expressions in OH-Induced HepG2 Cells
2.7. MiR-34a Inhibitor Activated SIRT1, p-AMPK/AMPK, and PPARα Signaling Pathways in OH-Induced HepG2 Cells
3. Discussion
4. Materials and Methods
4.1. Chemicals and Reagents
4.2. Preparation of MLE and CGA Isomers
4.3. Cell Culture
4.4. Cell Viability Assay
4.5. Nile Red Analysis
4.6. miRNA Extraction and Real-Time PCR
4.7. Cell Transfection
4.8. Western Immunoblotting
4.9. Statistical Analysis
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Chalasani, N.; Younossi, Z.; Lavine, J.E.; Charlton, M.; Cusi, K.; Rinella, M.; Harrison, S.A.; Brunt, E.M.; Sanyal, A.J. The diagnosis and management of nonalcoholic fatty liver disease: Practice guidance from the American Association for the Study of Liver Diseases. Hepatology 2018, 67, 328–357. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Robertson, R.P.; Harmon, J.; Tran, P.O.T.; Poitout, V. β-cell glucose toxicity, lipotoxicity, and chronic oxidative stress in type 2 diabetes. Diabetes 2004, 53, S119–S124. [Google Scholar] [CrossRef] [Green Version]
- Lambert, J.E.; Ramos-Roman, M.A.; Browning, J.D.; Parks, E.J. Increased de novo lipogenesis is a distinct characteristic of individuals with nonalcoholic fatty liver disease. Gastroenterology 2014, 146, 726–735. [Google Scholar] [CrossRef]
- Das, S.; Mohamed, I.N.; Teoh, S.L.; Thevaraj, T.; Ku Ahmad Nasir, K.N.; Zawawi, A.; Salim, H.H.; Zhou, D.K. Micro-RNA and the features of metabolic syndrome: A narrative review. Mini Rev. Med. Chem. 2020, 20, 626–635. [Google Scholar] [CrossRef]
- Aryal, B.; Singh, A.K.; Rotllan, N.; Price, N.; Fernández-Hernando, C. MicroRNAs and lipid metabolism. Curr. Opin. Lipidol. 2017, 28, 273. [Google Scholar] [CrossRef]
- Xu, Y.; Zalzala, M.; Xu, J.; Li, Y.; Yin, L.; Zhang, Y. A metabolic stress-inducible miR-34a-HNF4α pathway regulates lipid and lipoprotein metabolism. Nat. Commun. 2015, 6, 7466. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xu, Y.; Zhu, Y.; Hu, S.; Pan, X.; Bawa, F.C.; Wang, H.H.; Wang, D.Q.-H.; Yin, L.; Zhang, Y. Hepatocyte miR-34a is a key regulator in the development and progression of non-alcoholic fatty liver disease. Mol. Metab. 2021, 51, 101244. [Google Scholar] [CrossRef] [PubMed]
- Sud, N.; Zhang, H.; Pan, K.; Cheng, X.; Cui, J.; Su, Q. Aberrant expression of microRNA induced by high-fructose diet: Implications in the pathogenesis of hyperlipidemia and hepatic insulin resistance. J. Nutr. Biochem. 2017, 43, 125–131. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Horton, J.D.; Shah, N.A.; Warrington, J.A.; Anderson, N.N.; Park, S.W.; Brown, M.S.; Goldstein, J.L. Combined analysis of oligonucleotide microarray data from transgenic and knockout mice identifies direct SREBP target genes. Proc. Natl. Acad. Sci. USA 2003, 100, 12027–12032. [Google Scholar] [CrossRef] [Green Version]
- Liu, S.; Gao, Y.; Zhang, L.; Yin, Y.; Zhang, W. Rspo1/Rspo3-LGR4 signaling inhibits hepatic cholesterol synthesis through the AMPKα-SREBP2 pathway. FASEB J. 2020, 34, 14946–14959. [Google Scholar] [CrossRef]
- Price, N.L.; Gomes, A.P.; Ling, A.J.; Duarte, F.V.; Martin-Montalvo, A.; North, B.J.; Agarwal, B.; Ye, L.; Ramadori, G.; Teodoro, J.S. SIRT1 is required for AMPK activation and the beneficial effects of resveratrol on mitochondrial function. Cell Metab. 2012, 15, 675–690. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Serviddio, G.; Bellanti, F.; Vendemiale, G. Free radical biology for medicine: Learning from nonalcoholic fatty liver disease. Free. Radic. Biol. Med. 2013, 65, 952–968. [Google Scholar] [CrossRef] [Green Version]
- Chyau, C.-C.; Wang, H.-F.; Zhang, W.-J.; Chen, C.-C.; Huang, S.-H.; Chang, C.-C.; Peng, R.Y. Antrodan alleviates high-fat and high-fructose diet-induced fatty liver disease in C57BL/6 mice model via AMPK/Sirt1/SREBP-1c/PPARγ pathway. Int. J. Mol. Sci. 2020, 21, 360. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, Q.; Liu, S.; Zhai, A.; Zhang, B.; Tian, G. AMPK-mediated regulation of lipid metabolism by phosphorylation. Biol. Pharm. Bull. 2018, 41, 985–993. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Iskender, H.; Dokumacıoğlu, E.; Saral, S.; Yenice, G.; Sevim, Ç. NF-κB, TNF-α and IL-6 Levels in Liver and Kidney of High-Fructose-Fed Rats. Int. J. Res. Pharm. Biomed. Sci. 2018, 18, 1–7. [Google Scholar] [CrossRef]
- Baker, R.G.; Hayden, M.S.; Ghosh, S. NF-κB, inflammation, and metabolic disease. Cell Metab. 2011, 13, 11–22. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jin, K.; Liu, Y.; Shi, Y.; Zhang, H.; Sun, Y.; Zhangyuan, G.; Wang, F.; Yu, W.; Wang, J.; Tao, X. PTPROt aggravates inflammation by enhancing NF-κB activation in liver macrophages during nonalcoholic steatohepatitis. Theranostics 2020, 10, 5290. [Google Scholar] [CrossRef]
- Gryn-Rynko, A.; Bazylak, G.; Olszewska-Slonina, D. New potential phytotherapeutics obtained from white mulberry (Morus alba L.) leaves. Biomed. Pharmacother. 2016, 84, 628–636. [Google Scholar] [CrossRef]
- Zhang, D.-Y.; Wan, Y.; Hao, J.-Y.; Hu, R.-Z.; Chen, C.; Yao, X.-H.; Zhao, W.-G.; Liu, Z.-Y.; Li, L. Evaluation of the alkaloid, polyphenols, and antioxidant contents of various mulberry cultivars from different planting areas in eastern China. Ind. Crop. Prod. 2018, 122, 298–307. [Google Scholar] [CrossRef]
- Yu, M.-H.; Hung, T.-W.; Wang, C.-C.; Wu, S.-W.; Yang, T.-W.; Yang, C.-Y.; Tseng, T.-H.; Wang, C.-J. Neochlorogenic Acid Attenuates Hepatic Lipid Accumulation and Inflammation via Regulating miR-34a In Vitro. Int. J. Mol. Sci. 2021, 22, 13163. [Google Scholar] [CrossRef] [PubMed]
- Ding, J.; Li, M.; Wan, X.; Jin, X.; Chen, S.; Yu, C.; Li, Y. Effect of miR-34a in regulating steatosis by targeting PPARα expression in nonalcoholic fatty liver disease. Sci. Rep. 2015, 5, 13729. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lin, H.-Y.; Yang, Y.-L.; Wang, P.-W.; Wang, F.-S.; Huang, Y.-H. The emerging role of microRNAs in NAFLD: Highlight of microRNA-29a in modulating oxidative stress, inflammation, and beyond. Cells 2020, 9, 1041. [Google Scholar] [CrossRef] [Green Version]
- Hart, M.; Rheinheimer, S.; Leidinger, P.; Backes, C.; Menegatti, J.; Fehlmann, T.; Grässer, F.; Keller, A.; Meese, E. Identification of miR-34a-target interactions by a combined network based and experimental approach. Oncotarget 2016, 7, 34288. [Google Scholar] [CrossRef] [Green Version]
- Takahashi, Y.; Fukusato, T. Histopathology of nonalcoholic fatty liver disease/nonalcoholic steatohepatitis. World J. Gastroenterol. WJG 2014, 20, 15539. [Google Scholar] [CrossRef]
- Cohen, J.C.; Horton, J.D.; Hobbs, H.H. Human fatty liver disease: Old questions and new insights. Science 2011, 332, 1519–1523. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gjorgjieva, M.; Sobolewski, C.; Dolicka, D.; de Sousa, M.C.; Foti, M. miRNAs and NAFLD: From pathophysiology to therapy. Gut 2019, 68, 2065–2079. [Google Scholar] [CrossRef] [PubMed]
- Guo, W.-R.; Liu, J.; Cheng, L.-D.; Liu, Z.-Y.; Zheng, X.-B.; Liang, H.; Xu, F. Metformin Alleviates Steatohepatitis in Diet-Induced Obese Mice in a SIRT1-Dependent Way. Front. Pharmacol. 2021, 12, 704112. [Google Scholar] [CrossRef]
- Ratziu, V.; Harrison, S.A.; Francque, S.; Bedossa, P.; Lehert, P.; Serfaty, L.; Romero-Gomez, M.; Boursier, J.; Abdelmalek, M.; Caldwell, S. Elafibranor, an agonist of the peroxisome proliferator-activated receptor −α and −δ, induces resolution of nonalcoholic steatohepatitis without fibrosis worsening. Gastroenterology 2016, 150, 1147–1159. [Google Scholar] [CrossRef] [Green Version]
- Qu, Y.-L.; Deng, C.-H.; Luo, Q.; Shang, X.-Y.; Wu, J.-X.; Shi, Y.; Han, Z.-G. Arid1a protects against hepatic steatosis and insulin resistance via PPARα-mediated fatty acid oxidation. bioRxiv 2019, 507517. [Google Scholar] [CrossRef] [Green Version]
- Bougarne, N.; Weyers, B.; Desmet, S.J.; Deckers, J.; Ray, D.W.; Staels, B.; De Bosscher, K. Molecular actions of PPAR α in lipid metabolism and inflammation. Endocr. Rev. 2018, 39, 760–802. [Google Scholar] [CrossRef]
- Cong, W.-N.; Tao, R.-Y.; Tian, J.-Y.; Liu, G.-T.; Ye, F. The establishment of a novel non-alcoholic steatohepatitis model accompanied with obesity and insulin resistance in mice. Life Sci. 2008, 82, 983–990. [Google Scholar] [CrossRef]
- Moslehi, A.; Hamidi-Zad, Z. Role of SREBPs in liver diseases: A mini-review. J. Clin. Transl. Hepatol. 2018, 6, 332. [Google Scholar] [CrossRef] [Green Version]
- Kim, J.Y.; Garcia-Carbonell, R.; Yamachika, S.; Zhao, P.; Dhar, D.; Loomba, R.; Kaufman, R.J.; Saltiel, A.R.; Karin, M. ER stress drives lipogenesis and steatohepatitis via caspase-2 activation of S1P. Cell 2018, 175, 133–145. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dorn, C.; Riener, M.-O.; Kirovski, G.; Saugspier, M.; Steib, K.; Weiss, T.S.; Gäbele, E.; Kristiansen, G.; Hartmann, A.; Hellerbrand, C. Expression of fatty acid synthase in nonalcoholic fatty liver disease. Int. J. Clin. Exp. Pathol. 2010, 3, 505. [Google Scholar]
- Ren, R.; Gong, J.; Zhao, Y.; Zhuang, X.; Ye, Y.; Lin, W. Sulfated polysaccharides from Enteromorpha prolifera suppress SREBP-2 and HMG-CoA reductase expression and attenuate non-alcoholic fatty liver disease induced by a high-fat diet. Food Funct. 2017, 8, 1899–1904. [Google Scholar] [CrossRef] [PubMed]
- Zhou, W.; Zhu, Z.; Xiao, X.; Li, C.; Zhang, L.; Dang, Y.; Ge, G.; Ji, G.; Zhu, M.; Xu, H. Jiangzhi Granule attenuates non-alcoholic steatohepatitis by suppressing TNF/NFκB signaling pathway-a study based on network pharmacology. Biomed. Pharmacother. 2021, 143, 112181. [Google Scholar] [CrossRef] [PubMed]
- Park, H.J.; Lee, J.-Y.; Chung, M.-Y.; Park, Y.-K.; Bower, A.M.; Koo, S.I.; Giardina, C.; Bruno, R.S. Green tea extract suppresses NFκB activation and inflammatory responses in diet-induced obese rats with nonalcoholic steatohepatitis. J. Nutr. 2012, 142, 57–63. [Google Scholar] [CrossRef] [Green Version]
- Fontes-Cal, T.C.; Mattos, R.T.; Medeiros, N.I.; Pinto, B.F.; Belchior-Bezerra, M.; Roque-Souza, B.; Dutra, W.O.; Ferrari, T.C.; Vidigal, P.V.; Faria, L.C. Crosstalk Between Plasma Cytokines, Inflammation, and Liver Damage as a New Strategy to Monitoring NAFLD Progression. Front. Immunol. 2021, 12, 708959. [Google Scholar] [CrossRef]
- Shi, L.; Karrar, E.; Wang, X. Sesamol ameliorates hepatic lipid accumulation and oxidative stress in steatosis HepG2 cells via the PPAR signaling pathway. J. Food Biochem. 2021, 45, e13976. [Google Scholar] [CrossRef]
- Greenspan, P.; Mayer, E.P.; Fowler, S.D. Nile red: A selective fluorescent stain for intracellular lipid droplets. J. Cell Biol. 1985, 100, 965–973. [Google Scholar] [CrossRef] [PubMed]








Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Yang, T.-Y.; Yu, M.-H.; Wu, Y.-L.; Hong, C.-C.; Chen, C.-S.; Chan, K.-C.; Wang, C.-J. Mulberry Leaf (Morus alba L.) Extracts and Its Chlorogenic Acid Isomer Component Improve Glucolipotoxicity-Induced Hepatic Lipid Accumulation via Downregulating miR-34a and Decreased Inflammation. Nutrients 2022, 14, 4808. https://doi.org/10.3390/nu14224808
Yang T-Y, Yu M-H, Wu Y-L, Hong C-C, Chen C-S, Chan K-C, Wang C-J. Mulberry Leaf (Morus alba L.) Extracts and Its Chlorogenic Acid Isomer Component Improve Glucolipotoxicity-Induced Hepatic Lipid Accumulation via Downregulating miR-34a and Decreased Inflammation. Nutrients. 2022; 14(22):4808. https://doi.org/10.3390/nu14224808
Chicago/Turabian StyleYang, Tsung-Yuan, Meng-Hsun Yu, Yi-Liang Wu, Ching-Chun Hong, Chin-Shuh Chen, Kuei-Chuan Chan, and Chau-Jong Wang. 2022. "Mulberry Leaf (Morus alba L.) Extracts and Its Chlorogenic Acid Isomer Component Improve Glucolipotoxicity-Induced Hepatic Lipid Accumulation via Downregulating miR-34a and Decreased Inflammation" Nutrients 14, no. 22: 4808. https://doi.org/10.3390/nu14224808