
Interaction of Vitamin D with Peptide Hormones with Emphasis on Parathyroid Hormone, FGF23, and the Renin-Angiotensin-Aldosterone System


{kind=link}
{kind=link}
Abstract
1. Introduction
2. Brief Overview of Vitamin D Metabolism
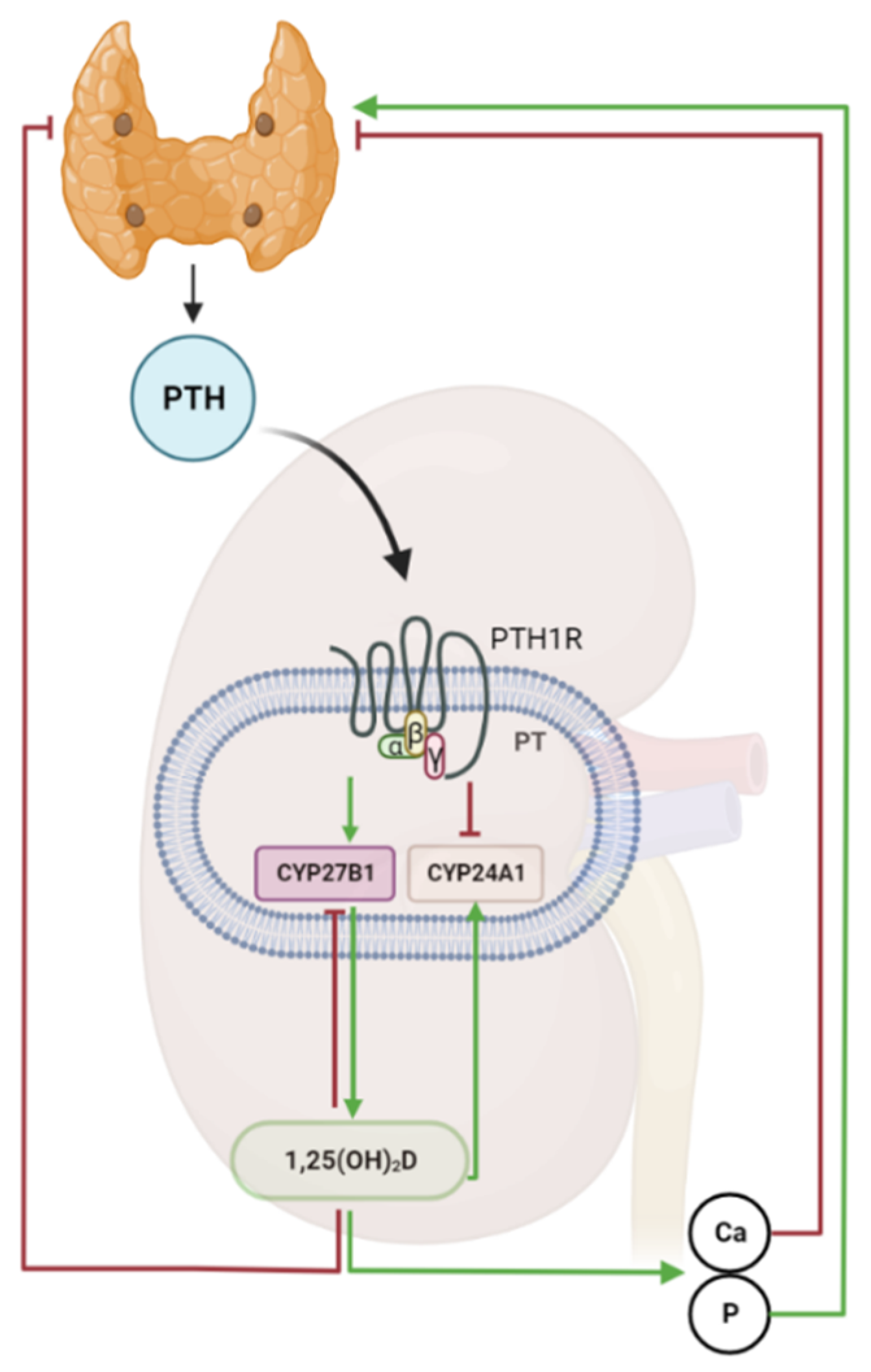
3. The Parathyroid-Kidney Axis in the Regulation of Vitamin D Metabolism
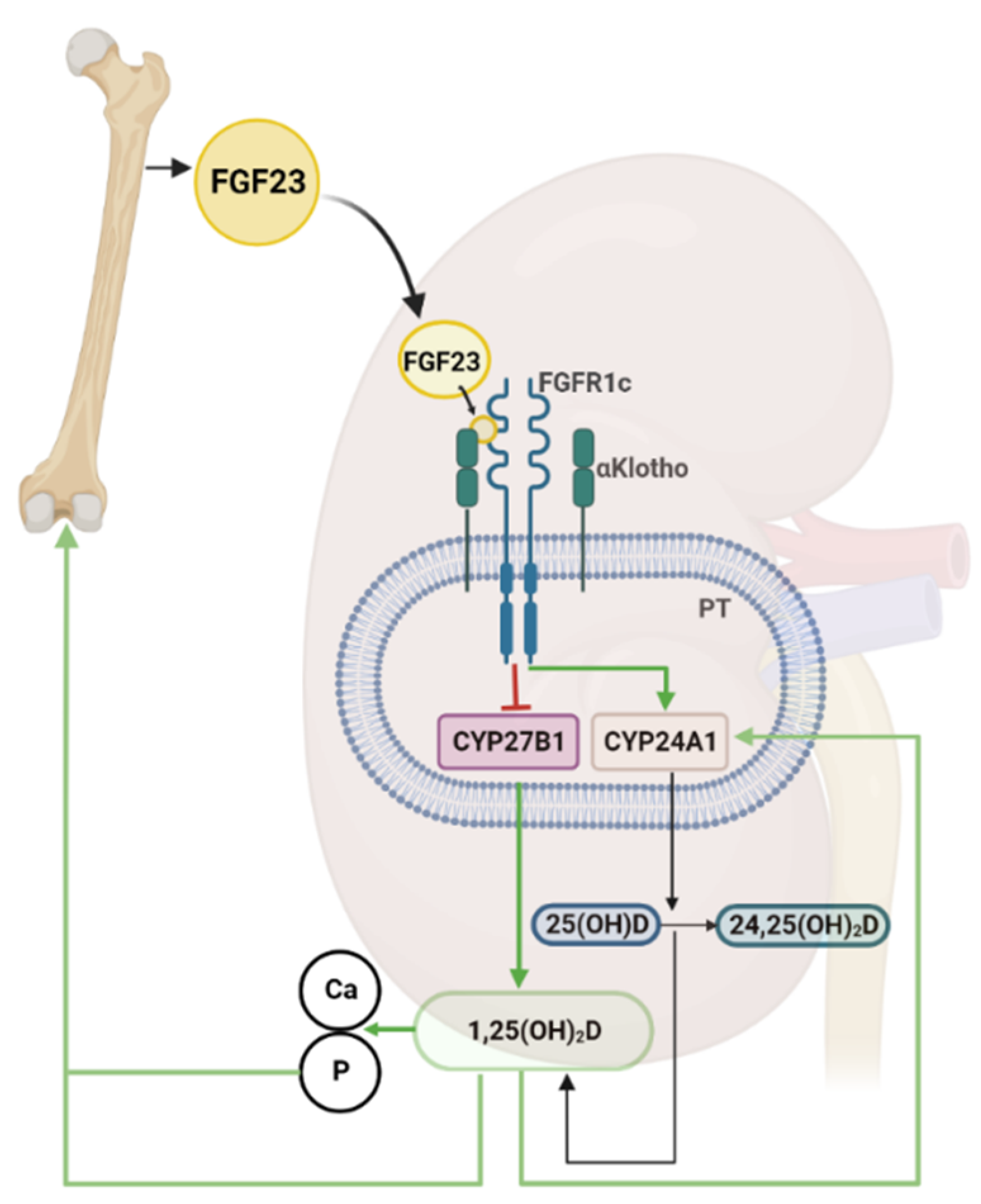
4. The Bone-Kidney Axis in the Regulation of Vitamin D Metabolism
5. Vitamin D and the Renin-Angiotensin-Aldosterone System (RAAS)
6. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Conflicts of Interest
References
- Hess, A.F.; Unger, L.J. The cure of infantile rickets by artificial light and by sunlight. Proc. Soc. Exp. Biol. Med. 1921, 18, 298. [Google Scholar] [CrossRef]
- McCollum, E.V.; Simmonds, N.; Becker, J.E.; Shipley, P.d. Studies on experimental rickets: XXI. An experimental demonstration of the existence of a vitamin which promotes calcium deposition. J. Biol. Chem. 1922, 53, 293–312. [Google Scholar] [CrossRef]
- Chick, H.; Dalyell, E.; Hume, M.; Smith, H.H.; Mackay, H.M.; Hams Wimberger, M.; Kinderklinik, R.T.T.U. The aetiology of rickets in infants: Prophylactic and curative observations at the Vienna University Kinderklinik. Lancet 1922, 200, 7–11. [Google Scholar] [CrossRef]
- Wolf, G. The Discovery of Vitamin D: The Contribution of Adolf Windaus. J. Nutr. 2004, 134, 1299–1302. [Google Scholar] [CrossRef]
- Kalra, S.; Baruah, M.P.; Sahay, R.; Sawhney, K. The history of parathyroid endocrinology. Indian J. Endocrinol. Metab. 2013, 17, 320–322. [Google Scholar] [CrossRef]
- Garabedian, M.; Holick, M.F.; Deluca, H.F.; Boyle, I.T. Control of 25-hydroxycholecalciferol metabolism by parathyroid glands. Proc. Natl. Acad. Sci. USA 1972, 69, 1673–1676. [Google Scholar] [CrossRef]
- Fraser, D.R.; Kodicek, E. Regulation of 25-Hydroxycholecalciferol-1-Hydroxylase Activity in Kidney by Parathyroid Hormone. Nat. New Biol. 1973, 241, 163–166. [Google Scholar] [CrossRef] [PubMed]
- Chertow, B.S.; Baylink, D.J.; Wergedal, J.E.; Su, M.H.; Norman, A.W. Decrease in serum immunoreactive parathyroid hormone in rats and in parathyroid hormone secretion in vitro by 1,25-dihydroxycholecalciferol. J. Clin. Investig. 1975, 56, 668–678. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Li, Y.C.; Kong, J.; Wei, M.; Chen, Z.-F.; Liu, S.Q.; Cao, L.-P. 1,25-Dihydroxyvitamin D3 is a negative endocrine regulator of the renin-angiotensin system. J. Clin. Investig. 2002, 110, 229–238. [Google Scholar] [CrossRef]
- Brunette, M.G.; Chan, M.; Ferriere, C.; Roberts, K.D. Site of 1,25(OH)2 vitamin D3 synthesis in the kidney. Nature 1978, 276, 287–289. [Google Scholar] [CrossRef]
- Fraser, D.R.; Kodicek, E. Unique Biosynthesis by Kidney of a Biologically Active Vitamin D Metabolite. Nature 1970, 228, 764–766. [Google Scholar] [CrossRef]
- Somjen, D.; Weisman, Y.; Kohen, F.; Gayer, B.; Limor, R.; Sharon, O.; Jaccard, N.; Knoll, E.; Stern, N. 25-hydroxyvitamin D3-1alpha-hydroxylase is expressed in human vascular smooth muscle cells and is upregulated by parathyroid hormone and estrogenic compounds. Circulation 2005, 111, 1666–1671. [Google Scholar] [CrossRef]
- Zehnder, D.; Bland, R.; Chana, R.S.; Wheeler, D.C.; Howie, A.J.; Williams, M.C.; Stewart, P.M.; Hewison, M. Synthesis of 1,25-Dihydroxyvitamin D3 by Human Endothelial Cells Is Regulated by Inflammatory Cytokines: A Novel Autocrine Determinant of Vascular Cell Adhesion. J. Am. Soc. Nephrol. 2002, 13, 621–629. [Google Scholar] [CrossRef] [PubMed]
- Bikle, D.D. Vitamin D Metabolism, Mechanism of Action, and Clinical Applications. Chem. Biol. 2014, 21, 319–329. [Google Scholar] [CrossRef]
- Bouillon, R.; Carmeliet, G.; Verlinden, L.; van Etten, E.; Verstuyf, A.; Luderer, H.F.; Lieben, L.; Mathieu, C.; Demay, M. Vitamin D and human health: Lessons from vitamin D receptor null mice. Endocr. Rev. 2008, 29, 726–776. [Google Scholar] [CrossRef]
- Bouillon, R.; Suda, T. Vitamin D: Calcium and bone homeostasis during evolution. Bonekey Rep. 2014, 3, 480. [Google Scholar] [CrossRef] [PubMed]
- Jones, G.; Prosser, D.E.; Kaufmann, M. 25-Hydroxyvitamin D-24-hydroxylase (CYP24A1): Its important role in the degradation of vitamin D. Arch. Biochem. Biophys. 2012, 523, 9–18. [Google Scholar] [CrossRef] [PubMed]
- St-Arnaud, R.; Arabian, A.; Travers, R.; Barletta, F.; Raval-Pandya, M.; Chapin, K.; Depovere, J.; Mathieu, C.; Christakos, S.; Demay, M.B.; et al. Deficient mineralization of intramembranous bone in vitamin D-24-hydroxylase-ablated mice is due to elevated 1,25-dihydroxyvitamin D and not to the absence of 24,25-dihydroxyvitamin D. Endocrinology 2000, 141, 2658–2666. [Google Scholar] [CrossRef]
- Nesterova, G.; Malicdan, M.C.; Yasuda, K.; Sakaki, T.; Vilboux, T.; Ciccone, C.; Horst, R.; Huang, Y.; Golas, G.; Introne, W.; et al. 1,25-(OH)2D-24 Hydroxylase (CYP24A1) Deficiency as a Cause of Nephrolithiasis. Clin. J. Am. Soc. Nephrol. 2013, 8, 649–657. [Google Scholar] [CrossRef]
- Slominski, A.T.; Kim, T.-K.; Li, W.; Postlethwaite, A.; Tieu, E.W.; Tang, E.K.Y.; Tuckey, R.C. Detection of novel CYP11A1-derived secosteroids in the human epidermis and serum and pig adrenal gland. Sci. Rep. 2015, 5, 14875. [Google Scholar] [CrossRef] [PubMed]
- Slominski, A.T.; Kim, T.K.; Shehabi, H.Z.; Semak, I.; Tang, E.K.; Nguyen, M.N.; Benson, H.A.; Korik, E.; Janjetovic, Z.; Chen, J.; et al. In vivo evidence for a novel pathway of vitamin D₃ metabolism initiated by P450scc and modified by CYP27B1. FASEB J. 2012, 26, 3901–3915. [Google Scholar] [CrossRef]
- Tang, E.K.; Chen, J.; Janjetovic, Z.; Tieu, E.W.; Slominski, A.T.; Li, W.; Tuckey, R.C. Hydroxylation of CYP11A1-derived products of vitamin D3 metabolism by human and mouse CYP27B1. Drug Metab. Dispos. 2013, 41, 1112–1124. [Google Scholar] [CrossRef]
- Verstuyf, A.; Carmeliet, G.; Bouillon, R.; Mathieu, C. Vitamin D: A pleiotropic hormone. Kidney Int. 2010, 78, 140–145. [Google Scholar] [CrossRef]
- Gensure, R.C.; Gardella, T.J.; Jüppner, H. Parathyroid hormone and parathyroid hormone-related peptide, and their receptors. Biochem. Biophys. Res. Commun. 2005, 328, 666–678. [Google Scholar] [CrossRef]
- Bienaime, F.; Prie, D.; Friedlander, G.; Souberbielle, J.C. Vitamin D metabolism and activity in the parathyroid gland. Mol. Cell. Endocrinol. 2011, 347, 30–41. [Google Scholar] [CrossRef]
- Sato, T.; Courbebaisse, M.; Ide, N.; Fan, Y.; Hanai, J.-i.; Kaludjerovic, J.; Densmore, M.J.; Yuan, Q.; Toka, H.R.; Pollak, M.R.; et al. Parathyroid hormone controls paracellular Ca2+ transport in the thick ascending limb by regulating the tight-junction protein Claudin14. Proc. Natl. Acad. Sci. USA 2017, 114, E3344–E3353. [Google Scholar] [CrossRef] [PubMed]
- Martin, A.; David, V.; Quarles, L.D. Regulation and function of the FGF23/klotho endocrine pathways. Physiol. Rev. 2012, 92, 131–155. [Google Scholar] [CrossRef]
- Goretti Penido, M.; Alon, U.S. Phosphate homeostasis and its role in bone health. Pediatr. Nephrol. 2012, 27, 2039–2048. [Google Scholar] [CrossRef] [PubMed]
- Meyer, M.B.; Benkusky, N.A.; Kaufmann, M.; Lee, S.M.; Onal, M.; Jones, G.; Pike, J.W. A kidney-specific genetic control module in mice governs endocrine regulation of the cytochrome P450 gene Cyp27b1 essential for vitamin D(3) activation. J. Biol. Chem. 2017, 292, 17541–17558. [Google Scholar] [CrossRef] [PubMed]
- Meyer, M.B.; Benkusky, N.A.; Lee, S.M.; Yoon, S.H.; Mannstadt, M.; Wein, M.N.; Pike, J.W. Rapid genomic changes by mineralotropic hormones and kinase SIK inhibition drive coordinated renal Cyp27b1 and Cyp24a1 expression via CREB modules. J. Biol. Chem. 2022, 298, 102559. [Google Scholar] [CrossRef]
- Meyer, M.B.; Pike, J.W. Mechanistic homeostasis of vitamin D metabolism in the kidney through reciprocal modulation of Cyp27b1 and Cyp24a1 expression. J. Steroid. Biochem. Mol. Biol. 2020, 196, 105500. [Google Scholar] [CrossRef]
- Silver, J.; Russell, J.; Sherwood, L.M. Regulation by vitamin D metabolites of messenger ribonucleic acid for preproparathyroid hormone in isolated bovine parathyroid cells. Proc. Natl. Acad. Sci. USA 1985, 82, 4270–4273. [Google Scholar] [CrossRef]
- Ritter, C.S.; Armbrecht, H.J.; Slatopolsky, E.; Brown, A.J. 25-Hydroxyvitamin D(3) suppresses PTH synthesis and secretion by bovine parathyroid cells. Kidney Int. 2006, 70, 654–659. [Google Scholar] [CrossRef]
- Silver, J.; Naveh-Many, T.; Mayer, H.; Schmelzer, H.J.; Popovtzer, M.M. Regulation by vitamin D metabolites of parathyroid hormone gene transcription in vivo in the rat. J. Clin. Investig. 1986, 78, 1296–1301. [Google Scholar] [CrossRef]
- Meir, T.; Levi, R.; Lieben, L.; Libutti, S.; Carmeliet, G.; Bouillon, R.; Silver, J.; Naveh-Many, T. Deletion of the vitamin D receptor specifically in the parathyroid demonstrates a limited role for the receptor in parathyroid physiology. Am. J. Physiol. Renal Physiol. 2009, 297, F1192–F1198. [Google Scholar] [CrossRef] [PubMed]
- Weber, K.; Bergow, C.; Hirmer, S.; Schüler, C.; Erben, R.G. Vitamin D-independent therapeutic effects of extracellular calcium in a mouse model of adult-onset secondary hyperparathyroidism. J. Bone Miner. Res. 2009, 24, 22–32. [Google Scholar] [CrossRef] [PubMed]
- Mackey, S.L.; Heymont, J.L.; Kronenberg, H.M.; Demay, M.B. Vitamin D receptor binding to the negative human parathyroid hormone vitamin D response element does not require the retinoid x receptor. Mol. Endocrinol. 1996, 10, 298–305. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Koszewski, N.J.; Ashok, S.; Russell, J. Turning a Negative into a Positive: Vitamin D Receptor Interactions with the Avian Parathyroid Hormone Response Element. Mol. Endocrinol. 1999, 13, 455–465. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Murayama, A.; Kim, M.S.; Yanagisawa, J.; Takeyama, K.; Kato, S. Transrepression by a liganded nuclear receptor via a bHLH activator through co-regulator switching. EMBO J. 2004, 23, 1598–1608. [Google Scholar] [CrossRef]
- Ritter, C.S.; Brown, A.J. Direct suppression of Pth gene expression by the vitamin D prohormones doxercalciferol and calcidiol requires the vitamin D receptor. J. Mol. Endocrinol. 2011, 46, 63–66. [Google Scholar] [CrossRef]
- Segersten, U.; Correa, P.; Hewison, M.; Hellman, P.; Dralle, H.; Carling, T.; Akerström, G.; Westin, G. 25-hydroxyvitamin D(3)-1alpha-hydroxylase expression in normal and pathological parathyroid glands. J. Clin. Endocrinol. Metab. 2002, 87, 2967–2972. [Google Scholar] [CrossRef] [PubMed]
- Cozzolino, M.; Covic, A.; Martinez-Placencia, B.; Xynos, K. Treatment Failure of Active Vitamin D Therapy in Chronic Kidney Disease: Predictive Factors. Am. J. Nephrol. 2015, 42, 228–236. [Google Scholar] [CrossRef] [PubMed]
- Coyne, D.W.; Goldberg, S.; Faber, M.; Ghossein, C.; Sprague, S.M. A randomized multicenter trial of paricalcitol versus calcitriol for secondary hyperparathyroidism in stages 3–4 CKD. Clin. J. Am. Soc. Nephrol. 2014, 9, 1620–1626. [Google Scholar] [CrossRef] [PubMed]
- Shah, A.; Aeddula, N.R. Renal Osteodystrophy. In StatPearls; StatPearls Publishing LLC: Treasure Island, FL, USA, 2022. [Google Scholar]
- Jamaluddin, E.J.; Gafor, A.H.; Yean, L.C.; Cader, R.; Mohd, R.; Kong, N.C.; Shah, S.A. Oral paricalcitol versus oral calcitriol in continuous ambulatory peritoneal dialysis patients with secondary hyperparathyroidism. Clin. Exp. Nephrol. 2014, 18, 507–514. [Google Scholar] [CrossRef]
- White, K.E.; Evans, W.E.; O’Riordan, J.L.H.; Speer, M.C.; Econs, M.J.; Lorenz-Depiereux, B.; Grabowski, M.; Meitinger, T.; Strom, T.M. Autosomal dominant hypophosphataemic rickets is associated with mutations in FGF23. Nat. Genet. 2000, 26, 345–348. [Google Scholar] [CrossRef]
- Shimada, T.; Mizutani, S.; Muto, T.; Yoneya, T.; Hino, R.; Takeda, S.; Takeuchi, Y.; Fujita, T.; Fukumoto, S.; Yamashita, T. Cloning and characterization of FGF23 as a causative factor of tumor-induced osteomalacia. Proc. Natl. Acad. Sci. USA 2001, 98, 6500–6505. [Google Scholar] [CrossRef]
- Yoshiko, Y.; Wang, H.; Minamizaki, T.; Ijuin, C.; Yamamoto, R.; Suemune, S.; Kozai, K.; Tanne, K.; Aubin, J.E.; Maeda, N. Mineralized tissue cells are a principal source of FGF23. Bone 2007, 40, 1565–1573. [Google Scholar] [CrossRef]
- Itoh, N.; Ohta, H.; Konishi, M. Endocrine FGFs: Evolution, Physiology, Pathophysiology, and Pharmacotherapy. Front. Endocrinol. 2015, 6, 154. [Google Scholar] [CrossRef] [PubMed]
- Tagliabracci, V.S.; Engel, J.L.; Wiley, S.E.; Xiao, J.; Gonzalez, D.J.; Nidumanda Appaiah, H.; Koller, A.; Nizet, V.; White, K.E.; Dixon, J.E. Dynamic regulation of FGF23 by Fam20C phosphorylation, GalNAc-T3 glycosylation, and furin proteolysis. Proc. Natl. Acad. Sci. USA 2014, 111, 5520–5525. [Google Scholar] [CrossRef]
- Urakawa, I.; Yamazaki, Y.; Shimada, T.; Iijima, K.; Hasegawa, H.; Okawa, K.; Fujita, T.; Fukumoto, S.; Yamashita, T. Klotho converts canonical FGF receptor into a specific receptor for FGF23. Nature 2006, 444, 770–774. [Google Scholar] [CrossRef] [PubMed]
- Chen, G.; Liu, Y.; Goetz, R.; Fu, L.; Jayaraman, S.; Hu, M.-C.; Moe, O.W.; Liang, G.; Li, X.; Mohammadi, M. α-Klotho is a non-enzymatic molecular scaffold for FGF23 hormone signalling. Nature 2018, 553, 461–466. [Google Scholar] [CrossRef] [PubMed]
- Goetz, R.; Ohnishi, M.; Ding, X.; Kurosu, H.; Wang, L.; Akiyoshi, J.; Ma, J.; Gai, W.; Sidis, Y.; Pitteloud, N.; et al. Klotho coreceptors inhibit signaling by paracrine fibroblast growth factor 8 subfamily ligands. Mol. Cell. Biol. 2012, 32, 1944–1954. [Google Scholar] [CrossRef] [PubMed]
- Ornitz, D.M.; Itoh, N. The Fibroblast Growth Factor signaling pathway. Wiley Interdiscip. Rev. Dev. Biol. 2015, 4, 215–266. [Google Scholar] [CrossRef]
- Ratsma, D.M.A.; Zillikens, M.C.; van der Eerden, B.C.J. Upstream Regulators of Fibroblast Growth Factor 23. Front. Endocrinol. 2021, 12, 588096. [Google Scholar] [CrossRef]
- Kolek, O.I.; Hines, E.R.; Jones, M.D.; LeSueur, L.K.; Lipko, M.A.; Kiela, P.R.; Collins, J.F.; Haussler, M.R.; Ghishan, F.K. 1α,25-Dihydroxyvitamin D3 upregulates FGF23 gene expression in bone: The final link in a renal-gastrointestinal-skeletal axis that controls phosphate transport. Am. J. Physiol.-Gastrointest. Liver Physiol. 2005, 289, G1036–G1042. [Google Scholar] [CrossRef] [PubMed]
- Ito, M.; Sakai, Y.; Furumoto, M.; Segawa, H.; Haito, S.; Yamanaka, S.; Nakamura, R.; Kuwahata, M.; Miyamoto, K. Vitamin D and phosphate regulate fibroblast growth factor-23 in K-562 cells. Am. J. Physiol. Endocrinol. Metab. 2005, 288, E1101–E1109. [Google Scholar] [CrossRef] [PubMed]
- Masuyama, R.; Stockmans, I.; Torrekens, S.; Van Looveren, R.; Maes, C.; Carmeliet, P.; Bouillon, R.; Carmeliet, G. Vitamin D receptor in chondrocytes promotes osteoclastogenesis and regulates FGF23 production in osteoblasts. J. Clin. Investig. 2006, 116, 3150–3159. [Google Scholar] [CrossRef]
- Nguyen-Yamamoto, L.; Karaplis, A.C.; St–Arnaud, R.; Goltzman, D. Fibroblast Growth Factor 23 Regulation by Systemic and Local Osteoblast-Synthesized 1,25-Dihydroxyvitamin D. J. Am. Soc. Nephrol. 2017, 28, 586–597. [Google Scholar] [CrossRef]
- Robinson-Cohen, C.; Bartz, T.M.; Lai, D.; Ikizler, T.A.; Peacock, M.; Imel, E.A.; Michos, E.D.; Foroud, T.M.; Akesson, K.; Taylor, K.D.; et al. Genetic Variants Associated with Circulating Fibroblast Growth Factor 23. J. Am. Soc. Nephrol. 2018, 29, 2583–2592. [Google Scholar] [CrossRef]
- Saini, R.K.; Kaneko, I.; Jurutka, P.W.; Forster, R.; Hsieh, A.; Hsieh, J.-C.; Haussler, M.R.; Whitfield, G.K. 1,25-Dihydroxyvitamin D3 Regulation of Fibroblast Growth Factor-23 Expression in Bone Cells: Evidence for Primary and Secondary Mechanisms Modulated by Leptin and Interleukin-6. Calcif. Tissue Int. 2013, 92, 339–353. [Google Scholar] [CrossRef]
- St John, H.C.; Bishop, K.A.; Meyer, M.B.; Benkusky, N.A.; Leng, N.; Kendziorski, C.; Bonewald, L.F.; Pike, J.W. The osteoblast to osteocyte transition: Epigenetic changes and response to the vitamin D3 hormone. Mol. Endocrinol. 2014, 28, 1150–1165. [Google Scholar] [CrossRef]
- Onal, M.; Carlson, A.H.; Thostenson, J.D.; Benkusky, N.A.; Meyer, M.B.; Lee, S.M.; Pike, J.W. A Novel Distal Enhancer Mediates Inflammation-, PTH-, and Early Onset Murine Kidney Disease-Induced Expression of the Mouse Fgf23 Gene. JBMR Plus 2018, 2, 32–47. [Google Scholar] [CrossRef] [PubMed]
- Lee, S.M.; Carlson, A.H.; Onal, M.; Benkusky, N.A.; Meyer, M.B.; Pike, J.W. A Control Region Near the Fibroblast Growth Factor 23 Gene Mediates Response to Phosphate, 1,25(OH)2D3, and LPS In Vivo. Endocrinology 2019, 160, 2877–2891. [Google Scholar] [CrossRef]
- Mattoo, R.L. The Roles of Fibroblast Growth Factor (FGF)-23, α-Klotho and Furin Protease in Calcium and Phosphate Homeostasis: A Mini-Review. Indian J. Clin. Biochem. 2014, 29, 8–12. [Google Scholar] [CrossRef]
- Knab, V.M.; Corbin, B.; Andrukhova, O.; Hum, J.M.; Ni, P.; Rabadi, S.; Maeda, A.; White, K.E.; Erben, R.G.; Juppner, H.; et al. Acute Parathyroid Hormone Injection Increases C-Terminal but Not Intact Fibroblast Growth Factor 23 Levels. Endocrinology 2017, 158, 1130–1139. [Google Scholar] [CrossRef] [PubMed]
- Andrukhova, O.; Smorodchenko, A.; Egerbacher, M.; Streicher, C.; Zeitz, U.; Goetz, R.; Shalhoub, V.; Mohammadi, M.; Pohl, E.E.; Lanske, B.; et al. FGF23 promotes renal calcium reabsorption through the TRPV5 channel. EMBO J. 2014, 33, 229–246. [Google Scholar] [CrossRef]
- Andrukhova, O.; Zeitz, U.; Goetz, R.; Mohammadi, M.; Lanske, B.; Erben, R.G. FGF23 acts directly on renal proximal tubules to induce phosphaturia through activation of the ERK1/2-SGK1 signaling pathway. Bone 2012, 51, 621–628. [Google Scholar] [CrossRef]
- Larsson, T.; Marsell, R.; Schipani, E.; Ohlsson, C.; Ljunggren, O.s.; Tenenhouse, H.S.; Jüppner, H.; Jonsson, K.B. Transgenic Mice Expressing Fibroblast Growth Factor 23 under the Control of the α1(I) Collagen Promoter Exhibit Growth Retardation, Osteomalacia, and Disturbed Phosphate Homeostasis. Endocrinology 2004, 145, 3087–3094. [Google Scholar] [CrossRef] [PubMed]
- Shimada, T.; Urakawa, I.; Yamazaki, Y.; Hasegawa, H.; Hino, R.; Yoneya, T.; Takeuchi, Y.; Fujita, T.; Fukumoto, S.; Yamashita, T. FGF-23 transgenic mice demonstrate hypophosphatemic rickets with reduced expression of sodium phosphate cotransporter type IIa. Biochem. Biophys. Res. Commun. 2004, 314, 409–414. [Google Scholar] [CrossRef]
- Shimada, T.; Hasegawa, H.; Yamazaki, Y.; Muto, T.; Hino, R.; Takeuchi, Y.; Fujita, T.; Nakahara, K.; Fukumoto, S.; Yamashita, T. FGF-23 Is a Potent Regulator of Vitamin D Metabolism and Phosphate Homeostasis. J. Bone Miner. Res. 2003, 19, 429–435. [Google Scholar] [CrossRef]
- Andrukhova, O.; Slavic, S.; Smorodchenko, A.; Zeitz, U.; Shalhoub, V.; Lanske, B.; Pohl, E.E.; Erben, R.G. FGF23 regulates renal sodium handling and blood pressure. EMBO Mol. Med. 2014, 6, 744–759. [Google Scholar] [CrossRef] [PubMed]
- Topaz, O.; Shurman, D.L.; Bergman, R.; Indelman, M.; Ratajczak, P.; Mizrachi, M.; Khamaysi, Z.; Behar, D.; Petronius, D.; Friedman, V.; et al. Mutations in GALNT3, encoding a protein involved in O-linked glycosylation, cause familial tumoral calcinosis. Nat. Genet. 2004, 36, 579–581. [Google Scholar] [CrossRef]
- Araya, K.; Fukumoto, S.; Backenroth, R.; Takeuchi, Y.; Nakayama, K.; Ito, N.; Yoshii, N.; Yamazaki, Y.; Yamashita, T.; Silver, J.; et al. A novel mutation in fibroblast growth factor 23 gene as a cause of tumoral calcinosis. J. Clin. Endocrinol. Metab. 2005, 90, 5523–5527. [Google Scholar] [CrossRef] [PubMed]
- Ichikawa, S.; Imel, E.A.; Kreiter, M.L.; Yu, X.; Mackenzie, D.S.; Sorenson, A.H.; Goetz, R.; Mohammadi, M.; White, K.E.; Econs, M.J. A homozygous missense mutation in human KLOTHO causes severe tumoral calcinosis. J. Clin. Investig. 2007, 117, 2684–2691. [Google Scholar] [CrossRef] [PubMed]
- Shimada, T.; Kakitani, M.; Yamazaki, Y.; Hasegawa, H.; Takeuchi, Y.; Fujita, T.; Fukumoto, S.; Tomizuka, K.; Yamashita, T. Targeted ablation of Fgf23 demonstrates an essential physiological role of FGF23 in phosphate and vitamin D metabolism. J. Clin. Investig. 2004, 113, 561–568. [Google Scholar] [CrossRef] [PubMed]
- Sitara, D.; Razzaque, M.S.; Hesse, M.; Yoganathan, S.; Taguchi, T.; Erben, R.G.; Juppner, H.; Lanske, B. Homozygous ablation of fibroblast growth factor-23 results in hyperphosphatemia and impaired skeletogenesis, and reverses hypophosphatemia in Phex-deficient mice. Matrix Biol. 2004, 23, 421–432. [Google Scholar] [CrossRef] [PubMed]
- Anour, R.; Andrukhova, O.; Ritter, E.; Zeitz, U.; Erben, R.G. Klotho lacks a vitamin D independent physiological role in glucose homeostasis, bone turnover, and steady-state PTH secretion in vivo. PLoS ONE 2012, 7, e31376. [Google Scholar] [CrossRef]
- Kuro-o, M.; Matsumura, Y.; Aizawa, H.; Kawaguchi, H.; Suga, T.; Utsugi, T.; Ohyama, Y.; Kurabayashi, M.; Kaname, T.; Kume, E.; et al. Mutation of the mouse klotho gene leads to a syndrome resembling ageing. Nature 1997, 390, 45–51. [Google Scholar] [CrossRef]
- Tsujikawa, H.; Kurotaki, Y.; Fujimori, T.; Fukuda, K.; Nabeshima, Y. Klotho, a gene related to a syndrome resembling human premature aging, functions in a negative regulatory circuit of vitamin D endocrine system. Mol. Endocrinol. 2003, 17, 2393–2403. [Google Scholar] [CrossRef]
- Yoshida, T.; Fujimori, T.; Nabeshima, Y. Mediation of unusually high concentrations of 1,25-dihydroxyvitamin D in homozygous klotho mutant mice by increased expression of renal 1alpha-hydroxylase gene. Endocrinology 2002, 143, 683–689. [Google Scholar] [CrossRef] [PubMed]
- Murali, S.K.; Roschger, P.; Zeitz, U.; Klaushofer, K.; Andrukhova, O.; Erben, R.G. FGF23 Regulates Bone Mineralization in a 1,25(OH)2 D3 and Klotho-Independent Manner. J. Bone Miner. Res. 2016, 31, 129–142. [Google Scholar] [CrossRef]
- Hesse, M.; Fröhlich, L.F.; Zeitz, U.; Lanske, B.; Erben, R.G. Ablation of vitamin D signaling rescues bone, mineral, and glucose homeostasis in Fgf-23 deficient mice. Matrix Biol. 2007, 26, 75–84. [Google Scholar] [CrossRef] [PubMed]
- Streicher, C.; Zeitz, U.; Andrukhova, O.; Rupprecht, A.; Pohl, E.; Larsson, T.E.; Windisch, W.; Lanske, B.; Erben, R.G. Long-term Fgf23 deficiency does not influence aging, glucose homeostasis, or fat metabolism in mice with a nonfunctioning vitamin D receptor. Endocrinology 2012, 153, 1795–1805. [Google Scholar] [CrossRef]
- Han, X.; Yang, J.; Li, L.; Huang, J.; King, G.; Quarles, L.D. Conditional Deletion of Fgfr1 in the Proximal and Distal Tubule Identifies Distinct Roles in Phosphate and Calcium Transport. PLoS ONE 2016, 11, e0147845. [Google Scholar] [CrossRef]
- Li, H.; Martin, A.; David, V.; Quarles, L.D. Compound deletion of Fgfr3 and Fgfr4 partially rescues the Hyp mouse phenotype. Am. J. Physiol. Endocrinol. Metab. 2011, 300, E508–E517. [Google Scholar] [CrossRef] [PubMed]
- Zhang, M.Y.; Ranch, D.; Pereira, R.C.; Armbrecht, H.J.; Portale, A.A.; Perwad, F. Chronic inhibition of ERK1/2 signaling improves disordered bone and mineral metabolism in hypophosphatemic (Hyp) mice. Endocrinology 2012, 153, 1806–1816. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Ranch, D.; Zhang, M.Y.; Portale, A.A.; Perwad, F. Fibroblast growth factor 23 regulates renal 1,25-dihydroxyvitamin D and phosphate metabolism via the MAP kinase signaling pathway in Hyp mice. J. Bone Miner. Res. 2011, 26, 1883–1890. [Google Scholar] [CrossRef]
- Shimada, T.; Yamazaki, Y.; Takahashi, M.; Hasegawa, H.; Urakawa, I.; Oshima, T.; Ono, K.; Kakitani, M.; Tomizuka, K.; Fujita, T.; et al. Vitamin D receptor-independent FGF23 actions in regulating phosphate and vitamin D metabolism. Am. J. Physiol. Renal Physiol. 2005, 289, F1088–F1095. [Google Scholar] [CrossRef]
- Erben, R.G. Fibroblast growth factor-23. In Encyclopedia of Signaling Molecules, 2nd ed.; Choi, S., Ed.; Springer: New York, NY, USA, 2018. [Google Scholar]
- Dorr, K.; Kammer, M.; Reindl-Schwaighofer, R.; Lorenz, M.; Prikoszovich, T.; Marculescu, R.; Beitzke, D.; Wielandner, A.; Erben, R.G.; Oberbauer, R. Randomized Trial of Etelcalcetide for Cardiac Hypertrophy in Hemodialysis. Circ. Res. 2021, 128, 1616–1625. [Google Scholar] [CrossRef]
- Scragg, R. Seasonality of cardiovascular disease mortality and the possible protective effect of ultra-violet radiation. Int. J. Epidemiol. 1981, 10, 337–341. [Google Scholar] [CrossRef]
- Latic, N.; Erben, R.G. Vitamin D and Cardiovascular Disease, with Emphasis on Hypertension, Atherosclerosis, and Heart Failure. Int. J. Mol. Sci. 2020, 21, 6483. [Google Scholar] [CrossRef] [PubMed]
- Walters, M.R.; Wicker, D.C.; Riggle, P.C. 1,25-Dihydroxyvitamin D<sub>3</sub> receptors identified in the rat heart. J. Mol. Cell. Cardiol. 1986, 18, 67–72. [Google Scholar] [CrossRef] [PubMed]
- Tishkoff, D.X.; Nibbelink, K.A.; Holmberg, K.H.; Dandu, L.; Simpson, R.U. Functional Vitamin D Receptor (VDR) in the T-Tubules of Cardiac Myocytes: VDR Knockout Cardiomyocyte Contractility. Endocrinology 2008, 149, 558–564. [Google Scholar] [CrossRef] [PubMed]
- Merke, J.; Milde, P.; Lewicka, S.; Hügel, U.; Klaus, G.; Mangelsdorf, D.J.; Haussler, M.R.; Rauterberg, E.W.; Ritz, E. Identification and regulation of 1,25-dihydroxyvitamin D3 receptor activity and biosynthesis of 1,25-dihydroxyvitamin D3. Studies in cultured bovine aortic endothelial cells and human dermal capillaries. J. Clin. Investig. 1989, 83, 1903–1915. [Google Scholar] [CrossRef] [PubMed]
- Al Mheid, I.; Patel, R.; Murrow, J.; Morris, A.; Rahman, A.; Fike, L.; Kavtaradze, N.; Uphoff, I.; Hooper, C.; Tangpricha, V.; et al. Vitamin D Status Is Associated With Arterial Stiffness and Vascular Dysfunction in Healthy Humans. J. Am. Coll. Cardiol. 2011, 58, 186–192. [Google Scholar] [CrossRef]
- Yiu, Y.-F.; Chan, Y.-H.; Yiu, K.-H.; Siu, C.-W.; Li, S.-W.; Wong, L.-Y.; Lee, S.W.L.; Tam, S.; Wong, E.W.K.; Cheung, B.M.Y.; et al. Vitamin D Deficiency Is Associated with Depletion of Circulating Endothelial Progenitor Cells and Endothelial Dysfunction in Patients with Type 2 Diabetes. J. Clin. Endocrinol. Metab. 2011, 96, E830–E835. [Google Scholar] [CrossRef]
- Lee, J.H.; Gadi, R.; Spertus, J.A.; Tang, F.; O’Keefe, J.H. Prevalence of Vitamin D Deficiency in Patients With Acute Myocardial Infarction. Am. J. Cardiol. 2011, 107, 1636–1638. [Google Scholar] [CrossRef]
- Holick, M.F. Vitamin D deficiency. N. Engl. J. Med. 2007, 357, 266–281. [Google Scholar] [CrossRef]
- Fountain, J.H.; Lappin, S.L. Physiology, Renin Angiotensin System. In StatPearls; StatPearls Publishing LLC: Treasure Island, FL, USA, 2022. [Google Scholar]
- Xanthakis, V.; Vasan, R.S. Aldosterone and the risk of hypertension. Curr. Hypertens. Rep. 2013, 15, 102–107. [Google Scholar] [CrossRef]
- Yuan, W.; Pan, W.; Kong, J.; Zheng, W.; Szeto, F.L.; Wong, K.E.; Cohen, R.; Klopot, A.; Zhang, Z.; Li, Y.C. 1,25-dihydroxyvitamin D3 suppresses renin gene transcription by blocking the activity of the cyclic AMP response element in the renin gene promoter. J. Biol. Chem. 2007, 282, 29821–29830. [Google Scholar] [CrossRef]
- Grundmann, S.M.; Schutkowski, A.; Schreier, B.; Rabe, S.; König, B.; Gekle, M.; Stangl, G.I. Vitamin D Receptor Deficiency Does Not Affect Blood Pressure and Heart Function. Front. Physiol. 2019, 10, 1118. [Google Scholar] [CrossRef]
- Jia, J.; Tao, X.; Tian, Z.; Liu, J.; Ye, X.; Zhan, Y. Vitamin D receptor deficiency increases systolic blood pressure by upregulating the renin-angiotensin system and autophagy. Exp. Ther. Med. 2022, 23, 314. [Google Scholar] [CrossRef] [PubMed]
- Zhou, C.; Lu, F.; Cao, K.; Xu, D.; Goltzman, D.; Miao, D. Calcium-independent and 1,25(OH)2D3-dependent regulation of the renin-angiotensin system in 1alpha-hydroxylase knockout mice. Kidney Int. 2008, 74, 170–179. [Google Scholar] [CrossRef] [PubMed]
- Andrukhova, O.; Slavic, S.; Zeitz, U.; Riesen, S.C.; Heppelmann, M.S.; Ambrisko, T.D.; Markovic, M.; Kuebler, W.M.; Erben, R.G. Vitamin D Is a Regulator of Endothelial Nitric Oxide Synthase and Arterial Stiffness in Mice. Mol. Endocrinol. 2014, 28, 53–64. [Google Scholar] [CrossRef] [PubMed]
- Ni, W.; Watts, S.W.; Ng, M.; Chen, S.; Glenn, D.J.; Gardner, D.G. Elimination of vitamin D receptor in vascular endothelial cells alters vascular function. Hypertension 2014, 64, 1290–1298. [Google Scholar] [CrossRef]
- Kota, S.K.; Kota, S.K.; Jammula, S.; Meher, L.K.; Panda, S.; Tripathy, P.R.; Modi, K.D. Renin-angiotensin system activity in vitamin D deficient, obese individuals with hypertension: An urban Indian study. Indian J. Endocrinol. Metab. 2011, 15 (Suppl. 4), S395–S401. [Google Scholar] [CrossRef] [PubMed]
- Resnick, L.M.; MÜLler, F.B.; Laragh, J.H. Calcium-Regulating Hormones in Essential Hypertension. Ann. Intern. Med. 1986, 105, 649–654. [Google Scholar] [CrossRef]
- Forman, J.P.; Williams, J.S.; Fisher, N.D. Plasma 25-hydroxyvitamin D and regulation of the renin-angiotensin system in humans. Hypertension 2010, 55, 1283–1288. [Google Scholar] [CrossRef]
- Vaidya, A.; Forman, J.P.; Hopkins, P.N.; Seely, E.W.; Williams, J.S. 25-Hydroxyvitamin D is associated with plasma renin activity and the pressor response to dietary sodium intake in Caucasians. J. Renin-Angiotensin-Aldosterone Syst. 2011, 12, 311–319. [Google Scholar] [CrossRef]
- Vaidya, A.; Sun, B.; Larson, C.; Forman, J.P.; Williams, J.S. Vitamin D3 therapy corrects the tissue sensitivity to angiotensin ii akin to the action of a converting enzyme inhibitor in obese hypertensives: An interventional study. J. Clin. Endocrinol. Metab. 2012, 97, 2456–2465. [Google Scholar] [CrossRef]
- McMullan, C.J.; Borgi, L.; Curhan, G.C.; Fisher, N.; Forman, J.P. The effect of vitamin D on renin-angiotensin system activation and blood pressure: A randomized control trial. J. Hypertens. 2017, 35, 822–829. [Google Scholar] [CrossRef] [PubMed]
- Manson, J.E.; Cook, N.R.; Lee, I.M.; Christen, W.; Bassuk, S.S.; Mora, S.; Gibson, H.; Gordon, D.; Copeland, T.; D’Agostino, D.; et al. Vitamin D Supplements and Prevention of Cancer and Cardiovascular Disease. N. Engl. J. Med. 2019, 380, 33–44. [Google Scholar] [CrossRef] [PubMed]
- Scragg, R.K.R. Overview of results from the Vitamin D Assessment (ViDA) study. J. Endocrinol. Investig. 2019, 42, 1391–1399. [Google Scholar] [CrossRef] [PubMed]
- Jiang, F.; Yang, J.; Zhang, Y.; Dong, M.; Wang, S.; Zhang, Q.; Liu, F.F.; Zhang, K.; Zhang, C. Angiotensin-converting enzyme 2 and angiotensin 1–7: Novel therapeutic targets. Nat. Rev. Cardiol. 2014, 11, 413–426. [Google Scholar] [CrossRef] [PubMed]
- Pagliaro, P.; Penna, C. ACE/ACE2 Ratio: A Key Also in 2019 Coronavirus Disease (Covid-19)? Front. Med. 2020, 7, 335. [Google Scholar] [CrossRef] [PubMed]
- Getachew, B.; Tizabi, Y. Vitamin D and COVID-19: Role of ACE2, age, gender, and ethnicity. J. Med. Virol. 2021, 93, 5285–5294. [Google Scholar] [CrossRef] [PubMed]
- Úri, K.; Fagyas, M.; Mányiné Siket, I.; Kertész, A.; Csanádi, Z.; Sándorfi, G.; Clemens, M.; Fedor, R.; Papp, Z.; Édes, I.; et al. New perspectives in the renin-angiotensin-aldosterone system (RAAS) IV: Circulating ACE2 as a biomarker of systolic dysfunction in human hypertension and heart failure. PLoS ONE 2014, 9, e87845. [Google Scholar] [CrossRef] [PubMed]
- Yang, J.; XU, J.; Zhang, H. Effect of Vitamin D on ACE2 and Vitamin D receptor expression in rats with LPS-induced acute lung injury. Chin. J. Emerg. Med. 2016, 25, 1284–1289. [Google Scholar] [CrossRef]
- Xu, J.; Yang, J.; Chen, J.; Luo, Q.; Zhang, Q.; Zhang, H. Vitamin D alleviates lipopolysaccharide-induced acute lung injury via regulation of the renin-angiotensin system. Mol. Med. Rep. 2017, 16, 7432–7438. [Google Scholar] [CrossRef] [PubMed]
- Kong, J.; Zhu, X.; Shi, Y.; Liu, T.; Chen, Y.; Bhan, I.; Zhao, Q.; Thadhani, R.; Li, Y.C. VDR Attenuates Acute Lung Injury by Blocking Ang-2-Tie-2 Pathway and Renin-Angiotensin System. Mol. Endocrinol. 2013, 27, 2116–2125. [Google Scholar] [CrossRef]
- Lin, M.; Gao, P.; Zhao, T.; He, L.; Li, M.; Li, Y.; Shui, H.; Wu, X. Calcitriol regulates angiotensin-converting enzyme and angiotensin converting-enzyme 2 in diabetic kidney disease. Mol. Biol. Rep. 2016, 43, 397–406. [Google Scholar] [CrossRef]
- Riera, M.; Anguiano, L.; Clotet, S.; Roca-Ho, H.; Rebull, M.; Pascual, J.; Soler, M.J. Paricalcitol modulates ACE2 shedding and renal ADAM17 in NOD mice beyond proteinuria. Am. J. Physiol.-Ren. Physiol. 2016, 310, F534–F546. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Q.; Lin, S.; Tang, R.; Veeraragoo, P.; Peng, W.; Wu, R. Role of Fosinopril and Valsartan on Klotho Gene Expression Induced by Angiotensin II in Rat Renal Tubular Epithelial Cells. Kidney Blood Press. Res. 2010, 33, 186–192. [Google Scholar] [CrossRef] [PubMed]
- Yoon, H.E.; Ghee, J.Y.; Piao, S.; Song, J.H.; Han, D.H.; Kim, S.; Ohashi, N.; Kobori, H.; Kuro-o, M.; Yang, C.W. Angiotensin II blockade upregulates the expression of Klotho, the anti-ageing gene, in an experimental model of chronic cyclosporine nephropathy. Nephrol. Dial. Transplant. 2011, 26, 800–813. [Google Scholar] [CrossRef] [PubMed]
- Saito, K.; Ishizaka, N.; Mitani, H.; Ohno, M.; Nagai, R. Iron chelation and a free radical scavenger suppress angiotensin II-induced downregulation of klotho, an anti-aging gene, in rat. FEBS Lett. 2003, 551, 58–62. [Google Scholar] [CrossRef]
- Mitani, H.; Ishizaka, N.; Aizawa, T.; Ohno, M.; Usui, S.-i.; Suzuki, T.; Amaki, T.; Mori, I.; Nakamura, Y.; Sato, M. In vivo klotho gene transfer ameliorates angiotensin II-induced renal damage. Hypertension 2002, 39, 838–843. [Google Scholar] [CrossRef]
- Mitobe, M.; Yoshida, T.; Sugiura, H.; Shirota, S.; Tsuchiya, K.; Nihei, H. Oxidative stress decreases klotho expression in a mouse kidney cell line. Nephron Exp. Nephrol. 2005, 101, e67–e74. [Google Scholar] [CrossRef]
- Sachse, A.; Wolf, G. Angiotensin II–induced reactive oxygen species and the kidney. J. Am. Soc. Nephrol. 2007, 18, 2439–2446. [Google Scholar] [CrossRef]
- Zuo, Z.; Lei, H.; Wang, X.; Wang, Y.; Sonntag, W.; Sun, Z. Aging-related kidney damage is associated with a decrease in klotho expression and an increase in superoxide production. Age 2011, 33, 261–274. [Google Scholar] [CrossRef][Green Version]
- Griendling, K.K.; Minieri, C.A.; Ollerenshaw, J.D.; Alexander, R.W. Angiotensin II stimulates NADH and NADPH oxidase activity in cultured vascular smooth muscle cells. Circ. Res. 1994, 74, 1141–1148. [Google Scholar] [CrossRef]
- Doran, D.E.; Weiss, D.; Zhang, Y.; Griendling, K.K.; Taylor, W.R. Differential effects of AT1 receptor and Ca2+ channel blockade on atherosclerosis, inflammatory gene expression, and production of reactive oxygen species. Atherosclerosis 2007, 195, 39–47. [Google Scholar] [CrossRef][Green Version]
- Dusso, A.; Arcidiacono, M.V.; Yang, J.; Tokumoto, M. Vitamin D inhibition of TACE and prevention of renal osteodystrophy and cardiovascular mortality. J. Steroid Biochem. Mol. Biol. 2010, 121, 193–198. [Google Scholar] [CrossRef]
- Chen, C.-D.; Podvin, S.; Gillespie, E.; Leeman, S.E.; Abraham, C.R. Insulin stimulates the cleavage and release of the extracellular domain of Klotho by ADAM10 and ADAM17. Proc. Natl. Acad. Sci. USA 2007, 104, 19796–19801. [Google Scholar] [CrossRef]
- Lautrette, A.; Li, S.; Alili, R.; Sunnarborg, S.W.; Burtin, M.; Lee, D.C.; Friedlander, G.; Terzi, F. Angiotensin II and EGF receptor cross-talk in chronic kidney diseases: A new therapeutic approach. Nat. Med. 2005, 11, 867–874. [Google Scholar] [CrossRef] [PubMed]
- Grund, A.; Sinha, M.D.; Haffner, D.; Leifheit-Nestler, M. Fibroblast Growth Factor 23 and Left Ventricular Hypertrophy in Chronic Kidney Disease-A Pediatric Perspective. Front. Pediatr. 2021, 9, 702719. [Google Scholar] [CrossRef] [PubMed]
- Christodoulou, M.; Aspray, T.J.; Schoenmakers, I. Vitamin D Supplementation for Patients with Chronic Kidney Disease: A Systematic Review and Meta-analyses of Trials Investigating the Response to Supplementation and an Overview of Guidelines. Calcif. Tissue Int. 2021, 109, 157–178. [Google Scholar] [CrossRef] [PubMed]


Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Latic, N.; Erben, R.G. Interaction of Vitamin D with Peptide Hormones with Emphasis on Parathyroid Hormone, FGF23, and the Renin-Angiotensin-Aldosterone System. Nutrients 2022, 14, 5186. https://doi.org/10.3390/nu14235186
Latic N, Erben RG. Interaction of Vitamin D with Peptide Hormones with Emphasis on Parathyroid Hormone, FGF23, and the Renin-Angiotensin-Aldosterone System. Nutrients. 2022; 14(23):5186. https://doi.org/10.3390/nu14235186
Chicago/Turabian StyleLatic, Nejla, and Reinhold G. Erben. 2022. "Interaction of Vitamin D with Peptide Hormones with Emphasis on Parathyroid Hormone, FGF23, and the Renin-Angiotensin-Aldosterone System" Nutrients 14, no. 23: 5186. https://doi.org/10.3390/nu14235186
APA StyleLatic, N., & Erben, R. G. (2022). Interaction of Vitamin D with Peptide Hormones with Emphasis on Parathyroid Hormone, FGF23, and the Renin-Angiotensin-Aldosterone System. Nutrients, 14(23), 5186. https://doi.org/10.3390/nu14235186