Prenatal Low-Protein Diet Affects Mitochondrial Structure and Function in the Skeletal Muscle of Adult Female Offspring

 and
and 
Abstract
:1. Introduction
2. Materials and Methods
2.1. Animals
2.2. Transmission Electron Microscopy (TEM)
2.3. Mitochondrial DNA Copy Number
2.4. Quantitative Real-Time qPCR
2.5. Mitochondrial Oxygen Consumption
2.6. Western Blot
2.7. Statistical Analysis
3. Results
3.1. Controls and LP Rats Had Similar Weights at 4 Months and Feed Intake
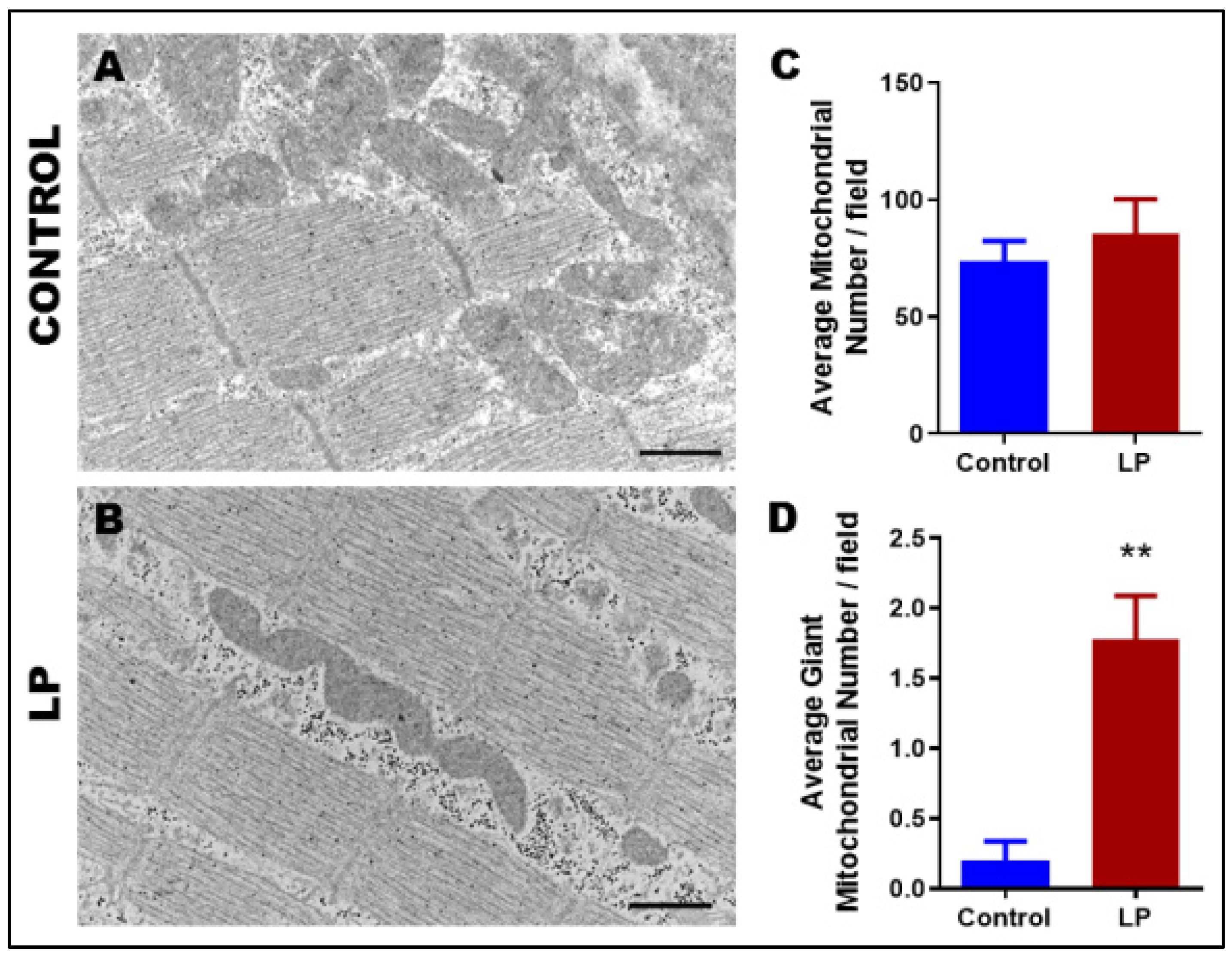
3.2. Maternal LP Diet Alters the Mitochondrial Morphology and Ultrastructure
3.3. LP Programming Did Not Alter mtDNA Copy Number in Gastrocnemius Muscles
3.4. LP Programming Reduced Oxygen Consumption in Skeletal Muscles
3.5. LP Programming Reduced the Expression of Mitochondrial Complex I
3.6. LP Programming Dysregulated Mitochondrial Dynamics and Biogenesis Genes
4. Discussion
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Hales, C.N.; Barker, D.J.P. Type 2 (non-insulin-dependent) diabetes mellitus: The thrifty phenotype hypothesis. Diabetologia 1992, 35, 595–601. [Google Scholar] [CrossRef] [PubMed]
- Chmurzynska, A. Fetal programming: Link between early nutrition, DNA methylation, and complex diseases. Nutr. Rev. 2010, 68, 87–98. [Google Scholar] [CrossRef] [PubMed]
- de Rooij, S.R.; Roseboom, T.J.; Painter, R.C. Famines in the Last 100 Years: Implications for Diabetes. Curr. Diabetes Rep. 2014, 14, 536. [Google Scholar] [CrossRef]
- Roseboom, T.J.; Van Der Meulen, J.H.P.; Ravelli, A.C.; Osmond, C.; Barker, D.J.P.; Bleker, O.P. Perceived health of adults after prenatal exposure to the Dutch famine. Paediatr. Périnat. Epidemiol. 2003, 17, 391–397. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; He, Y.; Qi, L.; Jaddoe, V.W.; Feskens, E.J.; Yang, X.; Ma, G.; Hu, F.B. Exposure to the Chinese Famine in Early Life and the Risk of Hyperglycemia and Type 2 Diabetes in Adulthood. Diabetes 2010, 59, 2400–2406. [Google Scholar] [CrossRef] [Green Version]
- Jia, Y.; Li, R.; Cong, R.; Yang, X.; Sun, Q.; Parvizi, N.; Zhao, R. Maternal Low-Protein Diet Affects Epigenetic Regulation of Hepatic Mitochondrial DNA Transcription in a Sex-Specific Manner in Newborn Piglets Associated with GR Binding to Its Promoter. PLoS ONE 2013, 8, e63855. [Google Scholar] [CrossRef] [Green Version]
- Zhu, M.J.; Ford, S.P.; Means, W.J.; Hess, B.W.; Nathanielsz, P.; Du, M. Maternal nutrient restriction affects properties of skeletal muscle in offspring. J. Physiol. 2006, 575, 241–250. [Google Scholar] [CrossRef]
- Jansson, N.; Pettersson, J.; Haafiz, A.; Ericsson, A.; Palmberg, I.; Tranberg, M.; Ganapathy, V.; Powell, T.L.; Jansson, T. Down-regulation of placental transport of amino acids precedes the development of intrauterine growth restriction in rats fed a low protein diet. J. Physiol. 2006, 576, 935–946. [Google Scholar] [CrossRef]
- Kelley, D.E.; Simoneau, J.A. Impaired free fatty acid utilization by skeletal muscle in non-insulin-dependent diabetes mellitus. J. Clin. Investig. 1994, 94, 2349–2356. [Google Scholar] [CrossRef]
- Kelley, D.E.; Mintun, M.A.; Watkins, S.; Simoneau, J.A.; Jadali, F.; Fredrickson, A.; Beattie, J.; Theriault, R. The effect of non-insulin-dependent diabetes mellitus and obesity on glucose transport and phosphorylation in skeletal muscle. J. Clin. Investig. 1996, 97, 2705–2713. [Google Scholar] [CrossRef]
- Mootha, V.K.; Lindgren, C.M.; Eriksson, K.-F.; Subramanian, A.; Sihag, S.; Lehar, J.; Puigserver, P.; Carlsson, E.; Ridderstråle, M.; Laurila, E.; et al. PGC-1α-responsive genes involved in oxidative phosphorylation are coordinately downregulated in human diabetes. Nat. Genet. 2003, 34, 267–273. [Google Scholar] [CrossRef] [PubMed]
- Shou, J.; Chen, P.-J.; Xiao, W.-H. Mechanism of increased risk of insulin resistance in aging skeletal muscle. Diabetol. Metab. Syndr. 2020, 12, 14. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shulman, G.I. Cellular mechanisms of insulin resistance. J. Clin. Investig. 2000, 106, 171–176. [Google Scholar] [CrossRef] [PubMed]
- Shulman, G.; Rothman, D.L.; Jue, T.; Stein, P.; DeFronzo, R.A.; Shulman, R.G. Quantitation of Muscle Glycogen Synthesis in Normal Subjects and Subjects with Non-Insulin-Dependent Diabetes by13C Nuclear Magnetic Resonance Spectroscopy. N. Engl. J. Med. 1990, 322, 223–228. [Google Scholar] [CrossRef] [PubMed]
- Hoeks, J.; Schrauwen, P. Muscle mitochondria and insulin resistance: A human perspective. Trends Endocrinol. Metab. 2012, 23, 444–450. [Google Scholar] [CrossRef] [PubMed]
- Wigmore, P.M.; Stickland, N.C. Muscle development in large and small pig fetuses. J. Anat. 1983, 137, 235–245. [Google Scholar] [PubMed]
- Patel, H.P.; Jameson, K.A.; Syddall, H.E.; Martin, H.J.; Stewart, C.; Sayer, A.A.; Cooper, C. Developmental Influences, Muscle Morphology, and Sarcopenia in Community-Dwelling Older Men. J. Gerontol. Ser. A 2011, 67, 82–87. [Google Scholar] [CrossRef] [Green Version]
- Confortim, H.D.; Jerônimo, L.C.; Centenaro, L.A.; Pinheiro, P.F.F.; Brancalhão, R.M.C.; Matheus, S.M.M.; Torrejais, M.M. Effects of aging and maternal protein restriction on the muscle fibers morphology and neuromuscular junctions of rats after nutritional recovery. Micron 2015, 71, 7–13. [Google Scholar] [CrossRef]
- Jousse, C.; Muranishi, Y.; Parry, L.; Montaurier, C.; Even, P.; Launay, J.-M.; Carraro, V.; Maurin, A.-C.; Averous, J.; Chaveroux, C.; et al. Perinatal Protein Malnutrition Affects Mitochondrial Function in Adult and Results in a Resistance to High Fat Diet-Induced Obesity. PLoS ONE 2014, 9, e104896. [Google Scholar] [CrossRef] [Green Version]
- Oreffo, R.O.; Lashbrooke, B.; Roach, H.I.; Clarke, N.M.; Cooper, C. Maternal protein deficiency affects mesenchymal stem cell activity in the developing offspring. Bone 2003, 33, 100–107. [Google Scholar] [CrossRef]
- Claycombe, K.J.; Roemmich, J.N.; Johnson, L.; Vomhof-DeKrey, E.E.; Johnson, W.T. Skeletal muscle Sirt3 expression and mitochondrial respiration are regulated by a prenatal low-protein diet. J. Nutr. Biochem. 2015, 26, 184–189. [Google Scholar] [CrossRef] [PubMed]
- Park, K.S.; Kim, S.K.; Kim, M.S.; Cho, E.Y.; Lee, J.H.; Lee, K.-U.; Pak, Y.; Lee, H.K. Fetal and Early Postnatal Protein Malnutrition Cause Long-Term Changes in Rat Liver and Muscle Mitochondria. J. Nutr. 2003, 133, 3085–3090. [Google Scholar] [CrossRef] [Green Version]
- Mortensen, O.H.; Olsen, H.L.; Frandsen, L.; Nielsen, P.E.; Nielsen, F.C.; Grunnet, N.; Quistorff, B. A maternal low protein diet has pronounced effects on mitochondrial gene expression in offspring liver and skeletal muscle; protective effect of taurine. J. Biomed. Sci. 2010, 17, S38. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zisman, A.; Peroni, O.D.; Abel, E.D.; Michael, M.D.; Mauvais-Jarvis, F.; Lowell, B.B.; Wojtaszewski, J.F.P.; Hirshman, M.F.; Virkamaki, A.; Goodyear, L.J.; et al. Targeted disruption of the glucose transporter 4 selectively in muscle causes insulin resistance and glucose intolerance. Nat. Med. 2000, 6, 924–928. [Google Scholar] [CrossRef] [PubMed]
- Blesson, C.S.; Schutt, A.K.; Balakrishnan, M.P.; Pautler, R.G.; Pedersen, S.E.; Sarkar, P.; Gonzales, D.; Zhu, G.; Marini, J.C.; Chacko, S.K.; et al. Novel lean type 2 diabetic rat model using gestational low-protein programming. Am. J. Obstet. Gynecol. 2016, 214, 540.e1–540.e7. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Blesson, C.S.; Chinnathambi, V.; Kumar, S.; Yallampalli, C. Gestational Protein Restriction Impairs Glucose Disposal in the Gastrocnemius Muscles of Female Rats. Endocrinology 2017, 158, 756–767. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Blesson, C.S.; Kumar, S.; Chinnathambi, V.; Yallampalli, C. Gestational Protein Restriction Impairs Insulin-Regulated Glucose Transport Mechanisms in Gastrocnemius Muscles of Adult Male Offspring. Endocrinology 2014, 155, 3036–3046. [Google Scholar] [CrossRef]
- Blesson, C.S.; Schutt, A.; Mathew, P.R.; Tanchico, D.; Balakrishnan, M.; Yallampalli, U.; Yallampalli, C. Folate treatment partially reverses gestational low-protein diet-induced glucose intolerance and the magnitude of reversal is age and sex dependent. Nutrition 2017, 49, 81–89. [Google Scholar] [CrossRef]
- Blesson, C.S.; Schutt, A.; Chacko, S.; Marini, J.C.; Mathew, P.R.; Tanchico, D.; Balakrishnan, M.; Yallampalli, C. Sex Dependent Dysregulation of Hepatic Glucose Production in Lean Type 2 Diabetic Rats. Front. Endocrinol. 2019, 10, 538. [Google Scholar] [CrossRef] [Green Version]
- Blesson, C.S.; Schutt, A.; Chacko, S.; Marini, J.; Balakrishanan, M.; Yallampalli, C. Sex Dependent Dysregulation of Hepatic Glucose Production in Gestational Low Protein Programmed Insulin Resistant Rat Offspring. In Reproductive Sciences; Sage Publications: Thousand Oaks, CA, USA, 2016; p. 233A. [Google Scholar]
- Chappell, N.R.; Zhou, B.; Schutt, A.K.; Gibbons, W.E.; Blesson, C.S. Prenatal androgen induced lean PCOS impairs mitochondria and mRNA profiles in oocytes. Endocr. Connect. 2020, 9, 261–270. [Google Scholar] [CrossRef] [Green Version]
- Chappell, N.R.; Zhou, B.; Hosseinzadeh, P.; Schutt, A.; Gibbons, W.E.; Blesson, C.S. Hyperandrogenemia alters mitochondrial structure and function in the oocytes of obese mouse with polycystic ovary syndrome. F&S Sci. 2021, 2, 101–112. [Google Scholar] [CrossRef]
- Schuh, R.A.; Jackson, K.C.; Khairallah, R.J.; Ward, C.W.; Spangenburg, E.E. Measuring mitochondrial respiration in intact single muscle fibers. Am. J. Physiol. Integr. Comp. Physiol. 2012, 302, R712–R719. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Blesson, C.S.; Schutt, A.K.; Vipin, V.A.; Tanchico, D.T.; Mathew, P.R.; Balakrishnan, M.; Bethancourt, A.; Yallampalli, C. In utero low-protein-diet-programmed type 2 diabetes in adult offspring is mediated by sex hormones in rats. Biol. Reprod. 2020, 103, 1110–1120. [Google Scholar] [CrossRef] [PubMed]
- Sivitz, W.I.; Yorek, M.A. Mitochondrial Dysfunction in Diabetes: From Molecular Mechanisms to Functional Significance and Therapeutic Opportunities. Antioxid. Redox Signal. 2010, 12, 537–577. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ong, S.-B.; Kalkhoran, S.B.; Cabrera-Fuentes, H.A.; Hausenloy, D.J. Mitochondrial fusion and fission proteins as novel therapeutic targets for treating cardiovascular disease. Eur. J. Pharmacol. 2015, 763, 104–114. [Google Scholar] [CrossRef] [Green Version]
- Szendroedi, J.; Phielix, E.; Roden, M. The role of mitochondria in insulin resistance and type 2 diabetes mellitus. Nat. Rev. Endocrinol. 2011, 8, 92–103. [Google Scholar] [CrossRef]
- Crescenzo, R.; Bianco, F.; Mazzoli, A.; Giacco, A.; Liverini, G.; Iossa, S. Mitochondrial efficiency and insulin resistance. Front. Physiol. 2015, 5, 512. [Google Scholar] [CrossRef] [Green Version]
- Ritov, V.B.; Menshikova, E.V.; Azuma, K.; Wood, R.; Toledo, F.; Goodpaster, B.H.; Ruderman, N.; Kelley, D.E. Deficiency of electron transport chain in human skeletal muscle mitochondria in type 2 diabetes mellitus and obesity. Am. J. Physiol. Metab. 2010, 298, E49–E58. [Google Scholar] [CrossRef] [Green Version]
- Patti, M.E.; Butte, A.J.; Crunkhorn, S.; Cusi, K.; Berria, R.; Kashyap, S.; Miyazaki, Y.; Kohane, I.; Costello, M.; Saccone, R.; et al. Coordinated reduction of genes of oxidative metabolism in humans with insulin resistance and diabetes: Potential role of PGC1 and NRF1. Proc. Natl. Acad. Sci. USA 2003, 100, 8466–8471. [Google Scholar] [CrossRef] [Green Version]
- Zorzano, A.; Hernández-Alvarez, M.I.; Palacín, M.; Mingrone, G. Alterations in the mitochondrial regulatory pathways constituted by the nuclear co-factors PGC-1α or PGC-1β and mitofusin 2 in skeletal muscle in type 2 diabetes. Biochim. Et Biophys. Acta 2010, 1797, 1028–1033. [Google Scholar] [CrossRef] [Green Version]
- Kim, J.; Wei, Y.; Sowers, J.R. Role of Mitochondrial Dysfunction in Insulin Resistance. Circ. Res. 2008, 102, 401–414. [Google Scholar] [CrossRef] [PubMed]
- Houzelle, A.; Jörgensen, J.A.; Schaart, G.; Daemen, S.; van Polanen, N.; Fealy, C.E.; Hesselink, M.K.C.; Schrauwen, P.; Hoeks, J. Human skeletal muscle mitochondrial dynamics in relation to oxidative capacity and insulin sensitivity. Diabetologia 2020, 64, 424–436. [Google Scholar] [CrossRef] [PubMed]
- Fealy, C.E.; Mulya, A.; Axelrod, C.L.; Kirwan, J.P. Mitochondrial dynamics in skeletal muscle insulin resistance and type 2 diabetes. Transl. Res. 2018, 202, 69–82. [Google Scholar] [CrossRef] [PubMed]
- Agnoux, A.M.; Antignac, J.-P.; Simard, G.; Poupeau, G.; Darmaun, D.; Parnet, P.; Alexandre-Gouabau, M.-C. Time window-dependent effect of perinatal maternal protein restriction on insulin sensitivity and energy substrate oxidation in adult male offspring. Am. J. Physiol. Integr. Comp. Physiol. 2014, 307, R184–R197. [Google Scholar] [CrossRef] [Green Version]
- Taylor, P.D.; McConnell, J.; Khan, I.Y.; Holemans, K.; Lawrence, K.M.; Asare-Anane, H.; Persaud, S.J.; Jones, P.M.; Petrie, L.; Hanson, M.A.; et al. Impaired glucose homeostasis and mitochondrial abnormalities in offspring of rats fed a fat-rich diet in pregnancy. Am. J. Physiol. Integr. Comp. Physiol. 2005, 288, R134–R139. [Google Scholar] [CrossRef]
- Latouche, C.; Heywood, S.E.; Henry, S.L.; Ziemann, M.; Lazarus, R.; El-Osta, A.; Armitage, J.A.; Kingwell, B.A. Maternal Overnutrition Programs Changes in the Expression of Skeletal Muscle Genes That Are Associated with Insulin Resistance and Defects of Oxidative Phosphorylation in Adult Male Rat Offspring. J. Nutr. 2013, 144, 237–244. [Google Scholar] [CrossRef] [Green Version]
- Schutt, A.K.; Blesson, C.S.; Hsu, J.W.; Valdes, C.T.; Gibbons, W.E.; Jahoor, F.; Yallampalli, C. Preovulatory exposure to a protein-restricted diet disrupts amino acid kinetics and alters mitochondrial structure and function in the rat oocyte and is partially rescued by folic acid. Reprod. Biol. Endocrinol. 2019, 17, 12. [Google Scholar] [CrossRef]
- Magwere, T.; Goodall, S.; Skepper, J.; Mair, W.; Brand, M.; Partridge, L. The Effect of Dietary Restriction on Mitochondrial Protein Density and Flight Muscle Mitochondrial Morphology in Drosophila. J. Gerontol. Ser. A 2006, 61, 36–47. [Google Scholar] [CrossRef]
- Rebelato, H.J.; Esquisatto, M.A.M.; Moraes, C.; Amaral, M.E.C.; Catisti, R. Gestational protein restriction induces alterations in placental morphology and mitochondrial function in rats during late pregnancy. Histochem. J. 2013, 44, 629–637. [Google Scholar] [CrossRef]
- Zorzano, A.; Liesa, M.; Palacín, M. Mitochondrial dynamics as a bridge between mitochondrial dysfunction and insulin resistance. Arch. Physiol. Biochem. 2009, 115, 1–12. [Google Scholar] [CrossRef]
- Mogensen, M.; Sahlin, K. Mitochondrial efficiency in rat skeletal muscle: Influence of respiration rate, substrate and muscle type. Acta Physiol. Scand. 2005, 185, 229–236. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schwerzmann, K.; Hoppeler, H.; Kayar, S.R.; Weibel, E.R. Oxidative capacity of muscle and mitochondria: Correlation of physiological, biochemical, and morphometric characteristics. Proc. Natl. Acad. Sci. USA 1989, 86, 1583–1587. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Glancy, B.; Balaban, R.S. Protein composition and function of red and white skeletal muscle mitochondria. Am. J. Physiol.-Cell Physiol. 2011, 300, C1280–C1290. [Google Scholar] [CrossRef] [PubMed]
- Leary, S.C.; Lyons, C.N.; Rosenberger, A.G.; Ballantyne, J.S.; Stillman, J.; Moyes, C.D. Fiber-type differences in muscle mitochondrial profiles. Am. J. Physiol. Integr. Comp. Physiol. 2003, 285, R817–R826. [Google Scholar] [CrossRef]
- Yadava, N.; Nicholls, D.G. Spare Respiratory Capacity Rather Than Oxidative Stress Regulates Glutamate Excitotoxicity after Partial Respiratory Inhibition of Mitochondrial Complex I with Rotenone. J. Neurosci. 2007, 27, 7310–7317. [Google Scholar] [CrossRef] [Green Version]
- Mogensen, M.; Sahlin, K.; Fernström, M.; Glintborg, D.; Vind, B.F.; Beck-Nielsen, H.; Højlund, K. Mitochondrial respiration is decreased in skeletal muscle of patients with type 2 diabetes. Diabetes 2007, 56, 1592–1599. [Google Scholar] [CrossRef] [Green Version]
- Phielix, E.; Schrauwen-Hinderling, V.B.; Mensink, M.; Lenaers, E.; Meex, R.; Hoeks, J.; Kooi, M.E.; Moonen-Kornips, E.; Sels, J.-P.; Hesselink, M.K.; et al. Lower Intrinsic ADP-Stimulated Mitochondrial Respiration Underlies In Vivo Mitochondrial Dysfunction in Muscle of Male Type 2 Diabetic Patients. Diabetes 2008, 57, 2943–2949. [Google Scholar] [CrossRef] [Green Version]
- Fern, R. Variations in spare electron transport chain capacity: The answer to an old riddle? J. Neurosci. Res. 2003, 71, 759–762. [Google Scholar] [CrossRef]
- Yamamoto, H.; Morino, K.; Mengistu, L.; Ishibashi, T.; Kiriyama, K.; Ikami, T.; Maegawa, H. Amla Enhances Mitochondrial Spare Respiratory Capacity by Increasing Mitochondrial Biogenesis and Antioxidant Systems in a Murine Skeletal Muscle Cell Line. Oxidative Med. Cell. Longev. 2016, 2016, 1735841. [Google Scholar] [CrossRef] [Green Version]
- Fiedorczuk, K.; Letts, J.; Degliesposti, G.; Kaszuba, K.; Skehel, G.D.M.; Sazanov, L.A. Atomic structure of the entire mammalian mitochondrial complex I. Nature 2016, 538, 406–410. [Google Scholar] [CrossRef] [Green Version]
- Fassone, E.; Rahman, S. Complex I deficiency: Clinical features, biochemistry and molecular genetics. J. Med. Genet. 2012, 49, 578–590. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Acin-Perez, R.; Silva, P.F.; Peleato, M.L.; Pérez-Martos, A.; Enriquez, J.A. Respiratory Active Mitochondrial Supercomplexes. Mol. Cell 2008, 32, 529–539. [Google Scholar] [CrossRef] [PubMed]
- Antoun, G.; McMurray, F.; Thrush, A.B.; Patten, D.A.; Peixoto, A.C.; Slack, R.; McPherson, R.; Dent, R.; Harper, M.-E. Impaired mitochondrial oxidative phosphorylation and supercomplex assembly in rectus abdominis muscle of diabetic obese individuals. Diabetologia 2015, 58, 2861–2866. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Frezza, C.; Cipolat, S.; De Brito, O.M.; Micaroni, M.; Beznoussenko, G.V.; Rudka, T.; Bartoli, D.; Polishuck, R.S.; Danial, N.N.; De Strooper, B.; et al. OPA1 Controls Apoptotic Cristae Remodeling Independently from Mitochondrial Fusion. Cell 2006, 126, 177–189. [Google Scholar] [CrossRef] [Green Version]
- Parra, V.; Verdejo, H.E.; Iglewski, M.; del Campo, A.; Troncoso, R.; Jones, D.; Zhu, Y.; Kuzmicic, J.; Pennanen, C.; Lopez-Crisosto, C.; et al. Insulin Stimulates Mitochondrial Fusion and Function in Cardiomyocytes via the Akt-mTOR-NFκB-Opa-1 Signaling Pathway. Diabetes 2013, 63, 75–88. [Google Scholar] [CrossRef] [Green Version]
- Eura, Y.; Ishihara, N.; Yokota, S.; Mihara, K. Two Mitofusin Proteins, Mammalian Homologues of FZO, with Distinct Functions Are Both Required for Mitochondrial Fusion. J. Biochem. 2003, 134, 333–344. [Google Scholar] [CrossRef]
- Bach, D.; Pich, S.; Soriano, F.X.; Vega, N.; Baumgartner, B.; Oriola, J.; Daugaard, J.R.; Lloberas, J.; Camps, M.; Zierath, J.R.; et al. Mitofusin-2 Determines Mitochondrial Network Architecture and Mitochondrial Metabolism: A novel regulatory mechanism altered in obesity. J. Biol. Chem. 2003, 278, 17190–17197. [Google Scholar] [CrossRef] [Green Version]
- Sebastián, D.; Hernández-Alvarez, M.I.; Segalés, J.; Sorianello, E.; Muñoz, J.P.; Sala, D.; Waget, A.; Liesa, M.; Paz, J.C.; Gopalacharyulu, P.; et al. Mitofusin 2 (Mfn2) links mitochondrial and endoplasmic reticulum function with insulin signaling and is essential for normal glucose homeostasis. Proc. Natl. Acad. Sci. USA 2012, 109, 5523–5528. [Google Scholar] [CrossRef] [Green Version]
- Hernandez-Alvarez, M.I.; Thabit, H.; Burns, N.; Shah, S.; Brema, I.; Hatunic, M.; Finucane, F.; Liesa, M.; Chiellini, C.; Naon, D.; et al. Subjects with Early-Onset Type 2 Diabetes Show Defective Activation of the Skeletal Muscle PGC-1 /Mitofusin-2 Regulatory Pathway in Response to Physical Activity. Diabetes Care 2009, 33, 645–651. [Google Scholar] [CrossRef] [Green Version]
- Kong, D.; Nie, Q.; Zhang, X.; Gan, K.; Song, G.; Wang, C.; Ma, H.; Ren, L. Overexpression of mitofusin 2 improves translocation of glucose transporter 4 in skeletal muscle of high-fat diet-fed rats through AMP-activated protein kinase signaling. Mol. Med. Rep. 2013, 8, 205–210. [Google Scholar] [CrossRef] [Green Version]
- Yu, R.; Jin, S.; Lendahl, U.; Nistér, M.; Zhao, J. Human Fis1 regulates mitochondrial dynamics through inhibition of the fusion machinery. EMBO J. 2019, 38, e99748. [Google Scholar] [CrossRef] [PubMed]
- Tilokani, L.; Nagashima, S.; Paupe, V.; Prudent, J. Mitochondrial dynamics: Overview of molecular mechanisms. Essays Biochem. 2018, 62, 341–360. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lin, J.; Tarr, P.T.; Yang, R.; Rhee, J.; Puigserver, P.; Newgard, C.B.; Spiegelman, B.M. PGC-1β in the Regulation of Hepatic Glucose and Energy Metabolism. J. Biol. Chem. 2003, 278, 30843–30848. [Google Scholar] [CrossRef] [Green Version]
- Lelliott, C.J.; Medina-Gómez, G.; Petrović, N.; Kis, A.; Feldmann, H.M.; Bjursell, M.; Parker, N.; Curtis, K.; Campbell, M.; Hu, P.; et al. Ablation of PGC-1β Results in Defective Mitochondrial Activity, Thermogenesis, Hepatic Function, and Cardiac Performance. PLoS Biol. 2006, 4, e369. [Google Scholar] [CrossRef] [PubMed]
- Wright, L.E.; Brandon, A.E.; Hoy, A.J.; Forsberg, G.-B.; Lelliott, C.J.; Reznick, J.; Löfgren, L.; Oscarsson, J.; Strömstedt, M.; Cooney, G.J.; et al. Amelioration of lipid-induced insulin resistance in rat skeletal muscle by overexpression of Pgc-1β involves reductions in long-chain acyl-CoA levels and oxidative stress. Diabetologia 2011, 54, 1417–1426. [Google Scholar] [CrossRef] [Green Version]
- Villena, J.A.; Kralli, A. ERRα: A metabolic function for the oldest orphan. Trends Endocrinol. Metab. 2008, 19, 269–276. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gleyzer, N.; Vercauteren, K.; Scarpulla, R.C. Control of Mitochondrial Transcription Specificity Factors (TFB1M and TFB2M) by Nuclear Respiratory Factors (NRF-1 and NRF-2) and PGC-1 Family Coactivators. Mol. Cell. Biol. 2005, 25, 1354–1366. [Google Scholar] [CrossRef] [Green Version]
- Uruno, A.; Yagishita, Y.; Katsuoka, F.; Kitajima, Y.; Nunomiya, A.; Nagatomi, R.; Pi, J.; Biswal, S.S.; Yamamoto, M. Nrf2-Mediated Regulation of Skeletal Muscle Glycogen Metabolism. Mol. Cell. Biol. 2016, 36, 1655–1672. [Google Scholar] [CrossRef] [Green Version]
- Santos, J.M.; Tewari, S.; Benite-Ribeiro, S.A. The effect of exercise on epigenetic modifications of PGC1: The impact on type 2 diabetes. Med. Hypotheses 2014, 82, 748–753. [Google Scholar] [CrossRef]
- Xie, X.; Lin, T.; Zhang, M.; Liao, L.; Yuan, G.; Gao, H.; Ning, Q.; Luo, X. IUGR with infantile overnutrition programs an insulin-resistant phenotype through DNA methylation of peroxisome proliferator–activated receptor-γ coactivator-1α in rats. Pediatr. Res. 2015, 77, 625–632. [Google Scholar] [CrossRef] [Green Version]
- Barres, R.; Osler, M.E.; Yan, J.; Rune, A.; Fritz, T.; Caidahl, K.; Krook, A.; Zierath, J.R. Non-CpG Methylation of the PGC-1α Promoter through DNMT3B Controls Mitochondrial Density. Cell Metab. 2009, 10, 189–198. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- St-Pierre, J.; Lin, J.; Krauss, S.; Tarr, P.T.; Yang, R.; Newgard, C.B.; Spiegelman, B.M. Bioenergetic Analysis of Peroxisome Proliferator-activated Receptor γ Coactivators 1α and 1β (PGC-1α and PGC-1β) in Muscle Cells. J. Biol. Chem. 2003, 278, 26597–26603. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Staiger, H.; Stefan, N.; Machicao, F.; Fritsche, A.; Häring, H.-U. PPARGC1A mRNA levels of in vitro differentiated human skeletal muscle cells are negatively associated with the plasma oleate concentrations of the donors. Diabetologia 2005, 49, 212–214. [Google Scholar] [CrossRef] [PubMed] [Green Version]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Gene | Primer (F = Forward; R = Reverse) |
|---|---|
| mtCox1 | F: 5′- ATCGCAATTCCTACAGGCGT-3′ R: 5′-TGTTAGGCCCCCTACTGTGA-3′ |
| mtCox2 | F: 5′-CAAGACGCCACATCACCTATC-3′ R: 5′-TTGGGCGTCTATTGTGCTTG-3′ |
| mtCox3 | F: 5′-GGAACATACCAAGGCCACCA-3′ R: 5′-TCGTGGGTAGGAACTAGGCT-3′ |
| Esrra | F: 5′- AAAGTCCTGGCCCATTTCTATG-3′ R: 5′-CCCTTGCCTCAGTCCATCAT-3′ |
| CoxIVa | F: 5′-CAAGGGCACCAATAGGTGGA-3′ R: 5′-GATGGGGCCATACACCTAGC-3′ |
| CoxIVb | F: 5′-CGTCTTCAGCTTGCAACTATGT-3′ R: 5′-ACATAGGGGGTCATCCTCCG-3′ |
| Cyc A | F: 5′-TATCTGCACTGCCAAGACTGAGTG-3′ R: 5′-CTTCTTGCTGGTCTTGCCATTCC-3′ |
| Fis1 | F: 5′-GTGCCTGGTTCGAAGCAAATA-3′ R: 5′-CATATTCCCGCTGCTCCTCTT-3′ |
| Mfn1 | F: 5′-ATCTTCGGCCAGTTACTGGAGTT-3′ R: 5′-AGATCATCCTCGGTTGCTATCC-3′ |
| Mfn2 | F: 5′-CCTTGAAGACACCCACAGGAATA-3′ R: 5′-CGCTGATTCCCCTGACCTT-3′ |
| Nrf1 | F: 5′-CTCTGCATCTCACCCTCCAAAC-3′ R: 5′-TCTTCCAGGATCATGCTCTTGTAC-3′ |
| Nrf2 | F: 5′-CATTTGTAGATGACCATGAGTCGC-3′ R: 5′-GAGCTATCGAGTGACTGAGCC-3′ |
| Opa1 | F: 5′-AAAAGCCCTTCCCAGTTCAGA-3′ R: 5′-TACCCGCAGTGAAGAAATCCTT-3′ |
| Pgc1a | F: 5′-CTACAATGAATGCAGCGGTCTT-3′ R: 5′-TGCTCCATGAATTCTCGGTCTT-3′ |
| Pgc1b | F: 5′-TCGGTGAAGGTCGTGTGGTATAC-3′ R: 5′-GCACTCGACTATCTCACCAAACA-3′ |
| Sirt1 | F: 5′-CTGTTTCCTGTGGGATACCTGACT-3′ R: 5′-ATCGAACATGGCTTGAGGATCT-3′ |
| Vdac1 | F: 5′-GTCACCGCCTCCGAGACCAT-3′ R: 5′-CCAATCCATTCTCGGACTTCGT-3′ |
| Tuba1a | F: 5′-ATGGTCTTGTCGCTTGGCAT-3′ R: 5′-CCCCTTTCCACAGCGTGAGT-3′ |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Vidyadharan, V.A.; Betancourt, A.; Smith, C.; Yallampalli, C.; Blesson, C.S. Prenatal Low-Protein Diet Affects Mitochondrial Structure and Function in the Skeletal Muscle of Adult Female Offspring. Nutrients 2022, 14, 1158. https://doi.org/10.3390/nu14061158
Vidyadharan VA, Betancourt A, Smith C, Yallampalli C, Blesson CS. Prenatal Low-Protein Diet Affects Mitochondrial Structure and Function in the Skeletal Muscle of Adult Female Offspring. Nutrients. 2022; 14(6):1158. https://doi.org/10.3390/nu14061158
Chicago/Turabian StyleVidyadharan, Vipin A., Ancizar Betancourt, Craig Smith, Chandrasekhar Yallampalli, and Chellakkan S. Blesson. 2022. "Prenatal Low-Protein Diet Affects Mitochondrial Structure and Function in the Skeletal Muscle of Adult Female Offspring" Nutrients 14, no. 6: 1158. https://doi.org/10.3390/nu14061158