
Effect of Exercise and Oral Niacinamide Mononucleotide on Improving Mitochondrial Autophagy in Alzheimer’s Disease

 , ,
, , {kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Oral NMN and Exercise Enhance NAD+ In Vivo
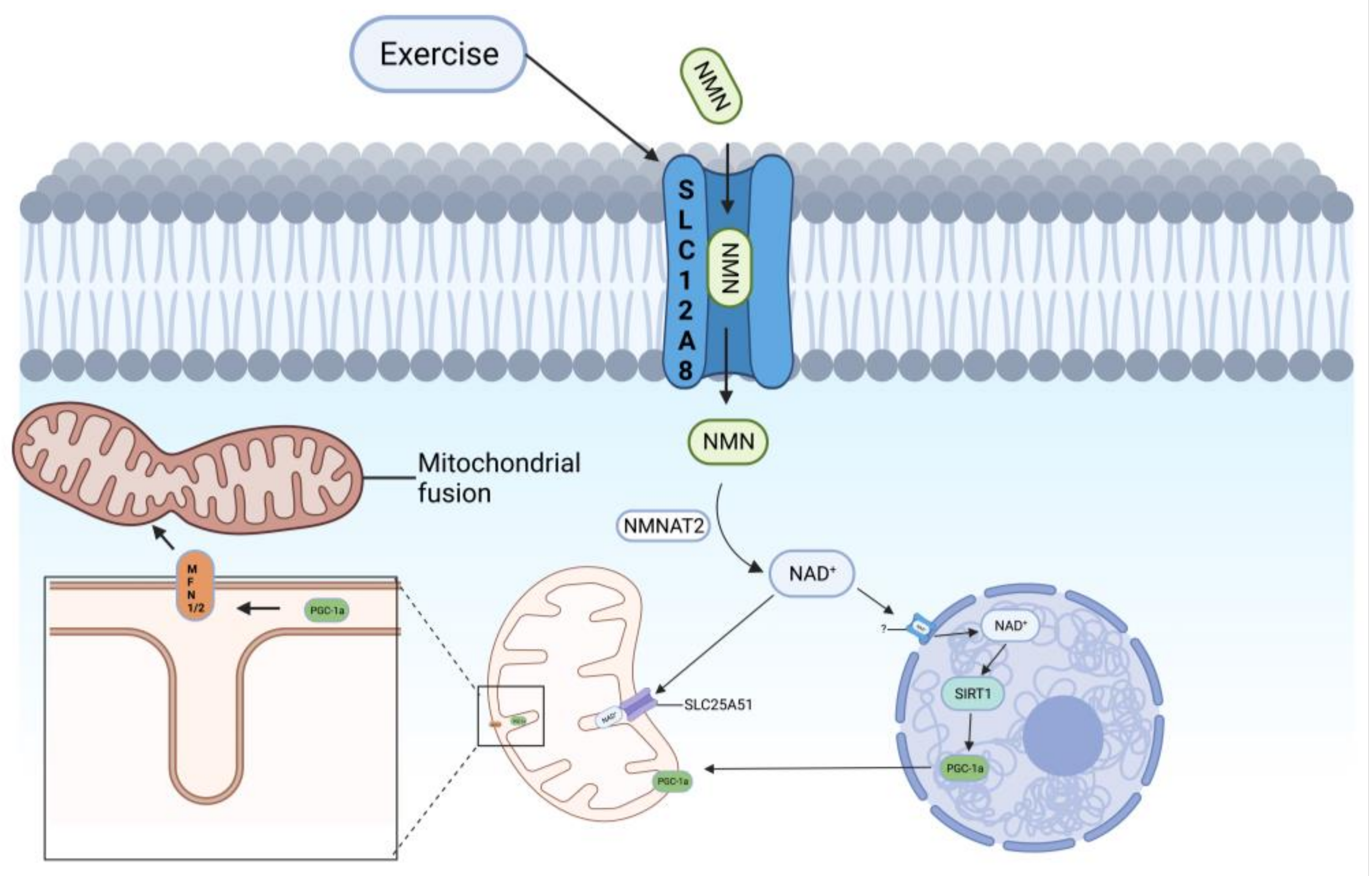
2.1. Pathway of NAD+ Synthesis by NMN
2.2. Oral NMN Can Improve the Level of NAD+ in All Tissues
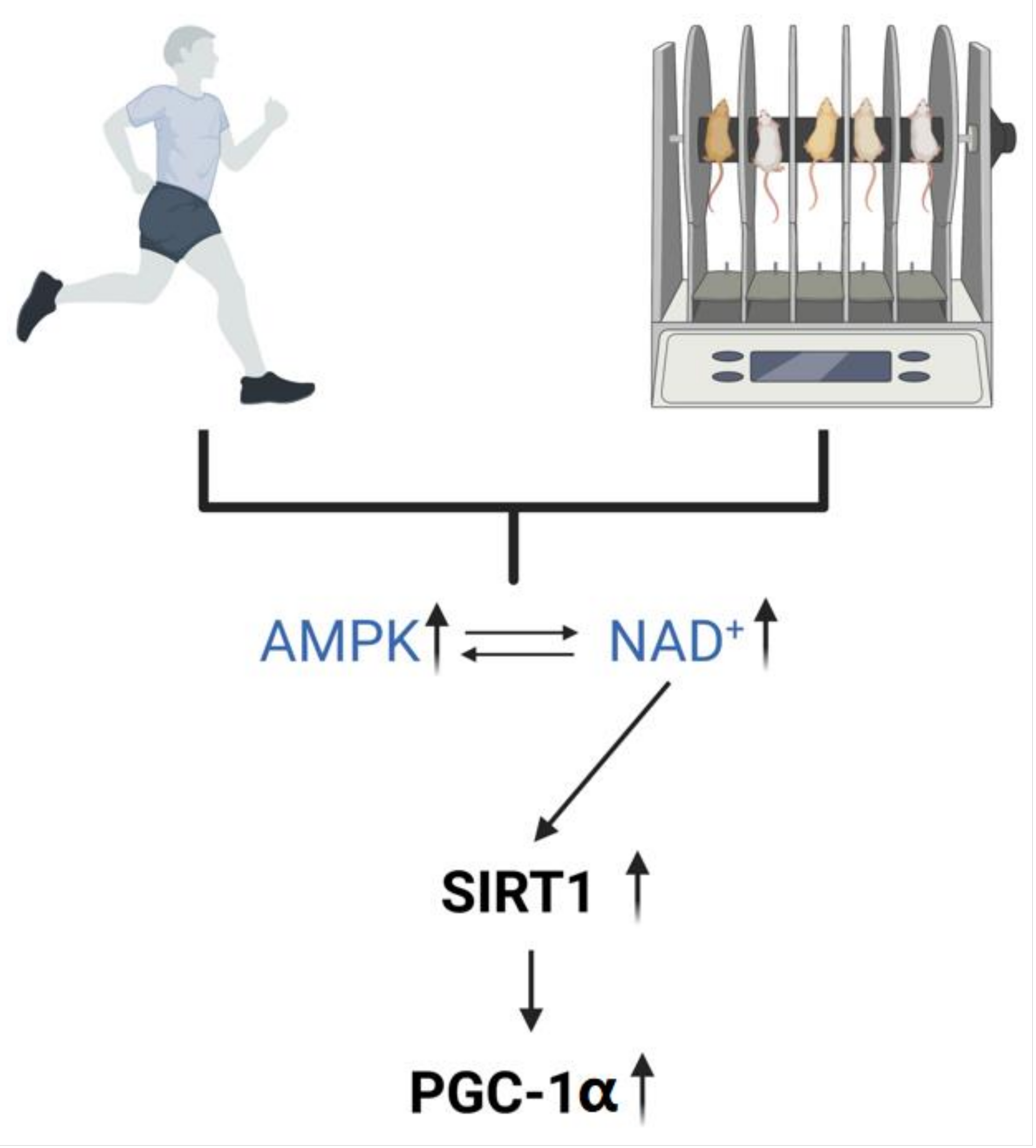
2.3. Exercise Regulates the Level of NAD+ In Vivo
3. NAD+ Ameliorates Abnormal Mitochondrial Autophagy
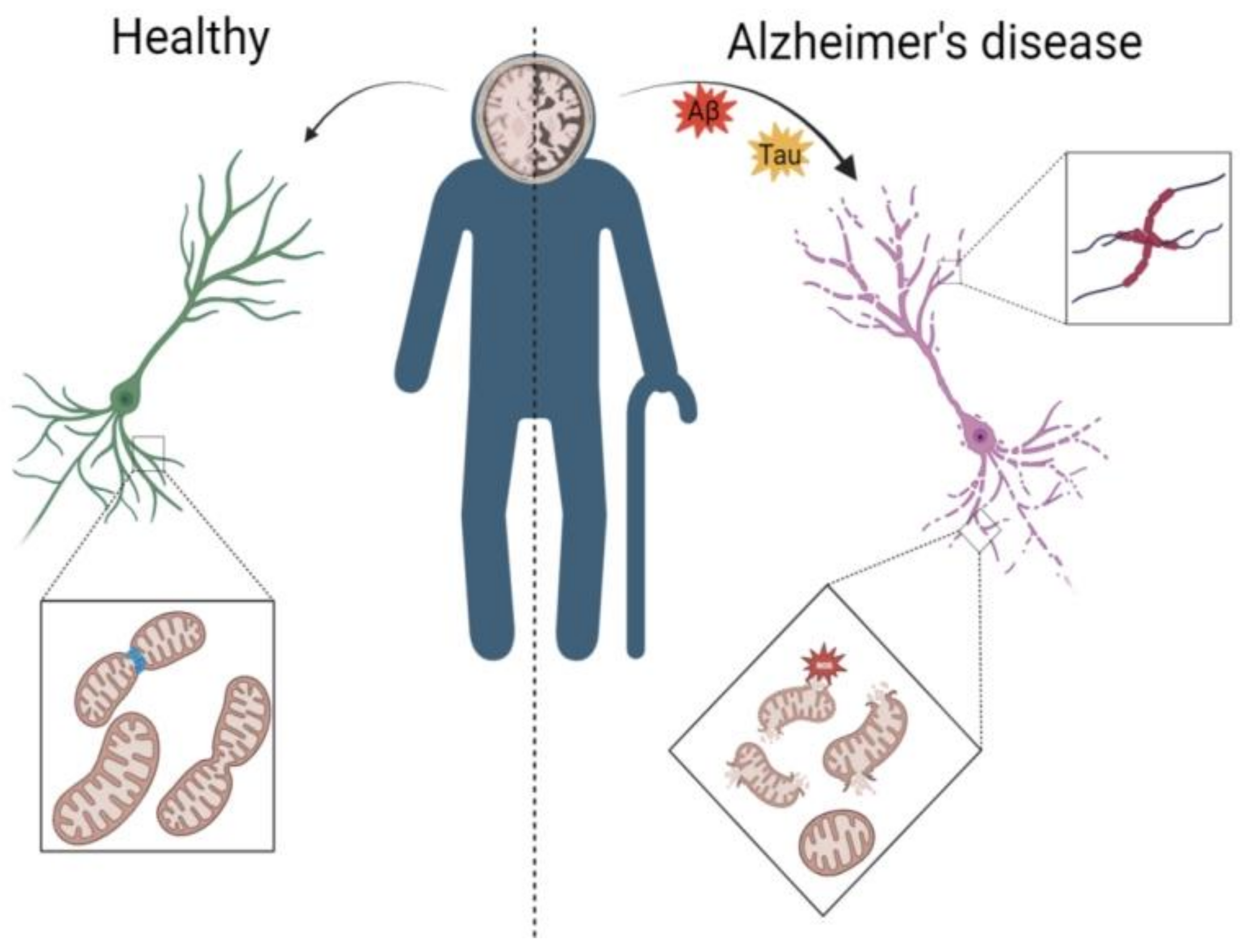
3.1. AD Leads to Abnormal Mitochondrial Autophagy in the Brain
3.2. Mechanisms by Which NAD+ Ameliorates Abnormal Mitochondrial Autophagy in AD
3.3. Exercise Ameliorates Abnormal Mitochondrial Autophagy in AD
4. Potential Mechanisms for Upregulation of NAD+ to Improve Mitochondrial Autophagy
4.1. SLC12A8—An NMN Transporter Protein on the Cell Membrane
4.2. SLC25A51—An NAD+ Transporter Protein on Mitochondria
4.3. SLC12A8 and SLC25A51 May Be Potential Therapeutic Targets for Improving Mitochondrial Autophagy
5. Summary and Outlook
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Jack, C.R., Jr. Advances in Alzheimer’s disease research over the past two decades. Lancet Neurol. 2022, 21, 866–869. [Google Scholar] [CrossRef]
- Swerdlow, R.H.; Khan, S.M. A “mitochondrial cascade hypothesis” for sporadic Alzheimer’s disease. Med. Hypotheses 2004, 63, 8–20. [Google Scholar] [CrossRef]
- Duyckaerts, C.; Delatour, B.; Potier, M.C. Classification and basic pathology of Alzheimer disease. Acta Neuropathol. 2009, 118, 5–36. [Google Scholar] [CrossRef]
- Manczak, M.; Kandimalla, R.; Yin, X.; Reddy, P.H. Hippocampal mutant APP and amyloid beta-induced cognitive decline, dendritic spine loss, defective autophagy, mitophagy and mitochondrial abnormalities in a mouse model of Alzheimer’s disease. Hum. Mol. Genet. 2018, 27, 1332–1342. [Google Scholar] [CrossRef] [Green Version]
- Cheignon, C.; Tomas, M.; Bonnefont-Rousselot, D.; Faller, P.; Hureau, C.; Collin, F. Oxidative stress and the amyloid beta peptide in Alzheimer’s disease. Redox Biol. 2018, 14, 450–464. [Google Scholar] [CrossRef] [PubMed]
- Ghavami, S.; Shojaei, S.; Yeganeh, B.; Ande, S.R.; Jangamreddy, J.R.; Mehrpour, M.; Christoffersson, J.; Chaabane, W.; Moghadam, A.R.; Kashani, H.H.; et al. Autophagy and apoptosis dysfunction in neurodegenerative disorders. Prog. Neurobiol. 2014, 112, 24–49. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bieganowski, P.; Brenner, C. Discoveries of nicotinamide riboside as a nutrient and conserved NRK genes establish a Preiss-Handler independent route to NAD+ in fungi and humans. Cell 2004, 117, 495–502. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ramanathan, C.; Lackie, T.; Williams, D.H.; Simone, P.S.; Zhang, Y.; Bloomer, R.J. Oral Administration of Nicotinamide Mononucleotide Increases Nicotinamide Adenine Dinucleotide Level in an Animal Brain. Nutrients 2022, 14, 300. [Google Scholar] [CrossRef]
- Chini, C.C.S.; Tarragó, M.G.; Chini, E.N. NAD and the aging process: Role in life, death and everything in between. Mol. Cell. Endocrinol. 2017, 455, 62–74. [Google Scholar] [CrossRef]
- Migliavacca, E.; Tay, S.K.H.; Patel, H.P.; Sonntag, T.; Civiletto, G.; McFarlane, C.; Forrester, T.; Barton, S.J.; Leow, M.K.; Antoun, E.; et al. Mitochondrial oxidative capacity and NAD(+) biosynthesis are reduced in human sarcopenia across ethnicities. Nat. Commun. 2019, 10, 5808. [Google Scholar] [CrossRef] [Green Version]
- Covarrubias, A.J.; Perrone, R.; Grozio, A.; Verdin, E. NAD(+) metabolism and its roles in cellular processes during ageing. Nat. Rev. Mol. Cell Biol. 2021, 22, 119–141. [Google Scholar] [CrossRef] [PubMed]
- Klimova, N.; Kristian, T. Multi-targeted Effect of Nicotinamide Mononucleotide on Brain Bioenergetic Metabolism. Neurochem. Res. 2019, 44, 2280–2287. [Google Scholar] [CrossRef] [PubMed]
- Klimova, N.; Fearnow, A.; Long, A.; Kristian, T. NAD(+) precursor modulates post-ischemic mitochondrial fragmentation and reactive oxygen species generation via SIRT3 dependent mechanisms. Exp. Neurol. 2020, 325, 113144. [Google Scholar] [CrossRef] [PubMed]
- Fang, E.F.; Hou, Y.; Palikaras, K.; Adriaanse, B.A.; Kerr, J.S.; Yang, B.; Lautrup, S.; Hasan-Olive, M.M.; Caponio, D.; Dan, X.; et al. Mitophagy inhibits amyloid-β and tau pathology and reverses cognitive deficits in models of Alzheimer’s disease. Nat. Neurosci. 2019, 22, 401–412. [Google Scholar] [CrossRef]
- Fang, E.F. Mitophagy and NAD(+) inhibit Alzheimer disease. Autophagy 2019, 15, 1112–1114. [Google Scholar] [CrossRef] [Green Version]
- De la Rosa, A.; Olaso-Gonzalez, G.; Arc-Chagnaud, C.; Millan, F.; Salvador-Pascual, A.; García-Lucerga, C.; Blasco-Lafarga, C.; Garcia-Dominguez, E.; Carretero, A.; Correas, A.G.; et al. Physical exercise in the prevention and treatment of Alzheimer’s disease. J. Sport Health Sci. 2020, 9, 394–404. [Google Scholar] [CrossRef]
- Chong, M.C.; Silva, A.; James, P.F.; Wu, S.S.X.; Howitt, J. Exercise increases the release of NAMPT in extracellular vesicles and alters NAD(+) activity in recipient cells. Aging Cell 2022, 21, e13647. [Google Scholar] [CrossRef]
- Ryu, K.W.; Nandu, T.; Kim, J.; Challa, S.; DeBerardinis, R.J.; Kraus, W.L. Metabolic regulation of transcription through compartmentalized NAD(+) biosynthesis. Science 2018, 360, eaan5780. [Google Scholar] [CrossRef] [Green Version]
- Grozio, A.; Mills, K.F.; Yoshino, J.; Bruzzone, S.; Sociali, G.; Tokizane, K.; Lei, H.C.; Cunningham, R.; Sasaki, Y.; Migaud, M.E.; et al. Slc12a8 is a nicotinamide mononucleotide transporter. Nat Metab. 2019, 1, 47–57. [Google Scholar]
- Kory, N.; Uit de Bos, J.; van der Rijt, S.; Jankovic, N.; Güra, M.; Arp, N.; Pena, I.A.; Prakash, G.; Chan, S.H.; Kunchok, T.; et al. MCART1/SLC25A51 is required for mitochondrial NAD transport. Sci. Adv. 2020, 6, abe5310. [Google Scholar] [CrossRef]
- Wang, T.; Zhang, F.; Peng, W.; Wang, L.; Zhang, J.; Dong, W.; Tian, X.; Ye, C.; Li, Y.; Gong, Y. Overexpression of NMNAT3 improves mitochondrial function and enhances antioxidative stress capacity of bone marrow mesenchymal stem cells via the NAD+-Sirt3 pathway. Biosci. Rep. 2022, 42, BSR20211005. [Google Scholar] [CrossRef]
- Irie, J.; Inagaki, E.; Fujita, M.; Nakaya, H.; Mitsuishi, M.; Yamaguchi, S.; Yamashita, K.; Shigaki, S.; Ono, T.; Yukioka, H.; et al. Effect of oral administration of nicotinamide mononucleotide on clinical parameters and nicotinamide metabolite levels in healthy Japanese men. Endocr. J. 2020, 67, 153–160. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Long, A.N.; Owens, K.; Schlappal, A.E.; Kristian, T.; Fishman, P.S.; Schuh, R.A. Effect of nicotinamide mononucleotide on brain mitochondrial respiratory deficits in an Alzheimer’s disease-relevant murine model. BMC Neurol. 2015, 15, 19. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mills, K.F.; Yoshida, S.; Stein, L.R.; Grozio, A.; Kubota, S.; Sasaki, Y.; Redpath, P.; Migaud, M.E.; Apte, R.S.; Uchida, K.; et al. Long-Term Administration of Nicotinamide Mononucleotide Mitigates Age-Associated Physiological Decline in Mice. Cell Metab. 2016, 24, 795–806. [Google Scholar] [CrossRef] [Green Version]
- Yoshino, J.; Mills, K.F.; Yoon, M.J.; Imai, S. Nicotinamide mononucleotide, a key NAD(+) intermediate, treats the pathophysiology of diet- and age-induced diabetes in mice. Cell Metab. 2011, 14, 528–536. [Google Scholar] [CrossRef] [Green Version]
- Peek, C.B.; Affinati, A.H.; Ramsey, K.M.; Kuo, H.Y.; Yu, W.; Sena, L.A.; Ilkayeva, O.; Marcheva, B.; Kobayashi, Y.; Omura, C.; et al. Circadian clock NAD+ cycle drives mitochondrial oxidative metabolism in mice. Science 2013, 342, 1243417. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stromsdorfer, K.L.; Yamaguchi, S.; Yoon, M.J.; Moseley, A.C.; Franczyk, M.P.; Kelly, S.C.; Qi, N.; Imai, S.; Yoshino, J. NAMPT-Mediated NAD(+) Biosynthesis in Adipocytes Regulates Adipose Tissue Function and Multi-organ Insulin Sensitivity in Mice. Cell Rep. 2016, 16, 1851–1860. [Google Scholar] [CrossRef] [Green Version]
- Karamanlidis, G.; Lee, C.F.; Garcia-Menendez, L.; Kolwicz, S.C., Jr.; Suthammarak, W.; Gong, G.; Sedensky, M.M.; Morgan, P.G.; Wang, W.; Tian, R. Mitochondrial complex I deficiency increases protein acetylation and accelerates heart failure. Cell Metab. 2013, 18, 239–250. [Google Scholar] [CrossRef] [Green Version]
- Martin, A.S.; Abraham, D.M.; Hershberger, K.A.; Bhatt, D.P.; Mao, L.; Cui, H.; Liu, J.; Liu, X.; Muehlbauer, M.J.; Grimsrud, P.A.; et al. Nicotinamide mononucleotide requires SIRT3 to improve cardiac function and bioenergetics in a Friedreich’s ataxia cardiomyopathy model. JCI Insight 2017, 2, e93885. [Google Scholar] [CrossRef] [Green Version]
- North, B.J.; Rosenberg, M.A.; Jeganathan, K.B.; Hafner, A.V.; Michan, S.; Dai, J.; Baker, D.J.; Cen, Y.; Wu, L.E.; Sauve, A.A.; et al. SIRT2 induces the checkpoint kinase BubR1 to increase lifespan. EMBO J. 2014, 33, 1438–1453. [Google Scholar] [CrossRef] [Green Version]
- Yamamoto, T.; Byun, J.; Zhai, P.; Ikeda, Y.; Oka, S.; Sadoshima, J. Nicotinamide mononucleotide, an intermediate of NAD+ synthesis, protects the heart from ischemia and reperfusion. PLoS ONE 2014, 9, e98972. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pirinen, E.; Auranen, M.; Khan, N.A.; Brilhante, V.; Urho, N.; Pessia, A.; Hakkarainen, A.; Kuula, J.; Heinonen, U.; Schmidt, M.S.; et al. Niacin Cures Systemic NAD(+) Deficiency and Improves Muscle Performance in Adult-Onset Mitochondrial Myopathy. Cell Metab. 2020, 31, 1078–1090.e1075. [Google Scholar] [CrossRef]
- Guan, Y.; Wang, S.R.; Huang, X.Z.; Xie, Q.H.; Xu, Y.Y.; Shang, D.; Hao, C.M. Nicotinamide Mononucleotide, an NAD(+) Precursor, Rescues Age-Associated Susceptibility to AKI in a Sirtuin 1-Dependent Manner. J. Am. Soc. Nephrol. JASN 2017, 28, 2337–2352. [Google Scholar] [CrossRef] [Green Version]
- Stein, L.R.; Imai, S. Specific ablation of Nampt in adult neural stem cells recapitulates their functional defects during aging. EMBO J. 2014, 33, 1321–1340. [Google Scholar] [CrossRef] [Green Version]
- Yoon, M.J.; Yoshida, M.; Johnson, S.; Takikawa, A.; Usui, I.; Tobe, K.; Nakagawa, T.; Yoshino, J.; Imai, S. SIRT1-Mediated eNAMPT Secretion from Adipose Tissue Regulates Hypothalamic NAD+ and Function in Mice. Cell Metab. 2015, 21, 706–717. [Google Scholar] [CrossRef] [Green Version]
- Liu, L.; Su, X.; Quinn, W.J., 3rd; Hui, S.; Krukenberg, K.; Frederick, D.W.; Redpath, P.; Zhan, L.; Chellappa, K.; White, E.; et al. Quantitative Analysis of NAD Synthesis-Breakdown Fluxes. Cell Metab. 2018, 27, 1067–1080.e1065. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Petley, A.; Macklin, B.; Renwick, A.G.; Wilkin, T.J. The pharmacokinetics of nicotinamide in humans and rodents. Diabetes 1995, 44, 152–155. [Google Scholar] [CrossRef] [PubMed]
- Igarashi, M.; Nakagawa-Nagahama, Y.; Miura, M.; Kashiwabara, K.; Yaku, K.; Sawada, M.; Sekine, R.; Fukamizu, Y.; Sato, T.; Sakurai, T.; et al. Chronic nicotinamide mononucleotide supplementation elevates blood nicotinamide adenine dinucleotide levels and alters muscle function in healthy older men. npj Aging 2022, 8, 5. [Google Scholar] [CrossRef]
- Campbell, J.M. Supplementation with NAD(+) and Its Precursors to Prevent Cognitive Decline across Disease Contexts. Nutrients 2022, 14, 3231. [Google Scholar] [CrossRef]
- Yoo, K.H.; Tang, J.J.; Rashid, M.A.; Cho, C.H.; Corujo-Ramirez, A.; Choi, J.; Bae, M.G.; Brogren, D.; Hawse, J.R.; Hou, X.; et al. Nicotinamide Mononucleotide Prevents Cisplatin-Induced Cognitive Impairments. Cancer Res. 2021, 81, 3727–3737. [Google Scholar] [CrossRef]
- Zapata-Pérez, R.; Tammaro, A.; Schomakers, B.V.; Scantlebery, A.M.L.; Denis, S.; Elfrink, H.L.; Giroud-Gerbetant, J.; Cantó, C.; López-Leonardo, C.; McIntyre, R.L.; et al. Reduced nicotinamide mononucleotide is a new and potent NAD(+) precursor in mammalian cells and mice. FASEB J. Off. Publ. Fed. Am. Soc. Exp. Biol. 2021, 35, e21456. [Google Scholar] [CrossRef] [PubMed]
- White, A.T.; Schenk, S. NAD(+)/NADH and skeletal muscle mitochondrial adaptations to exercise. Am. J. Physiology. Endocrinol. Metab. 2012, 303, E308–E321. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Graham, T.; Sjøgaard, G.; Löllgen, H.; Saltin, B. NAD in muscle of man at rest and during exercise. Pflug. Arch. Eur. J. Physiol. 1978, 376, 35–39. [Google Scholar] [CrossRef]
- Katz, A.; Sahlin, K. Effect of decreased oxygen availability on NADH and lactate contents in human skeletal muscle during exercise. Acta Physiol. Scand. 1987, 131, 119–127. [Google Scholar] [CrossRef]
- Hu, Q.; Wu, D.; Walker, M.; Wang, P.; Tian, R.; Wang, W. Genetically encoded biosensors for evaluating NAD(+)/NADH ratio in cytosolic and mitochondrial compartments. Cell Rep. Methods 2021, 1, 100116. [Google Scholar] [CrossRef]
- Wang, Y.; Stancliffe, E.; Fowle-Grider, R.; Wang, R.; Wang, C.; Schwaiger-Haber, M.; Shriver, L.P.; Patti, G.J. Saturation of the mitochondrial NADH shuttles drives aerobic glycolysis in proliferating cells. Mol. Cell 2022, 82, 3270–3283.e3279. [Google Scholar] [CrossRef]
- Mao, P.; Reddy, P.H. Aging and amyloid beta-induced oxidative DNA damage and mitochondrial dysfunction in Alzheimer’s disease: Implications for early intervention and therapeutics. Biochim. Biophys. Acta 2011, 1812, 1359–1370. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cunnane, S.C.; Trushina, E.; Morland, C.; Prigione, A.; Casadesus, G.; Andrews, Z.B.; Beal, M.F.; Bergersen, L.H.; Brinton, R.D.; de la Monte, S.; et al. Brain energy rescue: An emerging therapeutic concept for neurodegenerative disorders of ageing. Nat. Rev. Drug Discov. 2020, 19, 609–633. [Google Scholar] [CrossRef] [PubMed]
- De la Cueva, M.; Antequera, D.; Ordoñez-Gutierrez, L.; Wandosell, F.; Camins, A.; Carro, E.; Bartolome, F. Amyloid-β impairs mitochondrial dynamics and autophagy in Alzheimer’s disease experimental models. Sci. Rep. 2022, 12, 10092. [Google Scholar] [CrossRef]
- Zhu, X.; Smith, M.A.; Perry, G.; Aliev, G. Mitochondrial failures in Alzheimer’s disease. Am. J. Alzheimer’s Dis. Other Dement. 2004, 19, 345–352. [Google Scholar] [CrossRef]
- Carbonell, F.; Zijdenbos, A.P.; Bedell, B.J. Spatially Distributed Amyloid-β Reduces Glucose Metabolism in Mild Cognitive Impairment. J. Alzheimer’s Dis. JAD 2020, 73, 543–557. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jian, Y.; Yuan, S.; Yang, J.; Lei, Y.; Li, X.; Liu, W. Aerobic Exercise Alleviates Abnormal Autophagy in Brain Cells of APP/PS1 Mice by Upregulating AdipoR1 Levels. Int. J. Mol. Sci. 2022, 23, 9921. [Google Scholar] [CrossRef] [PubMed]
- Shen, H.; Hyrc, K.L.; Goldberg, M.P. Maintaining energy homeostasis is an essential component of Wld(S)-mediated axon protection. Neurobiol. Dis. 2013, 59, 69–79. [Google Scholar] [CrossRef] [Green Version]
- Reddy, P.H.; Yin, X.; Manczak, M.; Kumar, S.; Pradeepkiran, J.A.; Vijayan, M.; Reddy, A.P. Mutant APP and amyloid beta-induced defective autophagy, mitophagy, mitochondrial structural and functional changes and synaptic damage in hippocampal neurons from Alzheimer’s disease. Hum. Mol. Genet. 2018, 27, 2502–2516. [Google Scholar] [CrossRef]
- Palikaras, K.; Lionaki, E.; Tavernarakis, N. Coordination of mitophagy and mitochondrial biogenesis during ageing in C. elegans. Nature 2015, 521, 525–528. [Google Scholar] [CrossRef] [PubMed]
- Fang, E.F.; Scheibye-Knudsen, M.; Brace, L.E.; Kassahun, H.; SenGupta, T.; Nilsen, H.; Mitchell, J.R.; Croteau, D.L.; Bohr, V.A. Defective mitophagy in XPA via PARP-1 hyperactivation and NAD(+)/SIRT1 reduction. Cell 2014, 157, 882–896. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Motta, M.C.; Divecha, N.; Lemieux, M.; Kamel, C.; Chen, D.; Gu, W.; Bultsma, Y.; McBurney, M.; Guarente, L. Mammalian SIRT1 represses forkhead transcription factors. Cell 2004, 116, 551–563. [Google Scholar] [CrossRef] [Green Version]
- Eijkelenboom, A.; Burgering, B.M. FOXOs: Signalling integrators for homeostasis maintenance. Nat. Rev. Mol. Cell Biol. 2013, 14, 83–97. [Google Scholar] [CrossRef]
- Miller, K.N.; Clark, J.P.; Martin, S.A.; Howell, P.R.; Burhans, M.S.; Haws, S.A.; Johnson, N.B.; Rhoads, T.W.; Pavelec, D.M.; Eliceiri, K.W.; et al. PGC-1a integrates a metabolism and growth network linked to caloric restriction. Aging Cell 2019, 18, e12999. [Google Scholar] [CrossRef]
- Xiao, W.; Wang, R.S.; Handy, D.E.; Loscalzo, J. NAD(H) and NADP(H) Redox Couples and Cellular Energy Metabolism. Antioxid. Redox Signal. 2018, 28, 251–272. [Google Scholar] [CrossRef]
- Katsyuba, E.; Auwerx, J. Modulating NAD(+) metabolism, from bench to bedside. EMBO J. 2017, 36, 2670–2683. [Google Scholar] [CrossRef] [PubMed]
- Mouchiroud, L.; Houtkooper, R.H.; Moullan, N.; Katsyuba, E.; Ryu, D.; Cantó, C.; Mottis, A.; Jo, Y.S.; Viswanathan, M.; Schoonjans, K.; et al. The NAD(+)/Sirtuin Pathway Modulates Longevity through Activation of Mitochondrial UPR and FOXO Signaling. Cell 2013, 154, 430–441. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, H.; Ryu, D.; Wu, Y.; Gariani, K.; Wang, X.; Luan, P.; D’Amico, D.; Ropelle, E.R.; Lutolf, M.P.; Aebersold, R.; et al. NAD+ repletion improves mitochondrial and stem cell function and enhances life span in mice. Science 2016, 352, 1436–1443. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhu, X.H.; Lu, M.; Lee, B.Y.; Ugurbil, K.; Chen, W. In vivo NAD assay reveals the intracellular NAD contents and redox state in healthy human brain and their age dependences. Proc. Natl. Acad. Sci. USA 2015, 112, 2876–2881. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yoshino, J.; Baur, J.A.; Imai, S.I. NAD(+) Intermediates: The Biology and Therapeutic Potential of NMN and NR. Cell Metab. 2018, 27, 513–528. [Google Scholar] [CrossRef] [Green Version]
- Imai, S.; Yoshino, J. The importance of NAMPT/NAD/SIRT1 in the systemic regulation of metabolism and ageing. Diabetes Obes. Metab. 2013, 15 (Suppl. 3), 26–33. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yao, Z.; Yang, W.; Gao, Z.; Jia, P. Nicotinamide mononucleotide inhibits JNK activation to reverse Alzheimer disease. Neurosci. Lett. 2017, 647, 133–140. [Google Scholar] [CrossRef]
- Michishita, E.; Park, J.Y.; Burneskis, J.M.; Barrett, J.C.; Horikawa, I. Evolutionarily conserved and nonconserved cellular localizations and functions of human SIRT proteins. Mol. Biol. Cell 2005, 16, 4623–4635. [Google Scholar] [CrossRef] [Green Version]
- Funk, J.A.; Odejinmi, S.; Schnellmann, R.G. SRT1720 induces mitochondrial biogenesis and rescues mitochondrial function after oxidant injury in renal proximal tubule cells. J. Pharmacol. Exp. Ther. 2010, 333, 593–601. [Google Scholar] [CrossRef] [Green Version]
- Lee, I.H.; Cao, L.; Mostoslavsky, R.; Lombard, D.B.; Liu, J.; Bruns, N.E.; Tsokos, M.; Alt, F.W.; Finkel, T. A role for the NAD-dependent deacetylase Sirt1 in the regulation of autophagy. Proc. Natl. Acad. Sci. USA 2008, 105, 3374–3379. [Google Scholar] [CrossRef] [Green Version]
- Ou, X.; Lee, M.R.; Huang, X.; Messina-Graham, S.; Broxmeyer, H.E. SIRT1 positively regulates autophagy and mitochondria function in embryonic stem cells under oxidative stress. Stem Cells Dayt. Ohio 2014, 32, 1183–1194. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, X.; Cai, S.; Zhang, C.; Liu, Z.; Luo, J.; Xing, B.; Du, X. Deacetylation of NAT10 by Sirt1 promotes the transition from rRNA biogenesis to autophagy upon energy stress. Nucleic Acids Res. 2018, 46, 9601–9616. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lazarou, M.; Sliter, D.A.; Kane, L.A.; Sarraf, S.A.; Wang, C.; Burman, J.L.; Sideris, D.P.; Fogel, A.I.; Youle, R.J. The ubiquitin kinase PINK1 recruits autophagy receptors to induce mitophagy. Nature 2015, 524, 309–314. [Google Scholar] [CrossRef] [Green Version]
- Rasti, G.; Becker, M.; Vazquez, B.N.; Espinosa-Alcantud, M.; Fernández-Duran, I.; Gámez-García, A.; Ianni, A.; Gonzalez, J.; Bosch-Presegué, L.; Marazuela-Duque, A.; et al. SIRT1 regulates DNA damage signaling through the PP4 phosphatase complex. Nucleic Acids Res. 2023, gkad504. [Google Scholar] [CrossRef] [PubMed]
- Xu, Y.; Wan, W. Acetylation in the regulation of autophagy. Autophagy 2023, 19, 379–387. [Google Scholar] [CrossRef]
- Li, X.; Cao, G.; Liu, X.; Tang, T.S.; Guo, C.; Liu, H. Polymerases and DNA Repair in Neurons: Implications in Neuronal Survival and Neurodegenerative Diseases. Front. Cell. Neurosci. 2022, 16, 852002. [Google Scholar] [CrossRef]
- Xu, H.; Liu, Y.Y.; Li, L.S.; Liu, Y.S. Sirtuins at the Crossroads between Mitochondrial Quality Control and Neurodegenerative Diseases: Structure, Regulation, Modifications, and Modulators. Aging Dis. 2023, 14, 794–824. [Google Scholar] [CrossRef] [PubMed]
- Shah, S.A.; Yoon, G.H.; Chung, S.S.; Abid, M.N.; Kim, T.H.; Lee, H.Y.; Kim, M.O. Novel osmotin inhibits SREBP2 via the AdipoR1/AMPK/SIRT1 pathway to improve Alzheimer’s disease neuropathological deficits. Mol. Psychiatry 2017, 22, 407–416. [Google Scholar] [CrossRef] [PubMed]
- Min, S.W.; Cho, S.H.; Zhou, Y.; Schroeder, S.; Haroutunian, V.; Seeley, W.W.; Huang, E.J.; Shen, Y.; Masliah, E.; Mukherjee, C.; et al. Acetylation of tau inhibits its degradation and contributes to tauopathy. Neuron 2010, 67, 953–966. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shin, M.K.; Vázquez-Rosa, E.; Koh, Y.; Dhar, M.; Chaubey, K.; Cintrón-Pérez, C.J.; Barker, S.; Miller, E.; Franke, K.; Noterman, M.F.; et al. Reducing acetylated tau is neuroprotective in brain injury. Cell 2021, 184, 2715–2732.e2723. [Google Scholar] [CrossRef]
- Corpas, R.; Revilla, S.; Ursulet, S.; Castro-Freire, M.; Kaliman, P.; Petegnief, V.; Giménez-Llort, L.; Sarkis, C.; Pallàs, M.; Sanfeliu, C. SIRT1 Overexpression in Mouse Hippocampus Induces Cognitive Enhancement Through Proteostatic and Neurotrophic Mechanisms. Mol. Neurobiol. 2017, 54, 5604–5619. [Google Scholar] [CrossRef]
- Kitada, M.; Ogura, Y.; Monno, I.; Koya, D. Sirtuins and Type 2 Diabetes: Role in Inflammation, Oxidative Stress, and Mitochondrial Function. Front. Endocrinol. 2019, 10, 187. [Google Scholar] [CrossRef] [Green Version]
- Scarpulla, R.C. Transcriptional paradigms in mammalian mitochondrial biogenesis and function. Physiol. Rev. 2008, 88, 611–638. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gleyzer, N.; Vercauteren, K.; Scarpulla, R.C. Control of mitochondrial transcription specificity factors (TFB1M and TFB2M) by nuclear respiratory factors (NRF-1 and NRF-2) and PGC-1 family coactivators. Mol. Cell. Biol. 2005, 25, 1354–1366. [Google Scholar] [CrossRef] [Green Version]
- Hu, Y.; Chen, H.; Zhang, L.; Lin, X.; Li, X.; Zhuang, H.; Fan, H.; Meng, T.; He, Z.; Huang, H.; et al. The AMPK-MFN2 axis regulates MAM dynamics and autophagy induced by energy stresses. Autophagy 2021, 17, 1142–1156. [Google Scholar] [CrossRef]
- Hampel, H.; Vassar, R.; De Strooper, B.; Hardy, J.; Willem, M.; Singh, N.; Zhou, J.; Yan, R.; Vanmechelen, E.; De Vos, A.; et al. The β-Secretase BACE1 in Alzheimer’s Disease. Biol. Psychiatry 2021, 89, 745–756. [Google Scholar] [CrossRef]
- Wrann, C.D.; White, J.P.; Salogiannnis, J.; Laznik-Bogoslavski, D.; Wu, J.; Ma, D.; Lin, J.D.; Greenberg, M.E.; Spiegelman, B.M. Exercise induces hippocampal BDNF through a PGC-1α/FNDC5 pathway. Cell Metab. 2013, 18, 649–659. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Herzig, S.; Shaw, R.J. AMPK: Guardian of metabolism and mitochondrial homeostasis. Nat. Reviews. Mol. Cell Biol. 2018, 19, 121–135. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wiley, C.D.; Velarde, M.C.; Lecot, P.; Liu, S.; Sarnoski, E.A.; Freund, A.; Shirakawa, K.; Lim, H.W.; Davis, S.S.; Ramanathan, A.; et al. Mitochondrial Dysfunction Induces Senescence with a Distinct Secretory Phenotype. Cell Metab. 2016, 23, 303–314. [Google Scholar] [CrossRef] [Green Version]
- Godoy, J.A.; Zolezzi, J.M.; Braidy, N.; Inestrosa, N.C. Role of Sirt1 during the ageing process: Relevance to protection of synapses in the brain. Mol. Neurobiol. 2014, 50, 744–756. [Google Scholar] [CrossRef]
- Sung, B.; Chung, J.W.; Bae, H.R.; Choi, J.S.; Kim, C.M.; Kim, N.D. Humulus japonicus extract exhibits antioxidative and anti-aging effects via modulation of the AMPK-SIRT1 pathway. Exp. Ther. Med. 2015, 9, 1819–1826. [Google Scholar] [CrossRef] [Green Version]
- Salminen, A.; Kaarniranta, K.; Kauppinen, A. Crosstalk between Oxidative Stress and SIRT1: Impact on the Aging Process. Int. J. Mol. Sci. 2013, 14, 3834–3859. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lagouge, M.; Argmann, C.; Gerhart-Hines, Z.; Meziane, H.; Lerin, C.; Daussin, F.; Messadeq, N.; Milne, J.; Lambert, P.; Elliott, P.; et al. Resveratrol improves mitochondrial function and protects against metabolic disease by activating SIRT1 and PGC-1alpha. Cell 2006, 127, 1109–1122. [Google Scholar] [CrossRef] [PubMed]
- Gerhart-Hines, Z.; Rodgers, J.T.; Bare, O.; Lerin, C.; Kim, S.H.; Mostoslavsky, R.; Alt, F.W.; Wu, Z.; Puigserver, P. Metabolic control of muscle mitochondrial function and fatty acid oxidation through SIRT1/PGC-1alpha. EMBO J. 2007, 26, 1913–1923. [Google Scholar] [CrossRef]
- Halling, J.F.; Pilegaard, H. PGC-1α-mediated regulation of mitochondrial function and physiological implications. Appl. Physiol. Nutr. Metab. Physiol. Appl. Nutr. Metab. 2020, 45, 927–936. [Google Scholar] [CrossRef]
- Romanello, V.; Sandri, M. Mitochondrial Quality Control and Muscle Mass Maintenance. Front. Physiol. 2015, 6, 422. [Google Scholar] [CrossRef]
- Yan, Z.; Okutsu, M.; Akhtar, Y.N.; Lira, V.A. Regulation of exercise-induced fiber type transformation, mitochondrial biogenesis, and angiogenesis in skeletal muscle. J. Appl. Physiol. 2011, 110, 264–274. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Laker, R.C.; Drake, J.C.; Wilson, R.J.; Lira, V.A.; Lewellen, B.M.; Ryall, K.A.; Fisher, C.C.; Zhang, M.; Saucerman, J.J.; Goodyear, L.J.; et al. Ampk phosphorylation of Ulk1 is required for targeting of mitochondria to lysosomes in exercise-induced mitophagy. Nat. Commun. 2017, 8, 548. [Google Scholar] [CrossRef] [Green Version]
- Sarraf, S.A.; Sideris, D.P.; Giagtzoglou, N.; Ni, L.; Kankel, M.W.; Sen, A.; Bochicchio, L.E.; Huang, C.H.; Nussenzweig, S.C.; Worley, S.H.; et al. PINK1/Parkin Influences Cell Cycle by Sequestering TBK1 at Damaged Mitochondria, Inhibiting Mitosis. Cell Rep. 2019, 29, 225–235.e225. [Google Scholar] [CrossRef] [Green Version]
- Seabright, A.P.; Fine, N.H.F.; Barlow, J.P.; Lord, S.O.; Musa, I.; Gray, A.; Bryant, J.A.; Banzhaf, M.; Lavery, G.G.; Hardie, D.G.; et al. AMPK activation induces mitophagy and promotes mitochondrial fission while activating TBK1 in a PINK1-Parkin independent manner. FASEB J. Off. Publ. Fed. Am. Soc. Exp. Biol. 2020, 34, 6284–6301. [Google Scholar] [CrossRef] [Green Version]
- Richter, B.; Sliter, D.A.; Herhaus, L.; Stolz, A.; Wang, C.; Beli, P.; Zaffagnini, G.; Wild, P.; Martens, S.; Wagner, S.A.; et al. Phosphorylation of OPTN by TBK1 enhances its binding to Ub chains and promotes selective autophagy of damaged mitochondria. Proc. Natl. Acad. Sci. USA 2016, 113, 4039–4044. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Heo, J.M.; Ordureau, A.; Paulo, J.A.; Rinehart, J.; Harper, J.W. The PINK1-PARKIN Mitochondrial Ubiquitylation Pathway Drives a Program of OPTN/NDP52 Recruitment and TBK1 Activation to Promote Mitophagy. Mol. Cell 2015, 60, 7–20. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gagnon, K.B.; Delpire, E. Physiology of SLC12 transporters: Lessons from inherited human genetic mutations and genetically engineered mouse knockouts. Am. J. Physiology. Cell Physiol. 2013, 304, C693–C714. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lin, L.; Yee, S.W.; Kim, R.B.; Giacomini, K.M. SLC transporters as therapeutic targets: Emerging opportunities. Nat. Rev. Drug Discov. 2015, 14, 543–560. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bowcock, A.M.; Cookson, W.O. The genetics of psoriasis, psoriatic arthritis and atopic dermatitis. Hum. Mol. Genet. 2004, 13, R43–R55. [Google Scholar] [CrossRef] [PubMed]
- Ito, N.; Takatsu, A.; Ito, H.; Koike, Y.; Yoshioka, K.; Kamei, Y.; Imai, S.I. Slc12a8 in the lateral hypothalamus maintains energy metabolism and skeletal muscle functions during aging. Cell Rep. 2022, 40, 111131. [Google Scholar] [CrossRef]
- Barile, M.; Passarella, S.; Danese, G.; Quagliariello, E. Rat liver mitochondria can synthesize nicotinamide adenine dinucleotide from nicotinamide mononucleotide and ATP via a putative matrix nicotinamide mononucleotide adenylyltransferase. Biochem. Mol. Biol. Int. 1996, 38, 297–306. [Google Scholar]
- Yang, H.; Yang, T.; Baur, J.A.; Perez, E.; Matsui, T.; Carmona, J.J.; Lamming, D.W.; Souza-Pinto, N.C.; Bohr, V.A.; Rosenzweig, A.; et al. Nutrient-sensitive mitochondrial NAD+ levels dictate cell survival. Cell 2007, 130, 1095–1107. [Google Scholar] [CrossRef] [Green Version]
- Pittelli, M.; Formentini, L.; Faraco, G.; Lapucci, A.; Rapizzi, E.; Cialdai, F.; Romano, G.; Moneti, G.; Moroni, F.; Chiarugi, A. Inhibition of nicotinamide phosphoribosyltransferase: Cellular bioenergetics reveals a mitochondrial insensitive NAD pool. J. Biol. Chem. 2010, 285, 34106–34114. [Google Scholar] [CrossRef] [Green Version]
- Girardi, E.; Agrimi, G.; Goldmann, U.; Fiume, G.; Lindinger, S.; Sedlyarov, V.; Srndic, I.; Gürtl, B.; Agerer, B.; Kartnig, F.; et al. Epistasis-driven identification of SLC25A51 as a regulator of human mitochondrial NAD import. Nat. Commun. 2020, 11, 6145. [Google Scholar] [CrossRef]
- Todisco, S.; Agrimi, G.; Castegna, A.; Palmieri, F. Identification of the mitochondrial NAD+ transporter in Saccharomyces cerevisiae. J. Biol. Chem. 2006, 281, 1524–1531. [Google Scholar] [CrossRef] [Green Version]
- Luongo, T.S.; Eller, J.M.; Lu, M.J.; Niere, M.; Raith, F.; Perry, C.; Bornstein, M.R.; Oliphint, P.; Wang, L.; McReynolds, M.R.; et al. SLC25A51 is a mammalian mitochondrial NAD(+) transporter. Nature 2020, 588, 174–179. [Google Scholar] [CrossRef] [PubMed]
- Ryu, W.I.; Shen, M.; Lee, Y.; Healy, R.A.; Bormann, M.K.; Cohen, B.M.; Sonntag, K.C. Nicotinamide riboside and caffeine partially restore diminished NAD availability but not altered energy metabolism in Alzheimer’s disease. Aging Cell 2022, 21, e13658. [Google Scholar] [CrossRef] [PubMed]
- Wyckelsma, V.L.; McKenna, M.J. Effects of Age on Na(+),K(+)-ATPase Expression in Human and Rodent Skeletal Muscle. Front. Physiol. 2016, 7, 316. [Google Scholar] [CrossRef] [Green Version]
- Shats, I.; Williams, J.G.; Liu, J.; Makarov, M.V.; Wu, X.; Lih, F.B.; Deterding, L.J.; Lim, C.; Xu, X.; Randall, T.A.; et al. Bacteria Boost Mammalian Host NAD Metabolism by Engaging the Deamidated Biosynthesis Pathway. Cell Metab. 2020, 31, 564–579.e567. [Google Scholar] [CrossRef] [PubMed]
- Yuan, S.; Yang, J.; Jian, Y.; Lei, Y.; Yao, S.; Hu, Z.; Liu, X.; Tang, C.; Liu, W. Treadmill Exercise Modulates Intestinal Microbes and Suppresses LPS Displacement to Alleviate Neuroinflammation in the Brains of APP/PS1 Mice. Nutrients 2022, 14, 4134. [Google Scholar] [CrossRef] [PubMed]

Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wu, W.; Yuan, S.; Tang, Y.; Meng, X.; Peng, M.; Hu, Z.; Liu, W. Effect of Exercise and Oral Niacinamide Mononucleotide on Improving Mitochondrial Autophagy in Alzheimer’s Disease. Nutrients 2023, 15, 2851. https://doi.org/10.3390/nu15132851
Wu W, Yuan S, Tang Y, Meng X, Peng M, Hu Z, Liu W. Effect of Exercise and Oral Niacinamide Mononucleotide on Improving Mitochondrial Autophagy in Alzheimer’s Disease. Nutrients. 2023; 15(13):2851. https://doi.org/10.3390/nu15132851
Chicago/Turabian StyleWu, Weijia, Shunling Yuan, Yingzhe Tang, Xiangyuan Meng, Mei Peng, Zelin Hu, and Wenfeng Liu. 2023. "Effect of Exercise and Oral Niacinamide Mononucleotide on Improving Mitochondrial Autophagy in Alzheimer’s Disease" Nutrients 15, no. 13: 2851. https://doi.org/10.3390/nu15132851
APA StyleWu, W., Yuan, S., Tang, Y., Meng, X., Peng, M., Hu, Z., & Liu, W. (2023). Effect of Exercise and Oral Niacinamide Mononucleotide on Improving Mitochondrial Autophagy in Alzheimer’s Disease. Nutrients, 15(13), 2851. https://doi.org/10.3390/nu15132851