
ALDH1A Inhibition Suppresses Colitis and Alters α4β7 Integrin Expression on Activated T Cells in Mdr1a−/− Mice


{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Studies to Test Effects of ALDH1A Inhibition on Colitis
2.2. Serum Cytokines and Helicobacter Status
2.3. Immune Cell Characterization in Mdr1a−/− Mice
2.4. Statistical Analyses
3. Results
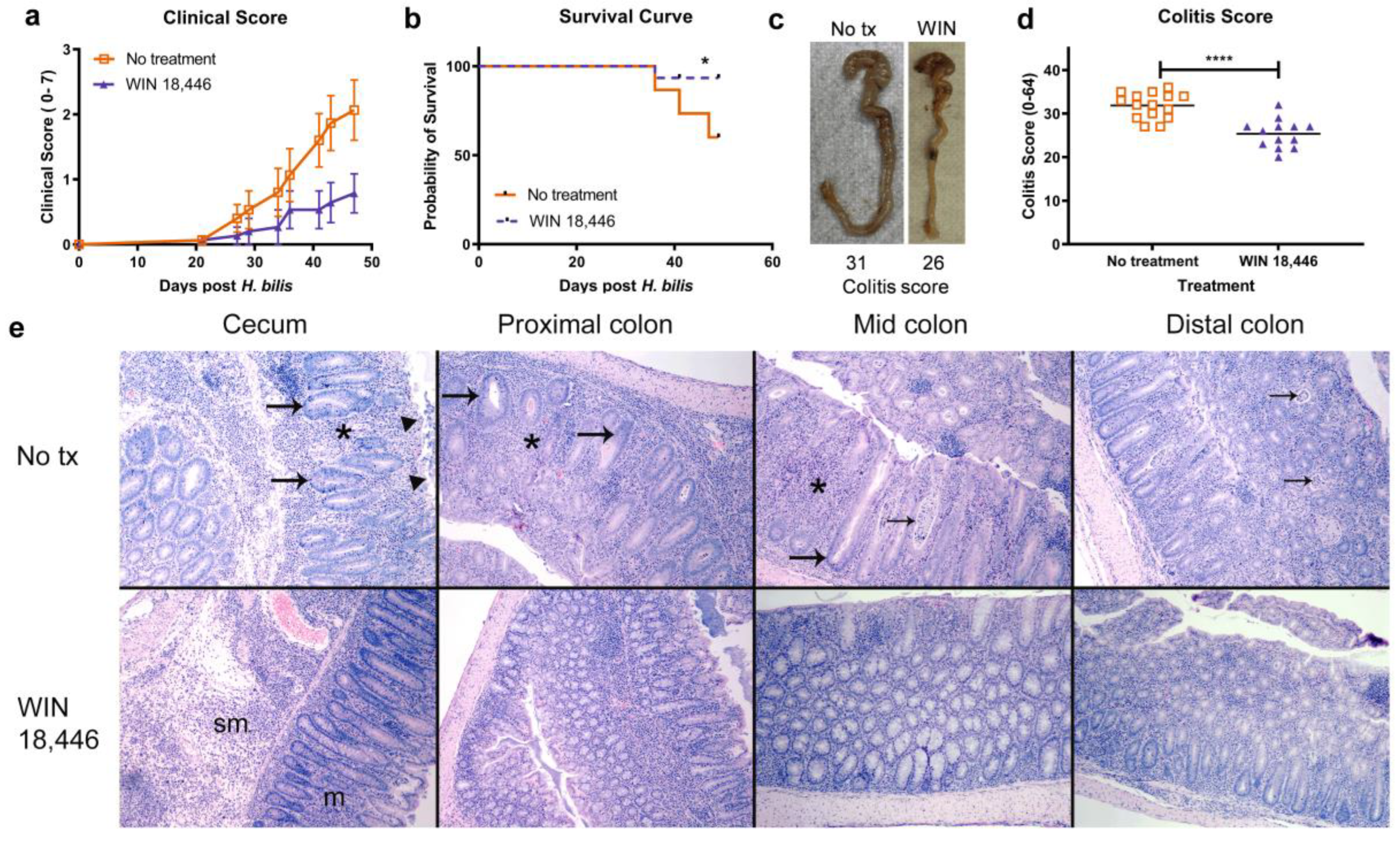
3.1. ALDH1A Inhibition Significantly Reduces the Severity of Colitis in Mdr1a−/− Mice but Not in Il10−/− Mice
3.2. Changes in Serum IL17E and IL22 Are Associated with WIN 18,446 Treatment and Colitis in Mdr1a−/− Mice
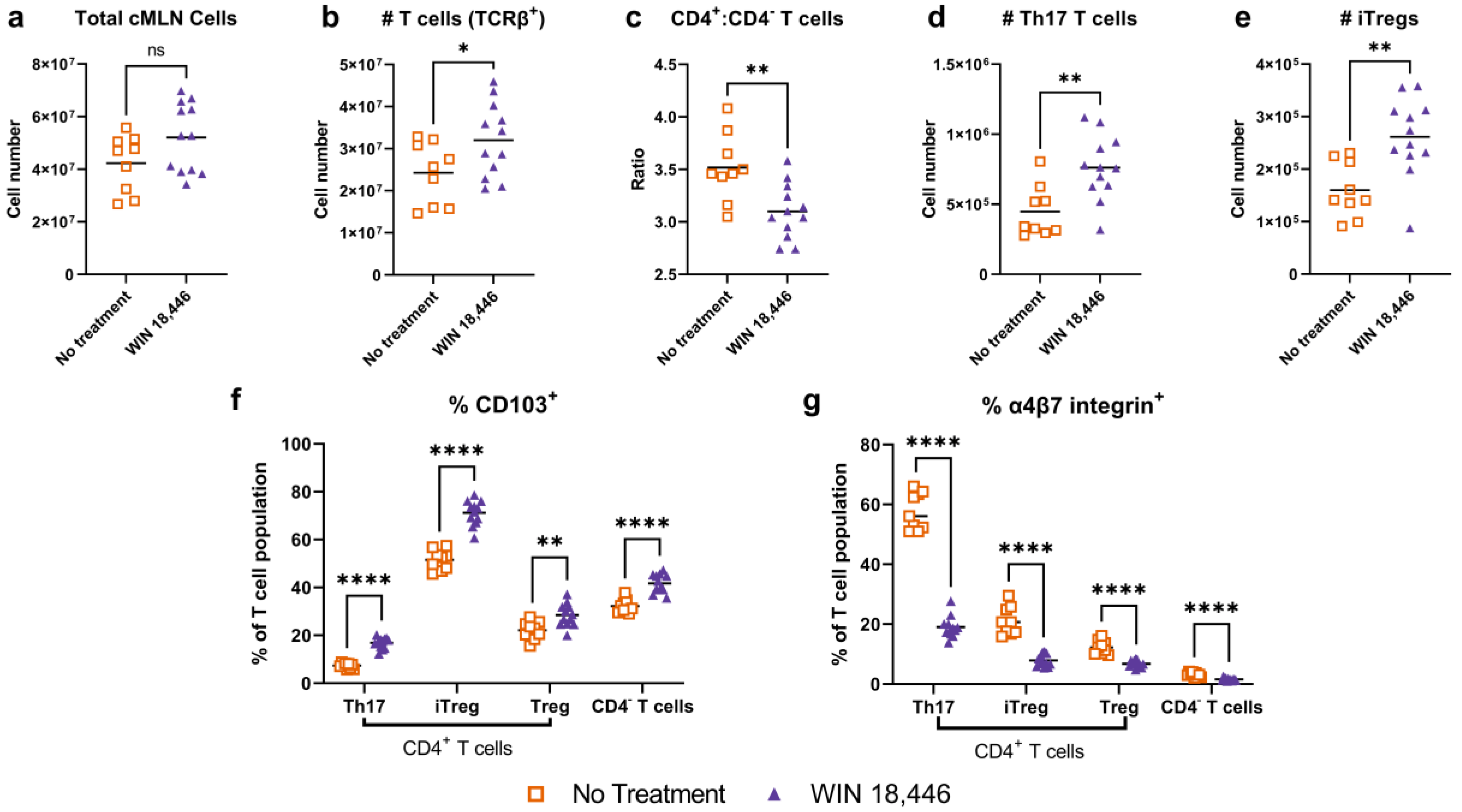
3.3. Colon-Draining MLN T Cell Subsets Are Altered with WIN 18,446 Treatment in Mdr1a−/− Mice
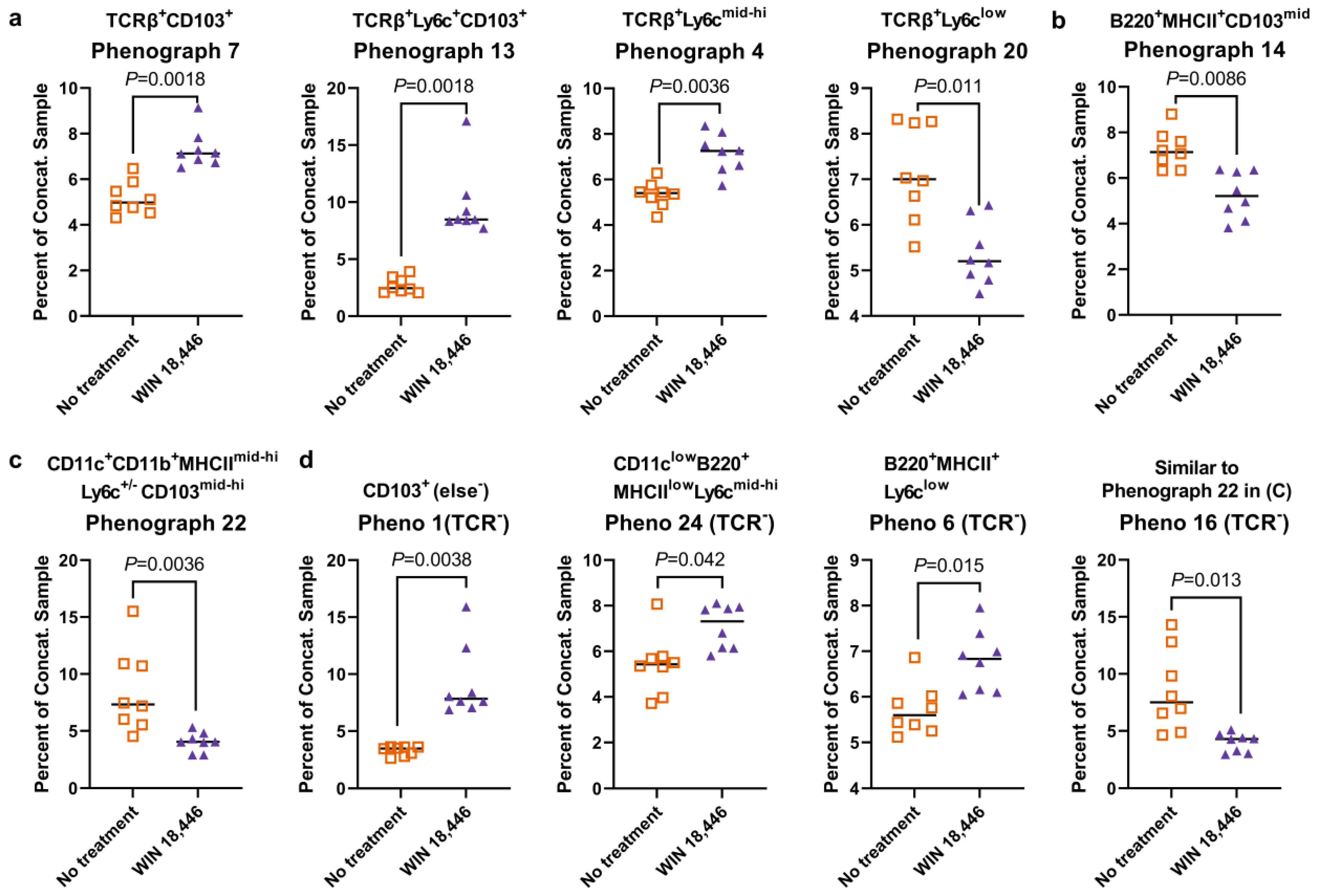
3.4. Colon-Draining MLN Leukocyte Cell Subsets Are Altered with Treatment with WIN 18,446 in Mdr1a−/− Mice
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Baumgart, D.C.; Le Berre, C. Newer Biologic and Small-Molecule Therapies for Inflammatory Bowel Disease. N. Engl. J. Med. 2021, 385, 1302–1315. [Google Scholar] [CrossRef] [PubMed]
- Eberhardson, M.; Hedin, C.; Carlson, M.; Tarnawski, L.; Levine, Y.A.; Olofsson, P.S. Toward Improved Control of Inflammatory Bowel Disease. Scand. J. Immunol. 2019, 89, e12745. [Google Scholar] [CrossRef] [PubMed]
- Iwata, M. Retinoic acid production by intestinal dendritic cells and its role in T-cell trafficking. Semin. Immunol. 2009, 21, 8–13. [Google Scholar] [CrossRef]
- O’Byrne, S.M.; Blaner, W.S. Retinol and retinyl esters: Biochemistry and physiology. J. Lipid Res. 2013, 54, 1731–1743. [Google Scholar] [CrossRef]
- Abdelhamid, L.; Luo, X.M. Retinoic Acid, Leaky Gut, and Autoimmune Diseases. Nutrients 2018, 10, 1016. [Google Scholar] [CrossRef]
- Jijon, H.B.; Suarez-Lopez, L.; Diaz, O.E.; Das, S.; De Calisto, J.; Parada-kusz, M.; Yaffe, M.B.; Pittet, M.J.; Mora, J.R.; Belkaid, Y.; et al. Intestinal epithelial cell-specific RARalpha depletion results in aberrant epithelial cell homeostasis and underdeveloped immune system. Mucosal Immunol. 2018, 11, 703–715. [Google Scholar] [CrossRef]
- Li, Y.; Gao, Y.; Cui, T.; Yang, T.; Liu, L.; Li, T.; Chen, J. Retinoic Acid Facilitates Toll-Like Receptor 4 Expression to Improve Intestinal Barrier Function through Retinoic Acid Receptor Beta. Cell Physiol. Biochem. 2017, 42, 1390–1406. [Google Scholar] [CrossRef]
- Iwata, M.; Hirakiyama, A.; Eshima, Y.; Kagechika, H.; Kato, C.; Song, S.Y. Retinoic acid imprints gut-homing specificity on T cells. Immunity 2004, 21, 527–538. [Google Scholar] [CrossRef]
- Mora, J.R.; Iwata, M.; von Andrian, U.H. Vitamin effects on the immune system: Vitamins A and D take centre stage. Nat. Rev. Immunol. 2008, 8, 685–698. [Google Scholar] [CrossRef]
- Mucida, D.; Park, Y.; Kim, G.; Turovskaya, O.; Scott, I.; Kronenberg, M.; Cheroutre, H. Reciprocal TH17 and regulatory T cell differentiation mediated by retinoic acid. Science 2007, 317, 256–260. [Google Scholar] [CrossRef]
- DePaolo, R.W.; Abadie, V.; Tang, F.; Fehlner-Peach, H.; Hall, J.A.; Wang, W.; Marietta, E.V.; Kasarda, D.D.; Waldmann, T.A.; Murray, J.A.; et al. Co-adjuvant effects of retinoic acid and IL-15 induce inflammatory immunity to dietary antigens. Nature 2011, 471, 220–224. [Google Scholar] [CrossRef]
- Bai, A.; Lu, N.; Guo, Y.; Liu, Z.; Chen, J.; Peng, Z. All-trans retinoic acid down-regulates inflammatory responses by shifting the Treg/Th17 profile in human ulcerative and murine colitis. J. Leukoc. Biol. 2009, 86, 959–969. [Google Scholar] [CrossRef] [PubMed]
- Collins, C.B.; Aherne, C.M.; Kominsky, D.; McNamee, E.N.; Lebsack, M.D.; Eltzschig, H.; Jedlicka, P.; Rivera-Nieves, J. Retinoic acid attenuates ileitis by restoring the balance between T-helper 17 and T regulatory cells. Gastroenterology 2011, 141, 1821–1831. [Google Scholar] [CrossRef] [PubMed]
- Oehlers, S.H.; Flores, M.V.; Hall, C.J.; Crosier, K.E.; Crosier, P.S. Retinoic acid suppresses intestinal mucus production and exacerbates experimental enterocolitis. Dis. Model. Mech. 2012, 5, 457–467. [Google Scholar] [CrossRef] [PubMed]
- Frey-Wagner, I.; Fischbeck, A.; Cee, A.; Leonardi, I.; Gruber, S.; Becker, E.; Atrott, K.; Lang, S.; Rogler, G. Effects of retinoids in mouse models of colitis: Benefit or danger to the gastrointestinal tract? Inflamm. Bowel Dis. 2013, 19, 2356–2365. [Google Scholar] [CrossRef]
- Hong, K.; Zhang, Y.; Guo, Y.; Xie, J.; Wang, J.; He, X.; Lu, N.; Bai, A. All-trans retinoic acid attenuates experimental colitis through inhibition of NF-kappaB signaling. Immunol. Lett. 2014, 162, 34–40. [Google Scholar] [CrossRef]
- Mielke, L.A.; Jones, S.A.; Raverdeau, M.; Higgs, R.; Stefanska, A.; Groom, J.R.; Misiak, A.; Dungan, L.S.; Sutton, C.E.; Streubel, G.; et al. Retinoic acid expression associates with enhanced IL-22 production by gammadelta T cells and innate lymphoid cells and attenuation of intestinal inflammation. J. Exp. Med. 2013, 210, 1117–1124. [Google Scholar] [CrossRef]
- Fransen, K.; Franzen, P.; Magnuson, A.; Elmabsout, A.A.; Nyhlin, N.; Wickbom, A.; Curman, B.; Torkvist, L.; D’Amato, M.; Bohr, J.; et al. Polymorphism in the retinoic acid metabolizing enzyme CYP26B1 and the development of Crohn’s Disease. PLoS ONE 2013, 8, e72739. [Google Scholar] [CrossRef]
- Rampal, R.; Wari, N.; Singh, A.K.; Das, U.; Bopanna, S.; Gupta, V.; Nayak, B.; Velapandian, T.; Kedia, S.; Kumar, D.; et al. Retinoic Acid Is Elevated in the Mucosa of Patients With Active Ulcerative Colitis and Displays a Proinflammatory Role by Augmenting IL-17 and IFNgamma Production. Inflamm. Bowel Dis. 2021, 27, 74–83. [Google Scholar] [CrossRef]
- Thangavelu, G.; Lee, Y.C.; Loschi, M.; Schaechter, K.M.; Feser, C.J.; Koehn, B.H.; Nowak, E.C.; Zeiser, R.; Serody, J.S.; Murphy, W.J.; et al. Dendritic Cell Expression of Retinal Aldehyde Dehydrogenase-2 Controls Graft-versus-Host Disease Lethality. J. Immunol. 2019, 202, 2795–2805. [Google Scholar] [CrossRef]
- Amory, J.K.; Muller, C.H.; Shimshoni, J.A.; Isoherranen, N.; Paik, J.; Moreb, J.S.; Amory, D.W., Sr.; Evanoff, R.; Goldstein, A.S.; Griswold, M.D. Suppression of spermatogenesis by bisdichloroacetyldiamines is mediated by inhibition of testicular retinoic acid biosynthesis. J. Androl. 2011, 32, 111–119. [Google Scholar] [CrossRef]
- Paik, J.; Haenisch, M.; Muller, C.H.; Goldstein, A.S.; Arnold, S.; Isoherranen, N.; Brabb, T.; Treuting, P.M.; Amory, J.K. Inhibition of retinoic acid biosynthesis by the bisdichloroacetyldiamine WIN 18,446 markedly suppresses spermatogenesis and alters retinoid metabolism in mice. J. Biol. Chem. 2014, 289, 15104–15117. [Google Scholar] [CrossRef]
- Seamons, A.; Haenisch, M.; Meeker, S.; Pershutkina, O.; Brabb, T.; Treuting, P.M.; Paik, J. Protective Effects of ALDH1A Enzyme Inhibition on Helicobacter-Induced Colitis in Smad3(−/−) Mice are Associated with Altered alpha4beta7 Integrin Expression on Activated T Cells. Nutrients 2020, 12, 2927. [Google Scholar] [CrossRef]
- Panwala, C.M.; Jones, J.C.; Viney, J.L. A novel model of inflammatory bowel disease: Mice deficient for the multiple drug resistance gene, mdr1a, spontaneously develop colitis. J. Immunol. 1998, 161, 5733–5744. [Google Scholar] [CrossRef] [PubMed]
- Willis, C.R.; Seamons, A.; Maxwell, J.; Treuting, P.M.; Nelson, L.; Chen, G.; Phelps, S.; Smith, C.L.; Brabb, T.; Iritani, B.M.; et al. Interleukin-7 receptor blockade suppresses adaptive and innate inflammatory responses in experimental colitis. J. Inflamm. 2012, 9, 39. [Google Scholar] [CrossRef] [PubMed]
- Maxwell, J.R.; Zhang, Y.; Brown, W.A.; Smith, C.L.; Byrne, F.R.; Fiorino, M.; Stevens, E.; Bigler, J.; Davis, J.A.; Rottman, J.B.; et al. Differential Roles for Interleukin-23 and Interleukin-17 in Intestinal Immunoregulation. Immunity 2015, 43, 739–750. [Google Scholar] [CrossRef] [PubMed]
- Nell, S.; Suerbaum, S.; Josenhans, C. The impact of the microbiota on the pathogenesis of IBD: Lessons from mouse infection models. Nat. Rev. Microbiol. 2010, 8, 564–577. [Google Scholar] [CrossRef]
- Maggio-Price, L.; Shows, D.; Waggie, K.; Burich, A.; Zeng, W.; Escobar, S.; Morrissey, P.; Viney, J.L. Helicobacter bilis infection accelerates and H. hepaticus infection delays the development of colitis in multiple drug resistance-deficient (mdr1a−/−) mice. Am. J. Pathol. 2002, 160, 739–751. [Google Scholar] [CrossRef]
- Meeker, S.; Seamons, A.; Paik, J.; Treuting, P.M.; Brabb, T.; Grady, W.M.; Maggio-Price, L. Increased dietary vitamin D suppresses MAPK signaling, colitis, and colon cancer. Cancer Res. 2014, 74, 4398–4408. [Google Scholar] [CrossRef]
- Levine, J.H.; Simonds, E.F.; Bendall, S.C.; Davis, K.L.; Amir, E.D.; Tadmor, M.D.; Litvin, O.; Fienberg, H.G.; Jager, A.; Zunder, E.R.; et al. Data-Driven Phenotypic Dissection of AML Reveals Progenitor-like Cells that Correlate with Prognosis. Cell 2015, 162, 184–197. [Google Scholar] [CrossRef]
- Oliveira, L.M.; Teixeira, F.M.E.; Sato, M.N. Impact of Retinoic Acid on Immune Cells and Inflammatory Diseases. Mediat. Inflamm. 2018, 2018, 3067126. [Google Scholar] [CrossRef]
- Feng, T.; Cong, Y.; Qin, H.; Benveniste, E.N.; Elson, C.O. Generation of mucosal dendritic cells from bone marrow reveals a critical role of retinoic acid. J. Immunol. 2010, 185, 5915–5925. [Google Scholar] [CrossRef]
- Vicente-Suarez, I.; Larange, A.; Reardon, C.; Matho, M.; Feau, S.; Chodaczek, G.; Park, Y.; Obata, Y.; Gold, R.; Wang-Zhu, Y.; et al. Unique lamina propria stromal cells imprint the functional phenotype of mucosal dendritic cells. Mucosal Immunol. 2015, 8, 141–151. [Google Scholar] [CrossRef]
- Torrence, A.E.; Brabb, T.; Viney, J.L.; Bielefeldt-Ohmann, H.; Treuting, P.; Seamons, A.; Drivdahl, R.; Zeng, W.; Maggio-Price, L. Serum biomarkers in a mouse model of bacterial-induced inflammatory bowel disease. Inflamm. Bowel Dis. 2008, 14, 480–490. [Google Scholar] [CrossRef]
- Borowczyk, J.; Shutova, M.; Brembilla, N.C.; Boehncke, W.H. IL-25 (IL-17E) in epithelial immunology and pathophysiology. J. Allergy Clin. Immunol. 2021, 148, 40–52. [Google Scholar] [CrossRef] [PubMed]
- Li, L.J.; Gong, C.; Zhao, M.H.; Feng, B.S. Role of interleukin-22 in inflammatory bowel disease. World J. Gastroenterol. 2014, 20, 18177–18188. [Google Scholar] [CrossRef] [PubMed]
- Saqui-Salces, M.; Keeley, T.M.; Grosse, A.S.; Qiao, X.T.; El-Zaatari, M.; Gumucio, D.L.; Samuelson, L.C.; Merchant, J.L. Gastric tuft cells express DCLK1 and are expanded in hyperplasia. Histochem. Cell Biol. 2011, 136, 191–204. [Google Scholar] [CrossRef]
- Seamons, A.; Paik, J. Immune Cell Subset Changes in Acute Helicobacter bilis-Induced Colitis in Smad3-/- Mice. University of Washington, Seattle, WA, USA. 2020, unpublished work. unpublished work.
- Roosenboom, B.; Wahab, P.J.; Smids, C.; Groenen, M.J.M.; van Koolwijk, E.; van Lochem, E.G.; Horjus Talabur Horje, C.S. Intestinal CD103+CD4+ and CD103+CD8+ T-Cell Subsets in the Gut of Inflammatory Bowel Disease Patients at Diagnosis and During Follow-up. Inflamm. Bowel Dis. 2019, 25, 1497–1509. [Google Scholar] [CrossRef] [PubMed]
- Noble, A.; Durant, L.; Hoyles, L.; McCartney, A.L.; Man, R.; Segal, J.; Costello, S.P.; Hendy, P.; Reddi, D.; Bouri, S.; et al. Deficient Resident Memory T Cell and CD8 T Cell Response to Commensals in Inflammatory Bowel Disease. J. Crohns Colitis 2020, 14, 525–537. [Google Scholar] [CrossRef]
- DeLong, J.H.; Hall, A.O.; Konradt, C.; Coppock, G.M.; Park, J.; Harms Pritchard, G.; Hunter, C.A. Cytokine- and TCR-Mediated Regulation of T Cell Expression of Ly6C and Sca-1. J. Immunol. 2018, 200, 1761–1770. [Google Scholar] [CrossRef]
- Hanninen, A.; Maksimow, M.; Alam, C.; Morgan, D.J.; Jalkanen, S. Ly6C supports preferential homing of central memory CD8+ T cells into lymph nodes. Eur. J. Immunol. 2011, 41, 634–644. [Google Scholar] [CrossRef]
- Zander, R.A.; Vijay, R.; Pack, A.D.; Guthmiller, J.J.; Graham, A.C.; Lindner, S.E.; Vaughan, A.M.; Kappe, S.H.I.; Butler, N.S. Th1-like Plasmodium-Specific Memory CD4(+) T Cells Support Humoral Immunity. Cell Rep. 2017, 21, 1839–1852. [Google Scholar] [CrossRef]
- Klebanoff, C.A.; Spencer, S.P.; Torabi-Parizi, P.; Grainger, J.R.; Roychoudhuri, R.; Ji, Y.; Sukumar, M.; Muranski, P.; Scott, C.D.; Hall, J.A.; et al. Retinoic acid controls the homeostasis of pre-cDC-derived splenic and intestinal dendritic cells. J. Exp. Med. 2013, 210, 1961–1976. [Google Scholar] [CrossRef] [PubMed]
- Zeng, R.; Bscheider, M.; Lahl, K.; Lee, M.; Butcher, E.C. Generation and transcriptional programming of intestinal dendritic cells: Essential role of retinoic acid. Mucosal Immunol. 2016, 9, 183–193. [Google Scholar] [CrossRef] [PubMed]
- Luciani, C.; Hager, F.T.; Cerovic, V.; Lelouard, H. Dendritic cell functions in the inductive and effector sites of intestinal immunity. Mucosal Immunol. 2022, 15, 40–50. [Google Scholar] [CrossRef] [PubMed]
- Musumeci, A.; Lutz, K.; Winheim, E.; Krug, A.B. What Makes a pDC: Recent Advances in Understanding Plasmacytoid DC Development and Heterogeneity. Front. Immunol. 2019, 10, 1222. [Google Scholar] [CrossRef]
- Evans, T.I.; Reeves, R.K. All-trans-retinoic acid imprints expression of the gut-homing marker alpha4beta7 while suppressing lymph node homing of dendritic cells. Clin. Vaccine Immunol. 2013, 20, 1642–1646. [Google Scholar] [CrossRef] [PubMed]

Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Seamons, A.; Staucean, O.; Snyder, J.M.; Brabb, T.; Hsu, C.C.; Paik, J. ALDH1A Inhibition Suppresses Colitis and Alters α4β7 Integrin Expression on Activated T Cells in Mdr1a−/− Mice. Nutrients 2023, 15, 3883. https://doi.org/10.3390/nu15183883
Seamons A, Staucean O, Snyder JM, Brabb T, Hsu CC, Paik J. ALDH1A Inhibition Suppresses Colitis and Alters α4β7 Integrin Expression on Activated T Cells in Mdr1a−/− Mice. Nutrients. 2023; 15(18):3883. https://doi.org/10.3390/nu15183883
Chicago/Turabian StyleSeamons, Audrey, Olesya Staucean, Jessica M. Snyder, Thea Brabb, Charlie C. Hsu, and Jisun Paik. 2023. "ALDH1A Inhibition Suppresses Colitis and Alters α4β7 Integrin Expression on Activated T Cells in Mdr1a−/− Mice" Nutrients 15, no. 18: 3883. https://doi.org/10.3390/nu15183883
APA StyleSeamons, A., Staucean, O., Snyder, J. M., Brabb, T., Hsu, C. C., & Paik, J. (2023). ALDH1A Inhibition Suppresses Colitis and Alters α4β7 Integrin Expression on Activated T Cells in Mdr1a−/− Mice. Nutrients, 15(18), 3883. https://doi.org/10.3390/nu15183883