
Uncovering Predictive Factors and Interventions for Restoring Microecological Diversity after Antibiotic Disturbance

 , , ,
, , , {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Study Populations and Samples Selection
2.2. Species-Level Taxonomic Profiling and Classification of Samples for All Cohorts
2.3. An Ensemble Learning Framework Used to Obtain p-RABs
2.4. Calculation of TD, FD, and FR within Samples
2.5. Metabolic Interaction Network of p-RABs
2.6. Microbial Food Web
2.7. A Mouse Model of Microbiome Recovery after Antibiotic Treatment
2.7.1. Strain Preparation
2.7.2. Animals and Design
2.7.3. Fecal Sample Collection and DNA Extraction
2.7.4. Taxonomic Profiling
2.7.5. Health Status Assessment
2.7.6. Gut Microbiota Co-Occurrence Network Construction and Characterization
3. Result
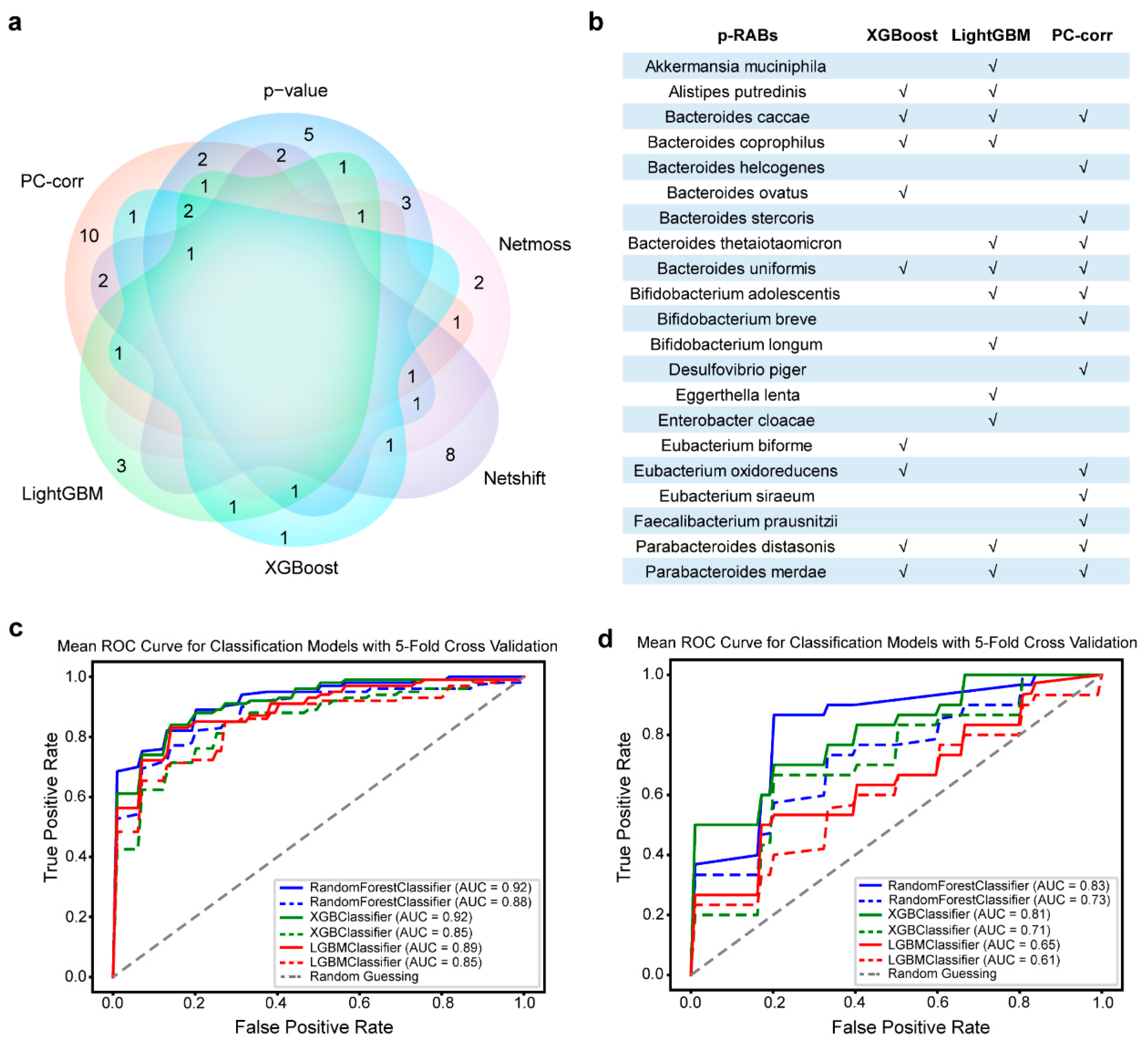
3.1. Better Predictive Recovery-Associated Bacterial Species Obtained from Different Methods
3.2. Within-Sample Taxonomic Diversity, Functional Diversity, and fprabs Predict Gut Microbiome Recovery under Antibiotic Disturbance
3.3. Akkermansia muciniphila Plays an Important Role in Gut Microbes Recovery after Antibiotic Disturbance
3.4. Synergy between Akkermansia muciniphila and Bacteroides uniformis Contributes to Rapid Reconstruction of Mice Intestinal Microecology after Antibiotics
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Data Availability Statement
Conflicts of Interest
References
- Ley, R.E.; Hamady, M.; Lozupone, C.; Turnbaugh, P.J.; Ramey, R.R.; Bircher, J.S.; Schlegel, M.L.; Tucker, T.A.; Schrenzel, M.D.; Knight, R.; et al. Evolution of mammals and their gut microbes. Science 2008, 320, 1647–1651. [Google Scholar] [CrossRef]
- Marchesi, J.R.; Adams, D.H.; Fava, F.; Hermes, G.D.; Hirschfield, G.M.; Hold, G.; Quraishi, M.N.; Kinross, J.; Smidt, H.; Tuohy, K.M.; et al. The gut microbiota and host health: A new clinical frontier. Gut 2016, 65, 330–339. [Google Scholar] [CrossRef] [PubMed]
- The Human Microbiome Project Consortium. Structure, function and diversity of the healthy human microbiome. Nature 2012, 486, 207–214. [Google Scholar] [CrossRef] [PubMed]
- Qin, J.; Li, Y.; Cai, Z.; Li, S.; Zhu, J.; Zhang, F.; Liang, S.; Zhang, W.; Guan, Y.; Shen, D.; et al. A metagenome-wide association study of gut microbiota in type 2 diabetes. Nature 2012, 490, 55–60. [Google Scholar] [CrossRef] [PubMed]
- Geng, J.F.; Ni, Q.Q.; Sun, W.; Li, L.G.; Feng, X.J. The links between gut microbiota and obesity and obesity related diseases. Biomed. Pharmacother. 2022, 147, 112678. [Google Scholar] [CrossRef]
- Gomes, A.C.; Hoffmann, C.; Mota, J.F. The human gut microbiota: Metabolism and perspective in obesity. Gut Microbes 2018, 9, 308–325. [Google Scholar] [CrossRef]
- Finucane, M.M.; Sharpton, T.J.; Laurent, T.J.; Pollard, K.S. A taxonomic signature of obesity in the microbiome? Getting to the guts of the matter. PLoS ONE 2014, 9, e84689. [Google Scholar] [CrossRef]
- Franzosa, E.A.; Sirota-Madi, A.; Avila-Pacheco, J.; Fornelos, N.; Haiser, H.J.; Reinker, S.; Vatanen, T.; Hall, A.B.; Mallick, H.; McIver, L.J.; et al. Gut microbiome structure and metabolic activity in inflammatory bowel disease. Nat. Microbiol. 2019, 4, 293–305. [Google Scholar] [CrossRef]
- Barberio, B.; Facchin, S.; Patuzzi, I.; Ford, A.C.; Massimi, D.; Valle, G.; Sattin, E.; Simionati, B.; Bertazzo, E.; Zingone, F.; et al. A specific microbiota signature is associated to various degrees of ulcerative colitis as assessed by a machine learning approach. Gut Microbes 2022, 14, 2028366. [Google Scholar] [CrossRef]
- Ai, D.M.; Pan, H.F.; Han, R.B.; Li, X.X.; Liu, G.; Xia, L.C. Using Decision Tree Aggregation with Random Forest Model to Identify Gut Microbes Associated with Colorectal Cancer. Genes 2019, 10, 112. [Google Scholar] [CrossRef]
- Yang, Y.Z.; Du, L.T.; Shi, D.B.; Kong, C.; Liu, J.Q.; Liu, G.; Li, X.X.; Ma, Y.L. Dysbiosis of human gut microbiome in young-onset colorectal cancer. Nat. Commun. 2021, 12, 6757. [Google Scholar] [CrossRef]
- Raymond, F.; Ouameur, A.A.; Deraspe, M.; Iqbal, N.; Gingras, H.; Dridi, B.; Leprohon, P.; Plante, P.L.; Giroux, R.; Berube, E.; et al. The initial state of the human gut microbiome determines its reshaping by antibiotics. ISME J. 2016, 10, 707–720. [Google Scholar] [CrossRef]
- Anthony, W.E.; Wang, B.; Sukhum, K.V.; D′Souza, A.W.; Hink, T.; Cass, C.; Seiler, S.; Reske, K.A.; Coon, C.; Dubberke, E.R.; et al. Acute and persistent effects of commonly used antibiotics on the gut microbiome and resistome in healthy adults. Cell Rep. 2022, 39, 110649. [Google Scholar] [CrossRef] [PubMed]
- Palleja, A.; Mikkelsen, K.H.; Forslund, S.K.; Kashani, A.; Allin, K.H.; Nielsen, T.; Hansen, T.H.; Liang, S.; Feng, Q.; Zhang, C.; et al. Recovery of gut microbiota of healthy adults following antibiotic exposure. Nat. Microbiol. 2018, 3, 1255–1265. [Google Scholar] [CrossRef]
- Smillie, C.S.; Smith, M.B.; Friedman, J.; Cordero, O.X.; David, L.A.; Alm, E.J. Ecology drives a global network of gene exchange connecting the human microbiome. Nature 2011, 480, 241–244. [Google Scholar] [CrossRef]
- D′Souza, A.W.; Moodley-Govender, E.; Berla, B.; Kelkar, T.; Wang, B.; Sun, X.; Daniels, B.; Coutsoudis, A.; Trehan, I.; Dantas, G. Cotrimoxazole Prophylaxis Increases Resistance Gene Prevalence and alpha-Diversity but Decreases β-Diversity in the Gut Microbiome of Human Immunodeficiency Virus-Exposed, Uninfected Infants. Clin. Infect. Dis. 2020, 71, 2858–2868. [Google Scholar] [CrossRef]
- Gasparrini, A.J.; Wang, B.; Sun, X.; Kennedy, E.A.; Hernandez-Leyva, A.; Ndao, I.M.; Tarr, P.I.; Warner, B.B.; Dantas, G. Persistent metagenomic signatures of early-life hospitalization and antibiotic treatment in the infant gut microbiota and resistome. Nat. Microbiol. 2019, 4, 2285–2297. [Google Scholar] [CrossRef] [PubMed]
- Cox, L.M.; Blaser, M.J. Antibiotics in early life and obesity. Nat. Rev. Endocrinol. 2015, 11, 182–190. [Google Scholar] [CrossRef] [PubMed]
- Buffie, C.G.; Jarchum, I.; Equinda, M.; Lipuma, L.; Gobourne, A.; Viale, A.; Ubeda, C.; Xavier, J.; Pamer, E.G. Profound alterations of intestinal microbiota following a single dose of clindamycin results in sustained susceptibility to Clostridium difficile-induced colitis. Infect. Immun. 2012, 80, 62–73. [Google Scholar] [CrossRef] [PubMed]
- Abeles, S.R.; Jones, M.B.; Santiago-Rodriguez, T.M.; Ly, M.; Klitgord, N.; Yooseph, S.; Nelson, K.E.; Pride, D.T. Microbial diversity in individuals and their household contacts following typical antibiotic courses. Microbiome 2016, 4, 39. [Google Scholar] [CrossRef] [PubMed]
- Chng, K.R.; Ghosh, T.S.; Tan, Y.H.; Nandi, T.; Lee, I.R.; Ng, A.H.Q.; Li, C.; Ravikrishnan, A.; Lim, K.M.; Lye, D.; et al. Metagenome-wide association analysis identifies microbial determinants of post-antibiotic ecological recovery in the gut. Nat. Ecol. Evol. 2020, 4, 1256–1267. [Google Scholar] [CrossRef] [PubMed]
- Lavelle, A.; Hoffmann, T.W.; Pham, H.P.; Langella, P.; Guedon, E.; Sokol, H. Baseline microbiota composition modulates antibiotic-mediated effects on the gut microbiota and host. Microbiome 2019, 7, 111. [Google Scholar] [CrossRef] [PubMed]
- Dethlefsen, L.; Relman, D.A. Incomplete recovery and individualized responses of the human distal gut microbiota to repeated antibiotic perturbation. Proc. Natl. Acad. Sci. USA 2011, 108 (Suppl. S1), 4554–4561. [Google Scholar] [CrossRef] [PubMed]
- Jakobsson, H.E.; Jernberg, C.; Andersson, A.F.; Sjolund-Karlsson, M.; Jansson, J.K.; Engstrand, L. Short-Term Antibiotic Treatment Has Differing Long-Term Impacts on the Human Throat and Gut Microbiome. PLoS ONE 2010, 5, e9836. [Google Scholar] [CrossRef]
- Raymond, F.; Deraspe, M.; Boissinot, M.; Bergeron, M.G.; Corbeil, J. Partial recovery of microbiomes after antibiotic treatment. Gut Microbes 2016, 7, 428–434. [Google Scholar] [CrossRef]
- Zaura, E.; Brandt, B.W.; Teixeira de Mattos, M.J.; Buijs, M.J.; Caspers, M.P.; Rashid, M.U.; Weintraub, A.; Nord, C.E.; Savell, A.; Hu, Y.; et al. Same Exposure but Two Radically Different Responses to Antibiotics: Resilience of the Salivary Microbiome versus Long-Term Microbial Shifts in Feces. mBio 2015, 6, e01693-15. [Google Scholar] [CrossRef]
- Kang, K.; Imamovic, L.; Misiakou, M.A.; Bornakke Sorensen, M.; Heshiki, Y.; Ni, Y.; Zheng, T.; Li, J.; Ellabaan, M.M.H.; Colomer-Lluch, M.; et al. Expansion and persistence of antibiotic-specific resistance genes following antibiotic treatment. Gut Microbes 2021, 13, 1900995. [Google Scholar] [CrossRef]
- Li, H.; Durbin, R. Fast and accurate long-read alignment with Burrows-Wheeler transform. Bioinformatics 2010, 26, 589–595. [Google Scholar] [CrossRef]
- Truong, D.T.; Franzosa, E.A.; Tickle, T.L.; Scholz, M.; Weingart, G.; Pasolli, E.; Tett, A.; Huttenhower, C.; Segata, N. MetaPhlAn2 for enhanced metagenomic taxonomic profiling. Nat. Methods 2015, 12, 902–903. [Google Scholar] [CrossRef]
- Quast, C.; Pruesse, E.; Yilmaz, P.; Gerken, J.; Schweer, T.; Yarza, P.; Peplies, J.; Glockner, F.O. The SILVA ribosomal RNA gene database project: Improved data processing and web-based tools. Nucleic Acids Res. 2013, 41, D590–D596. [Google Scholar] [CrossRef]
- Duran, C.; Ciucci, S.; Palladini, A.; Ijaz, U.Z.; Zippo, A.G.; Sterbini, F.P.; Masucci, L.; Cammarota, G.; Ianiro, G.; Spuul, P.; et al. Nonlinear machine learning pattern recognition and bacteria-metabolite multilayer network analysis of perturbed gastric microbiome. Nat. Commun. 2021, 12, 1926. [Google Scholar] [CrossRef]
- Xiao, L.W.; Zhang, F.Y.; Zhao, F.Q. Large-scale microbiome data integration enables robust biomarker identification. Nat. Comput. Sci. 2022, 2, 307–316. [Google Scholar] [CrossRef]
- Tian, L.; Wang, X.W.; Wu, A.K.; Fan, Y.; Friedman, J.; Dahlin, A.; Waldor, M.K.; Weinstock, G.M.; Weiss, S.T.; Liu, Y.Y. Deciphering functional redundancy in the human microbiome. Nat. Commun. 2020, 11, 6217. [Google Scholar] [CrossRef]
- Magnusdottir, S.; Heinken, A.; Kutt, L.; Ravcheev, D.A.; Bauer, E.; Noronha, A.; Greenhalgh, K.; Jager, C.; Baginska, J.; Wilmes, P.; et al. Generation of genome-scale metabolic reconstructions for 773 members of the human gut microbiota. Nat. Biotechnol. 2017, 35, 81–89. [Google Scholar] [CrossRef] [PubMed]
- Ravikrishnan, A.; Blank, L.M.; Srivastava, S.; Raman, K. Investigating metabolic interactions in a microbial co-culture through integrated modelling and experiments. Comput. Struct. Biotechnol. 2020, 18, 1249–1258. [Google Scholar] [CrossRef]
- Mathieu, B.; Sebastien, H.; Mathieu, J. Gephi: An Open Source Software for Exploring and Manipulating Networks. In Proceedings of the Third International ICWSM Conference, San Jose, CA, USA, 17–20 May 2009. [Google Scholar]
- Shaw, L.P.; Bassam, H.; Barnes, C.P.; Walker, A.S.; Klein, N.; Balloux, F. Modelling microbiome recovery after antibiotics using a stability landscape framework. ISME J. 2019, 13, 1845–1856. [Google Scholar] [CrossRef] [PubMed]
- Shannon, P.; Markiel, A.; Ozier, O.; Baliga, N.S.; Wang, J.T.; Ramage, D.; Amin, N.; Schwikowski, B.; Ideker, T. Cytoscape: A software environment for integrated models of biomolecular interaction networks. Genome Res. 2003, 13, 2498–2504. [Google Scholar] [CrossRef]
- Jang, H.M.; Lee, H.J.; Jang, S.E.; Han, M.J.; Kim, D.H. Evidence for interplay among antibacterial-induced gut microbiota disturbance, neuro-inflammation, and anxiety in mice. Mucosal Immunol. 2018, 11, 1386–1397. [Google Scholar] [CrossRef]
- Tian, P.; Wang, G.; Zhao, J.; Zhang, H.; Chen, W. Bifidobacterium with the role of 5-hydroxytryptophan synthesis regulation alleviates the symptom of depression and related microbiota dysbiosis. J. Nutr. Biochem. 2019, 66, 43–51. [Google Scholar] [CrossRef]
- Gupta, V.K.; Kim, M.; Bakshi, U.; Cunningham, K.Y.; Davis, J.M., 3rd; Lazaridis, K.N.; Nelson, H.; Chia, N.; Sung, J. A predictive index for health status using species-level gut microbiome profiling. Nat. Commun. 2020, 11, 4635. [Google Scholar] [CrossRef]
- Li, Y.; Chen, Y.; Fan, Y.; Chen, Y.; Chen, Y. Dynamic network modeling of gut microbiota during Alzheimer’s disease progression in mice. Gut Microbes 2023, 15, 2172672. [Google Scholar] [CrossRef] [PubMed]
- Ciucci, S.; Ge, Y.; Duran, C.; Palladini, A.; Jimenez-Jimenez, V.; Martinez-Sanchez, L.M.; Wang, Y.; Sales, S.; Shevchenko, A.; Poser, S.W.; et al. Enlightening discriminative network functional modules behind Principal Component Analysis separation in differential-omic science studies. Sci. Rep. 2017, 7, 43946. [Google Scholar] [CrossRef]
- Kuntal, B.K.; Chandrakar, P.; Sadhu, S.; Mande, S.S. ‘NetShift’: A methodology for understanding ‘driver microbes’ from healthy and disease microbiome datasets. ISME J. 2019, 13, 442–454. [Google Scholar] [CrossRef]
- Qin, J.; Li, R.; Raes, J.; Arumugam, M.; Burgdorf, K.S.; Manichanh, C.; Nielsen, T.; Pons, N.; Levenez, F.; Yamada, T.; et al. A human gut microbial gene catalogue established by metagenomic sequencing. Nature 2010, 464, 59–65. [Google Scholar] [CrossRef]
- Moya, A.; Ferrer, M. Functional Redundancy-Induced Stability of Gut Microbiota Subjected to Disturbance. Trends Microbiol. 2016, 24, 402–413. [Google Scholar] [CrossRef] [PubMed]
- Vieira-Silva, S.; Falony, G.; Darzi, Y.; Lima-Mendez, G.; Garcia Yunta, R.; Okuda, S.; Vandeputte, D.; Valles-Colomer, M.; Hildebrand, F.; Chaffron, S.; et al. Species-function relationships shape ecological properties of the human gut microbiome. Nat. Microbiol. 2016, 1, 16088. [Google Scholar] [CrossRef] [PubMed]
- Hardin, G. The competitive exclusion principle. Science 1960, 131, 1292–1297. [Google Scholar] [CrossRef]
- Sommer, F.; Anderson, J.M.; Bharti, R.; Raes, J.; Rosenstiel, P. The resilience of the intestinal microbiota influences health and disease. Nat. Rev. Microbiol. 2017, 15, 630–638. [Google Scholar] [CrossRef]
- Ingrisch, J.; Bahn, M. Towards a Comparable Quantification of Resilience. Trends Ecol. Evol. 2018, 33, 251–259. [Google Scholar] [CrossRef]
- Yeung, A.C.Y.; Richardson, J.S. Expanding Resilience Comparisons to Address Management Needs: A Response to Ingrisch and Bahn. Trends Ecol. Evol. 2018, 33, 647–649. [Google Scholar] [CrossRef]
- Ng, K.M.; Aranda-Diaz, A.; Tropini, C.; Frankel, M.R.; Van Treuren, W.; O’Loughlin, C.T.; Merrill, B.D.; Yu, F.B.; Pruss, K.M.; Oliveira, R.A.; et al. Recovery of the Gut Microbiota after Antibiotics Depends on Host Diet, Community Context, and Environmental Reservoirs. Cell Host Microbe 2020, 28, 628. [Google Scholar] [CrossRef] [PubMed]
- Dogra, S.K.; Dore, J.; Damak, S. Gut Microbiota Resilience: Definition, Link to Health and Strategies for Intervention. Front. Microbiol. 2020, 11, 572921. [Google Scholar] [CrossRef] [PubMed]
- Harvey, E.; Gounand, I.; Ward, C.L.; Altermatt, F. Bridging ecology and conservation: From ecological networks to ecosystem function. J. Appl. Ecol. 2017, 54, 371–379. [Google Scholar] [CrossRef]
- Bascompte, J.; Stouffer, D.B. The assembly and disassembly of ecological networks. Philos. Trans. R. Soc. Lond. B Biol. Sci. 2009, 364, 1781–1787. [Google Scholar] [CrossRef] [PubMed]
- Zou, M.L.; Yang, Z.H.; Fan, Y.; Gong, L.; Han, Z.Y.; Ji, L.; Hu, X.M.; Wu, D. Gut microbiota on admission as predictive biomarker for acute necrotizing pancreatitis. Front. Immunol. 2022, 13, 988326. [Google Scholar] [CrossRef] [PubMed]
- Marcos-Zambrano, L.J.; Karaduzovic-Hadziabdic, K.; Turukalo, T.L.; Przymus, P.; Trajkovik, V.; Aasmets, O.; Berland, M.; Gruca, A.; Hasic, J.; Hron, K.; et al. Applications of Machine Learning in Human Microbiome Studies: A Review on Feature Selection, Biomarker Identification, Disease Prediction and Treatment. Front. Microbiol. 2021, 12, 634511. [Google Scholar] [CrossRef]
- Cao, Y.; Geddes, T.A.; Yang, J.Y.H.; Yang, P. Ensemble deep learning in bioinformatics. Nat. Mach. Intell. 2020, 2, 500–508. [Google Scholar] [CrossRef]
- Ren, Y.; Zhang, L.; Suganthan, P.N. Ensemble Classification and Regression-Recent Developments, Applications and Future Directions. IEEE Comput. Intell. Mag. 2016, 11, 41–53. [Google Scholar] [CrossRef]
- Doan, T.; Arzika, A.M.; Ray, K.J.; Cotter, S.Y.; Kim, J.; Maliki, R.; Zhong, L.N.; Zhou, Z.X.; Porco, T.C.; Vanderschelden, B.; et al. Gut Microbial Diversity in Antibiotic-Naive Children After Systemic Antibiotic Exposure: A Randomized Controlled Trial. Clin. Infect. Dis. 2017, 64, 1147–1153. [Google Scholar] [CrossRef]
- Xiao, C.M.; Wang, J.T.; Su, C.; Miao, Z.L.; Tang, J.; Ouyang, Y.F.; Yan, Y.; Jiang, Z.L.; Fu, Y.Q.; Shuai, M.L.; et al. Associations of dietary diversity with the gut microbiome, fecal metabolites, and host metabolism: Results from 2 prospective Chinese cohorts. Am. J. Clin. Nutr. 2022, 116, 1049–1058. [Google Scholar] [CrossRef]
- Lin, H.; Wang, Q.; Yuan, M.; Liu, L.; Chen, Z.Y.; Zhao, Y.H.; Das, R.; Duan, Y.J.; Xu, X.M.; Xue, Y.G.; et al. The prolonged disruption of a single-course amoxicillin on mice gut microbiota and resistome, and recovery by inulin, Bifidobacterium longum and fecal microbiota transplantation. Environ. Pollut. 2020, 265, 114651. [Google Scholar] [CrossRef]
- Guo, H.; Yu, L.L.; Tian, F.W.; Zhao, J.X.; Zhang, H.; Chen, W.; Zhai, Q.X. Effects of Bacteroides-Based Microecologics against Antibiotic-Associated Diarrhea in Mice. Microorganisms 2021, 9, 2492. [Google Scholar] [CrossRef] [PubMed]
- Hugenholtz, F.; de Vos, W.M. Mouse models for human intestinal microbiota research: A critical evaluation. Cell. Mol. Life Sci. 2018, 75, 149–160. [Google Scholar] [CrossRef]
- Kim, J.S.; Kang, S.W.; Lee, J.H.; Park, S.H.; Lee, J.S. The evolution and competitive strategies of Akkermansia muciniphila in gut. Gut Microbes 2022, 14, 2025017. [Google Scholar] [CrossRef] [PubMed]
- Lee, H.B.; Do, M.H.; Jhun, H.; Ha, S.K.; Song, H.S.; Roh, S.W.; Chung, W.H.; Nam, Y.D.; Park, H.Y. Amelioration of Hepatic Steatosis in Mice through Bacteroides uniformis CBA7346-Mediated Regulation of High-Fat Diet-Induced Insulin Resistance and Lipogenesis. Nutrients 2021, 13, 2989. [Google Scholar] [CrossRef] [PubMed]
- Francino, M.P. Antibiotics and the Human Gut Microbiome: Dysbioses and Accumulation of Resistances. Front. Microbiol. 2015, 6, 1543. [Google Scholar] [CrossRef]
- Cho, I.; Blaser, M.J. The human microbiome: At the interface of health and disease. Nat. Rev. Genet. 2012, 13, 260–270. [Google Scholar] [CrossRef]
- Becattini, S.; Taur, Y.; Pamer, E.G. Antibiotic-Induced Changes in the Intestinal Microbiota and Disease. Trends Mol. Med. 2016, 22, 458–478. [Google Scholar] [CrossRef]
- Derrien, M.; Vaughan, E.E.; Plugge, C.M.; de Vos, W.M. Akkermansia muciniphila gen. nov., sp. nov., a human intestinal mucin-degrading bacterium. Int. J. Syst. Evol. Microbiol. 2004, 54, 1469–1476. [Google Scholar] [CrossRef]
- Sicard, J.F.; Le Bihan, G.; Vogeleer, P.; Jacques, M.; Harel, J. Interactions of Intestinal Bacteria with Components of the Intestinal Mucus. Front. Cell. Infect. Microbiol. 2017, 7, 387. [Google Scholar] [CrossRef]
- Flint, H.J.; Scott, K.P.; Duncan, S.H.; Louis, P.; Forano, E. Microbial degradation of complex carbohydrates in the gut. Gut Microbes 2012, 3, 289–306. [Google Scholar] [CrossRef]
- Belzer, C.; de Vos, W.M. Microbes inside-from diversity to function: The case of Akkermansia. ISME J. 2012, 6, 1449–1458. [Google Scholar] [CrossRef]
- Birchenough, G.; Schroeder, B.O.; Backhed, F.; Hansson, G.C. Dietary destabilisation of the balance between the microbiota and the colonic mucus barrier. Gut Microbes 2019, 10, 246–250. [Google Scholar] [CrossRef] [PubMed]
- Kim, C.H.; Park, J.; Kim, M. Gut microbiota-derived short-chain Fatty acids, T cells, and inflammation. Immune Netw. 2014, 14, 277–288. [Google Scholar] [CrossRef] [PubMed]
- Macfarlane, G.T.; Macfarlane, S. Fermentation in the human large intestine: Its physiologic consequences and the potential contribution of prebiotics. J. Clin. Gastroenterol. 2011, 45, S120–S127. [Google Scholar] [CrossRef]
- Everard, A.; Belzer, C.; Geurts, L.; Ouwerkerk, J.P.; Druart, C.; Bindels, L.B.; Guiot, Y.; Derrien, M.; Muccioli, G.G.; Delzenne, N.M.; et al. Cross-talk between Akkermansia muciniphila and intestinal epithelium controls diet-induced obesity. Proc. Natl. Acad. Sci. USA 2013, 110, 9066–9071. [Google Scholar] [CrossRef] [PubMed]
- Paone, P.; Cani, P.D. Mucus barrier, mucins and gut microbiota: The expected slimy partners? Gut 2020, 69, 2232–2243. [Google Scholar] [CrossRef]
- Wells, J.M.; Brummer, R.J.; Derrien, M.; MacDonald, T.T.; Troost, F.; Cani, P.D.; Theodorou, V.; Dekker, J.; Meheust, A.; de Vos, W.M.; et al. Homeostasis of the gut barrier and potential biomarkers. Am. J. Physiol. Gastrointest. Liver Physiol. 2017, 312, G171–G193. [Google Scholar] [CrossRef]
- Shan, M.; Gentile, M.; Yeiser, J.R.; Walland, A.C.; Bornstein, V.U.; Chen, K.; He, B.; Cassis, L.; Bigas, A.; Cols, M.; et al. Mucus enhances gut homeostasis and oral tolerance by delivering immunoregulatory signals. Science 2013, 342, 447–453. [Google Scholar] [CrossRef]






Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Chen, J.; Zhu, J.; Lu, W.; Wang, H.; Pan, M.; Tian, P.; Zhao, J.; Zhang, H.; Chen, W. Uncovering Predictive Factors and Interventions for Restoring Microecological Diversity after Antibiotic Disturbance. Nutrients 2023, 15, 3925. https://doi.org/10.3390/nu15183925
Chen J, Zhu J, Lu W, Wang H, Pan M, Tian P, Zhao J, Zhang H, Chen W. Uncovering Predictive Factors and Interventions for Restoring Microecological Diversity after Antibiotic Disturbance. Nutrients. 2023; 15(18):3925. https://doi.org/10.3390/nu15183925
Chicago/Turabian StyleChen, Jing, Jinlin Zhu, Wenwei Lu, Hongchao Wang, Mingluo Pan, Peijun Tian, Jianxin Zhao, Hao Zhang, and Wei Chen. 2023. "Uncovering Predictive Factors and Interventions for Restoring Microecological Diversity after Antibiotic Disturbance" Nutrients 15, no. 18: 3925. https://doi.org/10.3390/nu15183925