Abstract
Body sodium (Na) levels must be maintained within a narrow range for the correct functioning of the organism (Na homeostasis). Na disorders include not only elevated levels of this solute (hypernatremia), as in diabetes insipidus, but also reduced levels (hyponatremia), as in cerebral salt wasting syndrome. The balance in body Na levels therefore requires a delicate equilibrium to be maintained between the ingestion and excretion of Na. Salt (NaCl) intake is processed by receptors in the tongue and digestive system, which transmit the information to the nucleus of the solitary tract via a neural pathway (chorda tympani/vagus nerves) and to circumventricular organs, including the subfornical organ and area postrema, via a humoral pathway (blood/cerebrospinal fluid). Circuits are formed that stimulate or inhibit homeostatic Na intake involving participation of the parabrachial nucleus, pre-locus coeruleus, medial tuberomammillary nuclei, median eminence, paraventricular and supraoptic nuclei, and other structures with reward properties such as the bed nucleus of the stria terminalis, central amygdala, and ventral tegmental area. Finally, the kidney uses neural signals (e.g., renal sympathetic nerves) and vascular (e.g., renal perfusion pressure) and humoral (e.g., renin–angiotensin–aldosterone system, cardiac natriuretic peptides, antidiuretic hormone, and oxytocin) factors to promote Na excretion or retention and thereby maintain extracellular fluid volume. All these intake and excretion processes are modulated by chemical messengers, many of which (e.g., aldosterone, angiotensin II, and oxytocin) have effects that are coordinated at peripheral and central level to ensure Na homeostasis.
1. Introduction
Sodium (Na) is frequently considered antagonistic to health, given that a high-salt (NaCl) diet is harmful. However, the appetite for NaCl appears to be an innate mechanism, and an elevated NaCl intake may serve to protect against dehydration [1,2,3]. Maintenance of this ion within appropriate levels (Na balance) is not only an adaptive process necessary for survival but also an essential component of hydromineral homeostasis (Na and fluid balances).
Na is the primary cation in extracellular fluid (ECF) and, along with associated anions, constitutes 90% of ECF osmolality [4,5]. ECF, which contains slightly more than one-third of total body fluid, has a Na concentration of around 144 mOsm/L. Almost all the remaining body fluid is contained in intracellular fluid (ICF), which has a lower concentration of Na (10 mOsm/L) [6]. Given that the maintenance of these water and Na levels is essential for adequate cell functioning and because the cell membrane has low permeability to solutes, water moves by osmosis from areas of lower to higher solute concentration to equalize osmotic ECF and ICF concentrations [7,8]. Hence, Na is essential to maintain electrolyte and water balances (hydromineral regulation).
The Na balance is the difference between the amount of Na absorbed by the gut and the amount excreted via urine, feces, and skin [4,8]. This review does not address other functions associated with this cation, such as the generation of nerve impulses, heart activity, and some metabolic functions.
Two biological mechanisms can alter the Na balance and hydromineral homeostasis [7] (see Figure 1). The first is largely determined by the intake of NaCl, which accumulates in the extracellular space and leads to the displacement of water from cell to extracellular space, producing cell dehydration. In this situation, the appetite for Na is inhibited and its excretion is increased (natriuresis), while the intake of water is enhanced and the excretion of fluid is reduced (antidiuresis). Conversely, a reduction in ECF osmolality/natremia leads to the inhibition of water intake and promotion of diuresis, increasing the appetite for Na and suppressing natriuresis (Figure 1, upper). The second mechanism results from the loss of intravascular fluid (hypovolemia), one of the components of ECF (Figure 1, lower). Diarrhea, vomiting, hemorrhages, sweating, renal disease, and cardiovascular disorders are sometimes accompanied by the loss of intravascular volume. Hypovolemia triggers compensatory behavioral and physiological reactions to conserve Na and body fluids. These include a thirst and appetite for Na, especially for isotonic drinks containing the same sodium chloride concentration as intravascular fluid (0.15 M), as well as antidiuretic and antinatriuretic responses to retain water and Na. Conversely, ECF hypervolemia reduces the intake of water and Na and increases diuresis and natriuresis.
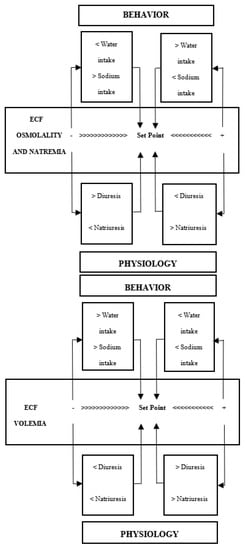
Figure 1.
Behavioral and physiological mechanisms to maintain the osmolality/natremia (upper) and volemia (lower) of extracellular fluid (ECF) (from Mahía and Bernal [7], with permission from Elsevier).
This review first addresses the NaCl intake process (behavioral mechanism) and the neural and humoral pathways that transmit information to the central nervous system (CNS). Next, it examines the brain circuits that monitor the Na balance and stimulate or inhibit NaCl intake. Finally, the process of renal Na excretion is described (physiological mechanism).
2. Behavioral Mechanism: Salt Intake
The Na ion [Na+] is an essential micronutrient that cannot be generated by endogenous processes and must be obtained through the diet [9]. The need to maintain an appropriate Na balance has led living organisms to develop mechanisms that identify the presence of Na the instant it enters the body [10]. The first level of detection is located in the oral cavity, from where it accesses the digestive system and is absorbed in the blood.
2.1. Detection and Processing in the Oral Cavity
Na is detected by the taste system within the oral cavity. The mineral generates a salty taste that guides and maintains its consumption [11,12,13,14]. The gustatory stimulus used as prototype to study the perception of this taste was table salt (NaCl), physiologically the most important salt in the diet, which induces the purest salty taste [11,15], and biologically the most relevant to maintain biochemical balance [16].
In mammals, gustatory receptors are observed throughout the oral cavity, especially on the tongue, where they are located on protuberances designated papillae. There are four types of papillae according to their shape and localization: fungiform (anterior two-thirds of the tongue), foliate (side toward the back), circumvallate (on the back), and filiform (anterior two-thirds of the tongue). Cells that detect NaCl are mainly located in fungiform papillae [12,17]. In general, gustatory papillae can contain several hundreds of taste buds, onion-shaped organs (Figure 2) that house taste receptor cells (50–100 cells each), i.e., the cells responsible for detecting and transducing taste stimuli (transforming chemical signals into neural activity). In the taste bud, these cells are mechanically held together by tight junctions that separate apical areas from the remaining basolateral region, forming an almost impermeable barrier. Only the apical membrane above the tight junctions (2–3% of the total membrane area of the cell) is exposed to saliva, while the basolateral area remains in a relatively constant interstitial milieu [12,13,14,18].
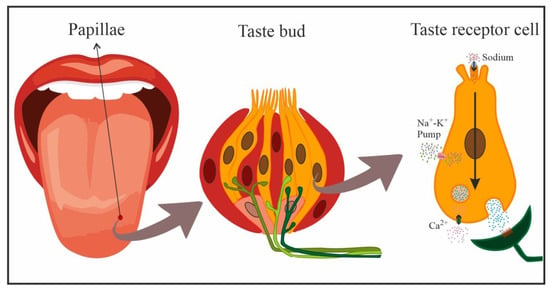
Figure 2.
Schematic representation of cells in a taste bud, and transduction of NaCl taste in a taste receptor cell (yellow: taste receptor cells, red: support cells, pink: immature precursor cells). Taste receptor cells form synapses with afferent nerve fibers that penetrate the taste bud, crossing the basal membrane.
Taste receptor cells are bipolar neuroepithelial cells with finger-like projections (microvilli) on their apical end that project towards the oral cavity. These microvillar processes protrude into the oral milieu through a small opening, the bud pore, in order to sample the chemical environment. When salty stimuli dissolved in saliva come into contact with these microvilli, the taste cells generate a biological signal that is transmitted to afferent neural fibers that penetrate the base of taste buds and extensively ramify. These afferent nerve fibers form synapses with taste receptor cells and respond to excitatory neurotransmitters released by the latter [19], relaying this information to the brain to code the taste sensation [10,14].
There appear to be two types of salty taste receptor cells in the lingual epithelium. One of them, specialized in coding the perceptual quality of “salty” taste, is highly selective for Na, being activated by low–medium concentrations of NaCl and blocked by lingual application of amiloride (a diuretic drug). The other type is activated by high (aversive) concentrations of NaCl (>150 mM), is amiloride-insensitive, and is non-selective for Na (other salts are equally effective). The latter type of taste receptor cells appears to be innervated by neural pathways associated more with aversion than with taste coding [12,14,19,20].
Taste reception begins when chemicals interact with taste receptors located at the apical segment of the taste cells. The receptors that mediate the transduction of salty taste are ion channels [12,19], designated epithelial Na channels (ENaCs), which are specialized in the detection or recognition of sodium chloride [11,12,15,16,19]. Interestingly, and as expected, it has been demonstrated that the gustatory response to Na in deficient animals can be augmented by some of the hormones involved in its regulation, e.g., aldosterone or angiotensin II (Ang II), through an increase in the number of active amiloride-sensitive Na channels in the apical membrane of taste cells [9,21,22].
With the increase in salivary Na concentration, this cation directly enters cells through amiloride-sensitive ENaCs on the apical membrane, following electrochemical gradients. The increase in positive charges inside the taste receptor cell creates a depolarization of the cell membrane that generates action potentials passively conducted towards the basolateral region of the taste receptor cell in a dose-dependent manner, i.e., the action potential frequency is dependent on the stimulus intensity. Here, the opening of voltage-activated Ca2+ gives rise to the influx of Ca2+ ions and the release of the corresponding neurotransmitter onto postsynaptic gustatory afferent fibers (Figure 2) that carry the taste signal to higher-order systems [10,16]. Detection by the nonspecific amiloride-insensitive mechanism is less well known and appears to take place below the apical membrane and taste cell tight junctions (basolateral detection) [13,16].
NaCl taste afferents are the peripheral processes of pseudounipolar neurons located in geniculate cranial ganglia. The chorda tympani nerve, with cell bodies in the geniculate ganglion, appears to be mainly responsible for transmitting information on the presence of Na in the oral cavity to the brain [18,23,24]. Thus, bilateral transection of this nerve severely disrupts the ability to discriminate Na from other salts [25]. In humans, the salty taste is generated at concentrations above 40 mM, a very similar threshold to that in rats (30 mM) but much higher than that in mice (10 mM). The rise in salivary Na+ concentration also increases the neural response (stimulus dependent), meaning that the Na concentration can be estimated as a function of the neural discharge rate. This capacity allows individuals to ingest the appropriate amount of NaCl to maintain homeostasis, i.e., around 1% NaCl (0.17 M), in the ECF [10,20,26].
Accordingly, the chorda tympani nerve appears to contain two types of fibers that differ in their sensitivity to NaCl; one type responds solely to salty taste (Na+ and Li+; NaCl specialist, highly specific fibers), while the other responds to a wide range of acid and salty stimuli (NaCl generalist), including Na [3,13,18]. Activity of the former but not the latter is blocked by applying amiloride on the tongue in various animal species, as expected [10].
Chorda tympani nerve fibers with oral saltiness information terminate in a highly specific region of the rostral nucleus of the solitary tract (rNST), especially at its most anterior pole and lateral division [18,27,28].
2.2. Sodium Detection and Processing in the Gastrointestinal System
Various studies have demonstrated that the most caudal levels of the digestive system are also prepared for Na detection (visceral sensory signals) [29,30,31,32,33].
Information on the entry of Na can be transmitted from the gastrointestinal system to the CNS in a rapid manner, via neural mechanisms, or more slowly, via the circulatory system (humoral pathway) [29,32,33,34,35,36].
The neural pathway is vagal in nature [37], and Na+ detection by subdiaphragmatic vagal afferents is well documented [33,38]. The presence of NaCl-sensitive receptors has been demonstrated in the gastrointestinal mucosa [39,40,41,42] and at hepatic level [43,44]. These receptors appear to be connected to small-diameter fibers, mostly unmyelinated vagal fibers that respond to the presence of hypertonic NaCl with a mean latency of around 10–20 s [39,40,41,42]. The first relay of visceral-vagal information from post-oral segments of the digestive system takes place in caudal segments of the nucleus of the solitary tract (NST), from which it is remitted to other brain structures [27,45,46].
Na absorption into the circulatory system largely takes place in the distal small bowel and the colon [47,48,49]. In these digestive segments, Na+ is taken up through absorptive cells of the epithelium, which connect on one side with the intestinal lumen (apical region, brush border) and on the other (basolateral region) with blood and lymphatic capillaries. In the small intestine, these cells are organized into digitiform projections or intestinal villi that significantly augment (around 600-fold) the absorption surface area; however, this organization is less marked towards distal ends of the intestine and is virtually absent at the most distal region of the colon [50].
Na absorption is transcellular in the intestine. In a first step, the ion penetrates from the intestinal lumen into the cytoplasm of the absorptive epithelial cell following electrochemical gradients and strongly assisted by Na-dependent transporters (e.g., Na+/glucose and Na+/cotransporter amino acids or Na–hydrogen exchangers (NHEs), or by Na reuptake through ENaCs mainly present in the distal colon [50,51]. Once within the epithelial cells, Na+ is actively transported towards paracellular spaces by the Na+/K+ ATPase pump in the basal and lateral membranes of the cells, avoiding retrograde diffusion, from where it enters the bloodstream [47,48,50].
The modality of Na+ absorption and the intestinal segment in which it occurs mainly depends on the circumstances. In this way, it appears to occur preferentially in the small intestine after meals, coupled with nutrient absorption, nutrient-dependent Na absorption, Na+/glucose, and Na+/cotransporter amino acids. In contrast, it largely takes place in the ileum and colon through NHE or ENaCs during interdigestive periods [50,52]. Under Na deficiency conditions, hormones such as aldosterone can increase Na uptake in the intestinal epithelium, stimulating the expression of the transporters that participate in its absorption (especially at colon level). Consequently, there is virtually no Na loss under these circumstances, with <0.5% of the intestinal content of Na being excreted via the feces under normal conditions [47,51,53,54].
Once in the circulatory system, Na is widely distributed throughout the organism, accessing circumventricular brain structures, which lack a blood–brain barrier, via fenestrated capillaries [55,56], and accessing cerebrospinal fluid (CSF) via ion transporters (e.g., ENaC) in epithelial cells of the choroid plexus [57,58,59,60]. Blood and CSF both have a [Na+] concentration of 144 mOsm/L [61].
3. Central Nervous System
The search for and consumption of water and NaCl depend on neuroanatomical circuits that are far from completely elucidated [8,55,62]. They include numerous interconnected structures widely distributed throughout the CNS, particularly the hypothalamus (HT) and brainstem (see Figure 3). Information from the blood and CSF is processed by circumventricular organs (CVOs) such as the subfornical organ (SFO), which lies in the midline of the third ventricle (IIIV), and the area postrema (AP), which lies in the midline of the fourth ventricle (IVV); information from the chorda tympani nerve originating in the oral cavity is processed by the rNST; and information from the vagus nerve originating in the digestive system is processed by the caudal NST (cNST). Accordingly, these three structures constitute the first information relay in the CNS.
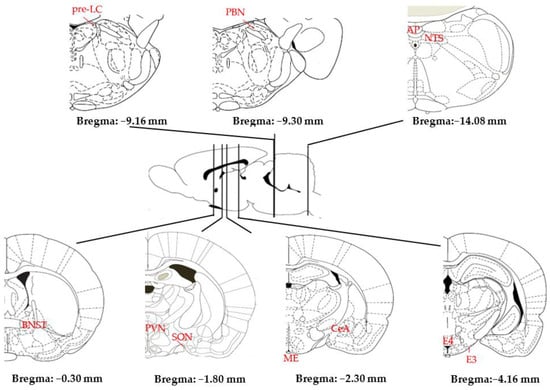
Figure 3.
Main structures of the rat brain involved in Na homeostasis (AP: area postrema; BNST: bed nucleus of the stria terminalis; CeA: central amygdala; E3–E4: medial tuberomammillary nuclei; ME: median eminence; NST: nucleus of the solitary tract; PBN: parabrachial nucleus; pre-LC: pre-locus coeruleus; PVN: paraventricular nucleus; SFO: subfornical organ; SON: supraoptic nucleus) (adapted from [63]).
3.1. Circumventricular Organs and Nucleus of the Solitary Tract
CVOs are characterized by an extensive network of fenestrated capillaries that permit the entry of Na. These structures also express receptors for hormones that stimulate NaCl intake such as Ang II [64], which is generated by activity of the renin–angiotensin system (RAS) (see Section 4) [8,55,65,66,67].
The SFO is the main CVO for sampling the plasma and CSF. It contains glutamatergic neurons [55,57] with Ang II-sensitive AT1 receptors that stimulate Na intake in Na+-depleted and hypovolemic states [8]. In this way, NaCl intake is abolished in AT1a-KO mice or after the intracerebroventricular administration of losartan, an AT1 blocker, under Na-depleted conditions [65]. The activity of these Ang II-sensitive glutamatergic neurons is suppressed in states of cell dehydration by a set of GABAergic interneurons of the SFO [8,55,57]. These GABAergic interneurons are activated by ependymal cells that separate the SFO of IIIV and monitor Na+ ([Na+]) increases in CSF and by astrocytes that monitor [Na+] increases in plasma [57].
The CVO AP is located ventromedial to the fourth ventricle [46,68,69], but its involvement in salt intake is less well understood than that of SFO [58]. The activation of c-Fos in the AP after hypertonic saline injections but not in an Na-deprived state [70] suggests that it forms part of NaCl intake-inhibiting circuits [8]. This inhibitory function may be mediated by serotoninergic neurons that express ENaCs and may detect increases in plasma [Na+] to generate a feeling of satiety for this electrolyte [71].
The NST constitutes the first central relay of both gustatory information from the oral cavity and visceral information from the digestive system. The cNST receives visceral-vagal information originating in post-oral segments of the digestive system as well as information from the AP [27,45,46]. The cNST contains neurons sensitive to aldosterone, a lipophilic hormone that crosses the blood–brain barrier. These neurons are activated by chronic dietary Na+ deprivation, driving NaCl appetite, and are inactivated after NaCl intake [8]. Conversely, the rNST appears to be involved in the inhibition of NaCl intake. It is the first brain relay for information on oral saltiness carried by the chorda tympani nerve from the tongue and orofacial cavity and possesses a degree of sensorial and viscerotropic organization, i.e., neurons responding to receptors in the anterior tongue are more likely found in the anterior half and those responding to receptors in the posterior oral cavity are in a more caudal position [18,27,46].
Hence, circuits that stimulate or inhibit NaCl intake originate in the SFO, AP, and NST (Figure 4) (see [8] for a review).
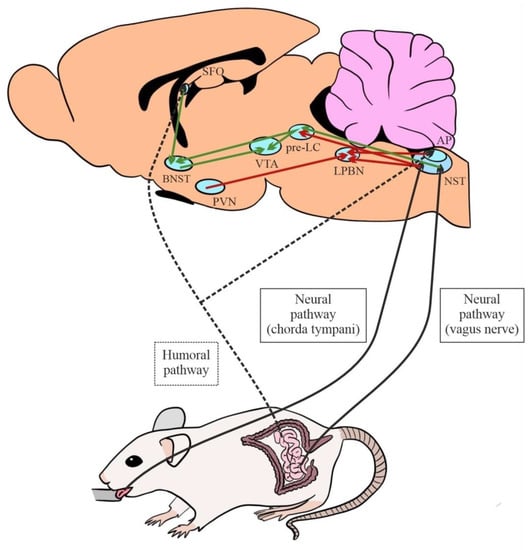
Figure 4.
Afferent neural and humoral pathways involved in salt intake and main central excitatory (green) and inhibitory (red) circuits (AP: area postrema; BNST: bed nucleus of the stria terminalis; LPBN: lateral parabrachial nucleus; NST: nucleus of the solitary tract; pre-LC: pre-locus coeruleus; PVN: paraventricular nucleus; SFO: subfornical organ; VTA: ventral tegmental area).
3.2. Excitatory and Inhibitory Circuits
SFO glutamatergic efferents activated by Ang II [72] and aldosterone-sensitive cNST neurons [73] project to the ventral region of the bed nucleus of the stria terminalis (vBNST), which therefore appears to integrate both humoral and neural signals [55,69]. This circuit stimulates Na intake, and bilateral electrolytic lesions in the vBNST were found to markedly decrease 0.3M NaCl intake by mice under Na-depleted conditions [65]. vBNST neurons project to the ventral tegmental area (VTA) for the regulation of appetitive motivation [74,75,76].
Other efferents that originate in the cNST have proven important to stimulate NaCl intake. One is directed towards the pre-locus coeruleus (pre-LC), where the activation of neurons can stimulate the intake of Na concentrations that are usually aversive [77]. This motivational system can be promoted by projections from the pre-LC towards the vBNST and VTA, among other structures [76]. These pre-LC neurons are inhibited by rNST projections related to the oral detection of Na [8].
An increase in physiological Na levels triggers the activation of inhibitory circuits that limit intake of this electrolyte. In this regard, a decisive role has been described for serotoninergic pathways that project from the AP to the lateral parabrachial nucleus (lPBN) [78]. In this way, pharmacological blockade by the intraparabrachial administration of 5-HT antagonists was found to increase Na consumption [79,80,81,82,83,84,85,86]. Other afferents to the lPBN derive from the rNST. Signals from receptor cells of the tongue that are activated by NaCl intake and project to the rNST were also found to converge in the lNPB, inhibiting its intake [46,87,88]. In addition, lPBN activity and Na intake suppression appear to be enhanced by oxytocinergic afferents from the parvocellular division of the hypothalamic paraventricular nucleus (pPVN) [89]. Inhibition of the lPBN has been related to the intake of highly concentrated (usually aversive) NaCl solutions, associated with endogenous opioid signaling and the central amygdala (see [8]).
3.3. Posterior Hypothalamus in Sodium Intake Regulation
The posterior HT is much less explored but no less important for Na homeostasis. Clinical and experimental findings have shown that lesions of mediobasal structures of the posterior HT produce major imbalances in body Na levels and osmolality [90,91,92,93]. Accordingly, Bacic et al. [94] described severe hyponatremia in soldiers with bullet wound injuries to the base of the brain and/or hypothalamic areas adjacent to the IIIV. In some cases, patients are diagnosed with cerebral salt wasting (CSW) syndrome, characterized by hyponatremia and hypovolemia, due to the early natriuresis/diuresis resulting from increased natriuretic peptide activity The most common approach to the restoration of volume and sodium levels in patients with CSW is to administer hypertonic saline solutions and/or salt tablets and limit the free intake of water to minimize the pathological natriuresis, diuresis and polydipsia observed in this disorder [95,96].
These data may suggest a parallelism between CSW patients and animals with lesions in mediobasal hypothalamic structures [97,98,99,100,101,102]. Thus, damage to the median eminence (ME) may interrupt some of the brain systems proposed to be involved in hydromineral regulation (e.g., posterior hypothalamus–hypophyseal axis), causing major derangements in the neuroendocrine control and regulation of water and sodium metabolism [92,103].
Transient activation of the affected neural tissue produced by the application of electric current during surgery [104,105] can generate the increased natriuretic response [92] observed in animals with CSW syndrome [95,96,106]. This mechanism may explain the early excretion of Na (hyponatremia) and hypovolemia developed by ME-lesioned animals during the first few hours post-surgery [92]. It is likely, as in patients with CSW, that the greater preference for hypertonic saline solutions (1.5% NaCl, aversive taste) without a proportional enhancement in water intake observed in ME rats a few hours after the lesion may constitute optimal strategies to restore the volume and composition of extracellular body fluid, of which sodium is one of the main constituents [102].
Other animal studies reported that disruption of the posterior hypothalamus–pituitary axis can cause hypothalamic CDI [92,100,101,103,107]. The altered fluid retention mechanisms observed in these animals can also be accompanied by hypernatremia and/or plasma hyperosmolality [92,108].
Continuing in the clinical setting, findings in patients with schizophrenia included significantly elevated plasma Na levels (hypernatremia) and polydipsia (5–20 L daily), whereas the value of other electrolytes (K+, Ca2+) remained within normal ranges [109,110,111]. Postmortem studies in these patients revealed a lower bilateral neuronal density (34%) in regions of the posterior HT with respect to individuals without this disorder [112]. Various clinical studies have observed hypernatremia symptoms after injuries to mediobasal hypothalamic areas that affect mechanisms responsible for the conservation of fluids during the chronic phase of central diabetes insipidus (CDI) [97,113].
These different clinical findings are in agreement with the experimental data recorded in our laboratory after electrolytic lesions of tuberomammillary nuclei of the posterior hypothalamus [93].
Tuberomammillary System
The tuberomammillary (TM) system is formed by five nuclei (designated E1–E5) located in the posterior HT on both sides of the mammillary recess [114,115,116,117]. It contains neurons with dendritic processes that extend throughout the ependymal layer of the IIIV, a position that facilitates the response to certain molecules and neuroactive substances in CSF [118,119,120]. Some of its efferents project to the magnocellular division of the paraventricular nucleus (mPVN) and supraoptic nucleus, hypothalamic structures that are responsible for the synthesis of antidiuretic hormone (ADH) and oxytocin (OXY) [7]. Thus, activation of TM cells was found to produce the depolarization of mPVN and SON [121,122,123,124,125,126] and the release of ADH and OXY into the posterior pituitary, from where they accessed the general circulation to modulate kidney function [127,128,129]. Conversely, pharmacologic blockade of TM cells significantly decreased the secretion of ADH [124] and OXY [123,130] in response to osmotic stimuli.
Electrolytic lesions of the medial TM (mTM) nuclei E3 and E4 produce a polydipsic response apparently related to increases in plasmatic Na and difficulties in its excretion [103]. In a study of rats, intraperitoneal administration of hypertonic NaCl produced an exaggerated polydipsic response in these animals [100,101]. Conversely, the administration of OXY, a natriuretic hormone (see Section 4), was found to favor Na excretion and abolish the polydipsia generated by lesion of the E3 nucleus [93]. According to these findings, mTM lesions generate a polydipsic response that may be explained by hydromineral imbalance in the mTM-mPVN/SON–kidney axis, which is involved in hydromineral regulation and is responsible for the secretion of ADH and OXY.
4. Physiological Mechanism: Kidney and Sodium Excretion
The third component of Na balance is renal Na excretion (natriuresis), which is dependent on kidney function. Although some Na is lost in feces and sweat, most of the Na in the body is excreted via the kidney. Hence, the kidney plays a central role in achieving a balance between the intake and excretion of NaCl and water, ensuring that ECF volume and cardiovascular homeostasis are maintained at a level consistent with normal physiology [131,132].
Each human kidney contains more than a million nephrons, its functional units (Figure 5). Blood flows into the kidney through the afferent arteriole, is filtered in a network of small blood vessels, the glomerulus, and is deposited in Bowman‘s capsule, the first element of the nephron. The glomerular filtrate successively passes from Bowman‘s capsule through the proximal tubule, the loop of Henle, with its descending and ascending limb, the distal tubule, and the collecting duct. The walls of eight or ten collecting ducts unite to form a larger single collecting duct that secretes urine. Around 65% of the Na that enters the nephrons is reabsorbed from the renal tubules into interstitial fluid and through peritubular capillaries into the general circulation. The ascending limb of the loop of Henle actively secretes Na into the interstitial space but is impermeable to water, so that the interstitial space between ascending and descending limb is hypertonic (see Figure 5). This hypertonicity creates an osmotic gradient between the fluid within the descending limb and the interstitial space, which removes the water from the descending limb and thereby increases the fluid tonicity within it. Given that the ascending limb is impermeable to water, Na continues to be actively pumped out [132,133].
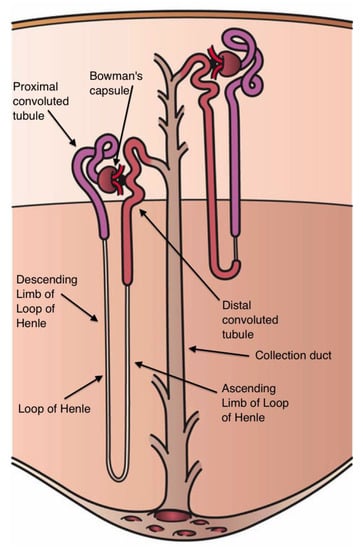
Figure 5.
Components of the nephron (From Holly Fischer, Wikipedia commons).
Natriuresis therefore depends on the ratio between glomerular filtration rate, Na+ tubular reabsorption, and secretion of the fluid containing Na+ into the nephron tubular lumen [132,134].
Na excretion can be modulated by the kidney via renal sympathetic nerves, arterial pressure, and endocrine factors such as the RAS, aldosterone, cardiac natriuretic peptides, ADH, and OXY [4,131,135].
4.1. Renin–Angiotensin System
At the end of the 19th century, Tigerstedt and Bergmann [136] demonstrated that injections of renal extracts elevate the blood pressure of normotensive animals. This was the first evidence that the kidney, long acknowledged to be an exocrine organ, might also possess important endocrine functions in the regulation of cardiovascular and body fluid homeostasis. Tigerstedt and Bergmann designated the biologically active substance isolated from the kidney as “renin”. This enzyme is synthesized by specialized granular cells (“juxtaglomerular cells”) of the afferent arteriole of each glomerulus. Renin converts plasma angiotensinogen [137,138,139] into angiotensin I, which is in turn transformed by angiotensin-converting enzyme into Ang II [140].
The main role of the RAS is to maintain the volume of ECF in hypovolemic situations, sustaining arterial pressure and exerting direct and indirect effects on renal Na excretion [141,142,143]. Indirect effects of the RAS include a decrease in peritubular capillary pressure (efferent arteriolar constriction) and increase in Na transport. Direct effects include the action of Ang II on AT1 receptors in the kidney to mediate hemodynamic and tubular function, including afferent and efferent arteriolar vasoconstriction and the enhancement of Na and fluid reabsorption [142,144]. Activation of AT1 receptors increases activities of the proximal tubule apical NHE and basolateral Na-bicarbonate cotransporter. Ang II also enhances activities of the Na-chloride cotransporter in the distal tubule and the ENaC in the collecting duct [145,146,147,148]. Hence, Ang II directly increases the receptor-mediated reabsorption of NaCl (antinatriuresis) and water (antidiuresis) in multiple nephron segments in order to conserve ECF volume [142,149]. In addition, as mentioned above, Ang II acts on AT1 receptors in the SFO to stimulate Na intake [8].
Ang II also stimulates the secretion of aldosterone and the sympathetic nervous system and elevates the blood pressure [150,151].
4.2. Aldosterone
In 1936, Curt Richter reported that Na excretion is reduced by the administration of adrenal cortical extracts, whereas extreme Na loss results from adrenalectomy [152]. This antinatriuretic effect is mediated by the action of aldosterone [153], a mineralocorticoid synthesized from zona glomerulosa cells of the adrenal cortex [154]. Its synthesis depends on the hypothalamic–pituitary–adrenal neurosecretory axis (hypothalamic corticotropin-releasing hormone and anterior pituitary adrenocorticotropin hormone), while the strongest stimuli triggering its secretion are hyperkalemia and Ang II [155,156,157,158]. In fact, zona glomerulosa cells are more responsive to an increase in plasma potassium levels (hyperkalemia), while aldosterone accounts for only a small percentage of the Na reabsorbed in the nephron [159,160].
After its release, aldosterone acts on mineralocorticoid receptors located in the distal tubule and collecting duct of the nephron to increase apical Na influx, Na reabsorption, and potassium excretion through Na/ENaCs and potassium channels by Na+/K+ ATPase [161,162,163,164,165,166]. The blood pressure is also increased by these changes [167].
4.3. Pressure Natriuresis
In 1854 and 1949, respectively, Goll and Selkurt et al. observed that acute pressure natriuresis can occur without major changes in renal blood flow or glomerular filtration rate [131,165]. The nephron sites most sensitive to renal perfusion pressure changes are the proximal tubules and/or descending limb of the loop of Henle in deep nephrons [168,169].
Increases in renal perfusion pressure [150,170] appear to reduce Ang II levels and increase nitric oxide (NO) production, directly inhibiting tubular Na reabsorption. Although the action mechanisms of NO are not well known, the presence of various isoforms of its synthesis enzyme, NO synthase, has been identified in endothelial cells of the renal vasculature and glomerular capillaries, renal sympathetic nerves, renal tubules, macula densa, and medullary regions [171]. Conversely, a reduction in renal perfusion pressure produces an increase in tubular Na reabsorption and a decrease in Na excretion [150].
Acute increases in arterial pressure restore Na excretion to normal values when the amount of Na excreted via pressure natriuresis is greater than its intake [150,167]. A rise in renal perfusion pressure depresses proximal tubular fluid reabsorption, increases fluid excretion, and reduces ECF volume, producing a short-term reduction in blood pressure to its baseline value [131]. Consequently, a balance is achieved between pressure natriuresis and ECF volume [150,167].
4.4. Sympathetic Nervous System
Muller and Barajas described kidney innervation by sympathetic post-ganglionic fibers in 1972. Norepinephrine (NE)-containing renal sympathetic nerve terminals are in direct contact with all renal tubular segments and with juxtaglomerular granular cells [172].
Renal sympatho-excitation produces vasoconstriction, antinatriuresis, and antidiuresis. Conversely, renal sympatho-inhibition results in vasorelaxation, natriuresis, and diuresis [171,172,173,174]. Sympathetic NE acts on the kidney at the following primary sites: renal vasculature, causing vasoconstriction; epithelial cells of proximal tubules, ascending thick limb of the loop of Henle and distal tubules, stimulating water and Na reabsorption; and the juxtaglomerular apparatus, producing the release of renin [131,171,172,174,175]. These effects result from the action of NE on G proteins coupled kidney adrenoceptors. When stimulated, β1-adrenoceptors of juxtaglomerular granular cells cause the release of renin [176], while α1-adrenoceptors of vascular smooth muscle cells produce a contraction that reduces the renal blood flow and/or glomerular filtration rate [177], and α1-adrenoceptors in nephron tubules enhance the cAMP accumulation induced by ADH to promote Na and water reabsorption, acting via the Na+/K+ ATPase pump [171,178]. Conversely, activation of tubular α2-adrenoceptors reverses ADH-induced Na and water retention [179]. Furthermore, presynaptic sympathetic α2-adrenoceptors decrease the amount of NE released by subsequent action potentials [171].
The sympathetic nervous system interacts with the RAS and with cardiac natriuretic peptides [180,181]. Ang II was found to enhance NE neurotransmission via AT1 receptors [171,182] and the blockade of AT1 receptors blunted renal nerve-mediated reductions in urinary Na excretion and flow rates [183]. It was also reported that atrial natriuretic peptide (ANP) can negatively modulate noradrenergic neurotransmission and reduce NE release from the rat adrenal medulla [184].
4.5. Cardiac Natriuretic Peptides
In 1981, De Bold et al. observed natriuresis and diuresis after the injection of an atrial homogenate [185], later identified as ANP, revealing the heart to be an endocrine organ [186,187,188,189]. ANP is also found in the brain, and perikarya containing elevated ANP levels were observed in the preoptic-hypothalamic area and magnocellular hypothalamic nuclei of rats [184]. Cardiac tissue also produces brain natriuretic peptide (BNP), initially identified in the brain [190,191]. ANP and BNP are secreted in response to cardiac wall stretching and extracellular volume expansion [192], while hypovolemia reduces the release of these natriuretic peptides [193,194].
In the kidney, ANP and BNP act on natriuretic peptide receptor A (NPR-A) in juxtaglomerular cells, proximal tubule, thin and thick ascending loop of Henle, and collecting duct [195,196,197,198,199]. ANP and BNP generate diuresis and natriuresis by inducing vasodilation of the afferent glomerular arteriole and vasoconstriction of the efferent glomerular arteriole, increasing the glomerular filtration rate [200]. The NPR-A also modulates activity of the cGMP-gated cation channel that mediates electrogenic Na absorption [195,201], inhibiting water and Na reabsorption [202,203,204,205,206].
As expected, natriuretic peptides have antagonistic effects on the RAS, given that ANP decreases renin secretion from juxtaglomerular cells, reduces Ang II, blocks aldosterone synthesis and release, and suppresses ADH secretion [207,208,209,210].
4.6. Antidiuretic Hormone
The role of the pituitary gland in body fluid homeostasis was first noted in the early 1900s after observing that the exogenous administration of a pituitary extract reduced the urine flow [211].
The hormone responsible for these effects is ADH, which is synthesized in the somata of magnocellular neurons of hypothalamic paraventricular (mPVN), supraoptic (SON), and accessory neurosecretory nuclei. Their axons pass through the median eminence (ME), forming the neurohypophyseal stalk, and terminate in the posterior pituitary, entering the circulation thereafter [7,212].
Increases in ADH release are more sensitive in response to a rise in plasma Na concentration than to hypovolemia or hypotension [213,214]. It acts via two types of receptors in the kidney: receptor V2, which mediates both antidiuresis and antinatriuresis; and receptor V1a, which mediates natriuretic effects. ADH increases water reabsorption by acting on V2 receptors in the collecting duct of the nephron [193], promoting the production and expression of water aquaporin 2 (AQP2) channels that permit water reabsorption [215]. Consequently, tubular fluid traversing this segment of the nephron equilibrates osmotically with the hyperosmotic interstitium [134,216]. In this way, the kidneys can process urine that has a concentration higher than serum osmolality [133], with the collecting ducts playing a key role in both the dilution and concentration of urine (excretion and conservation of water). Conversely, this region is impermeable to water under conditions of normal osmotic equilibrium in the absence of ADH, resulting in the continuing dilution of luminal fluid and excretion of diluted urine [135]. V2 receptors are also involved in the action of ADH to reduce renal Na excretion [217]. ADH Na reabsorption is mediated by ENaCs in the thick ascending limb [147,218], and ADH increases the number of ENaCs [219] and the apical Na+ reabsorption [220]. This action also produces a reduction in water excretion [214,221,222,223,224]. In this way, the activation of V2 favors antidiuresis associated with antinatriuresis in the presence of hypovolemia [225,226,227]. On the other hand, when ADH is introduced at higher doses, its activation of V1a receptors in connecting tubules and collecting ducts inhibits renal Na reabsorption and favors natriuresis [134,217,228]. These findings suggest that V1a and V2 receptors are activated in states of hypernatremia, promoting natriuretic and antidiuretic effects [134,225,226,227].
ADH is the only hormone traditionally related to diabetes insipidus (DI), characterized primarily by the excretion of a large volume of diluted, “tasteless” urine and secondarily by excessive water intake (polydipsia) and the presence of hypernatremia [229,230,231]. Two main DI types can be distinguished: central, neurogenic, neurohypophyseal, or hypothalamic DI (CDI), associated with deficient secretion of ADH, and nephrogenic DI, characterized by renal insensitivity to ADH.
CDI has been treated with ADH analogs [232], although nephrectomy does not prevent the polyuric response [233], indicating the implication of additional factors. In this regard, most alterations caused by CDI affect the secretion of both ADH and OXY [7,212,232,234,235,236], suggesting the possible involvement of OXY.
4.7. Oxytocin
Besides the aforementioned vasopressinergic neurons, the magnocellular neurosecretory system also contains cells responsible for OXY synthesis [237]. As in the case of ADH, OXY is generated in response to increased plasmatic osmolality (hyperosmolality and hypernatremia) and hypovolemia [7,212]. OXY is especially involved in the excretion of body Na [229,238,239,240,241,242], even at physiological plasma concentrations [243]. This physiological secretion appears to be triggered directly by increases in glomerular filtration rate [244] and reductions in tubular Na reabsorption [245] and indirectly by the cardiac secretion of ANP [246]. In addition, ADH and OXY have been reported to exert synergic natriuretic effects [247,248].
Urine excretion, water intake [229], and Na appetite [240] are increased by the natriuretic effects of OXY in food-deprived rats and in rats fed ad libitum with a low-sodium diet. Hence, OXY administration and low-sodium diets can produce negative Na and water balances [243] that can be counteracted by consuming Na in the diet or by hypertonic NaCl administration [239,240].
Over the past few decades, the role of OXY and Na balance disorders has been investigated in patients and animal models of CDI [212,234]. As a result, treatments of this disease have started to involve not only ADH but also OXY administration and food-deprivation and low sodium diets, both in animals [92,101,242,249,250,251] and humans [252,253,254]. The aim is to exert natriuretic and antidiuretic effects, potentially reducing the characteristic hypernatremia in CDI [92,253,255,256].
As already noted above, intraperitoneal administration of OXY was found to favor Na excretion and reduce the polydipsic response to the lesion of medial TM nuclei [93].
4.8. Salt Sensitivity and Hypertension
As already noted, kidney function can affect not only sodium excretion but also blood pressure, and some individuals respond to high salt intake with an increase in blood pressure [257]. In this context, salt sensitivity refers to changes in blood pressure levels parallel to changes in salt intake [258,259].
The kidney responds to increases in salt intake by activating natriuretic mechanisms (pressure natriuresis and cardiac natriuretic peptides) and inhibiting antinatriuretic ones (RAS-aldosterone and sympathetic nervous system). The former mechanisms are not sufficiently activated in salt-sensitive individuals, and the latter are not effectively suppressed [258,260]. Salt intake is of major clinical relevance in salt-sensitive individuals as a key element in the pathophysiology of hypertension [257]. The gut microbiota has also been related to salt sensitivity and hypertension development [261,262].
The trillions of bacteria, viruses, protozoa, archaea. and fungi that reside on the mucosal surface of the human gastrointestinal tract (gut microbiota) have proven crucial to maintain the homeostasis, metabolism, and immunity of the host [263,264]. The involvement of the gut microbiome in these functions would explain why its alteration (dysbiosis) is associated with numerous diseases, including hypertension [264,265].
The diet is an important determinant of the composition of the gut microbiota [264]. Recent findings demonstrated that a high salt intake is associated with major changes in the gut microbiome (modifications in bacterial taxa, dysbiosis) that are in turn related to salt sensitivity in hypertension [262,265]. A significant increase in Firmicutes, with an overall increase in the Firmicutes/Bacteroidetes ratio, has been described in humans with hypertension [266], leading to the proposal of the microbiota as therapeutic target in this disease. In this way, various prebiotics, probiotics, and postbiotics have been proposed as potential treatments due their antihypertensive effects [265].
The mechanisms by which gut microbiota disorders may cause hypertension are not fully elucidated, although the production of different circulating metabolites has been related to renal sodium regulation, vascular function, and immune processes [265].
5. Concluding Remarks
Na is essential for life, and its regulation depends on the balance between its intake (behavioral component) and its loss, mainly controlled by the kidney (physiological component). Numerous brain structures are involved in monitoring Na levels and forming circuits to stimulate or inhibit its intake in accordance with homeostatic and hedonic factors.
Many of the chemical substances involved in the above processes act in a coordinated manner. They are responsible for intrabrain effects as neurotransmitters that regulate the behavioral component and for hormonal effects on the oral cavity and gastrointestinal tract, modulating Na hunger, and on the kidney, regulating the physiological component (natriuresis). In Na-deficient animals, aldosterone enhances the gustatory response to Na by acting on taste cells [22] and increases its uptake in the intestinal epithelium [47,51,53,54]. In the CNS, aldosterone stimulates Na intake by modulating the activity of the NST [73]. Finally, it increases Na reabsorption by acting on nephrons [161,162,163,164,165,166]. Ang II also increases the gustatory response to Na in Na-deficient animals [22], activates glutamatergic neurons of the SFO that stimulate Na intake [8], and has renal effects that enhance Na reabsorption [142,147]. Accordingly, aldosterone and Ang II protect the organism against hypovolemic and hyponatremic states, whereas other chemical substances protect the organism against hypervolemia and hypernatremia. This is the case of OXY, which acts on the lPBN to inhibit Na intake [89] and acts on the kidney to stimulate Na excretion [243].
Besides Na homeostasis, these and other brain structures and chemical messengers participate in the regulation of other closely related processes (e.g., hydric homeostasis and cardiovascular regulation), hampering differentiation between direct and indirect effects. In this way, variations in body Na levels affect not only the intake and excretion of this solute but also the intake and excretion of fluids, as observed in Figure 1.
Author Contributions
All authors listed have made a substantial, direct and intellectual contribution to the work and approved it for publication. All authors have read and agreed to the published version of the manuscript.
Funding
This research was funded by Spanish Ministry of Economy, Industry and Competitiveness grant number PSI2017-89324-C2-1-P and The APC was funded by University of Granada.
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
Not applicable.
Acknowledgments
Conflicts of Interest
The authors declare no conflict of interest.
References
- Fessler, D.M.T. An Evolutionary Explanation of the Plasticity of Salt Preferences: Prophylaxis against Sudden Dehydration. Med. Hypotheses 2003, 61, 412–415. [Google Scholar] [CrossRef] [PubMed]
- Leshem, M. Salt Need Needs Investigation. Br. J. Nutr. 2020, 123, 1312–1320. [Google Scholar] [CrossRef] [PubMed]
- Schulkin, J. Sodium Hunger: The Search for a Salty Taste; Cambridge University Press: Cambridge, UK, 1991; ISBN 978-0-521-35368-7. [Google Scholar]
- Bie, P. Mechanisms of Sodium Balance: Total Body Sodium, Surrogate Variables, and Renal Sodium Excretion. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2018, 315, R945–R962. [Google Scholar] [CrossRef] [PubMed]
- Strazzullo, P.; Leclercq, C. Sodium. Adv. Nutr. 2014, 5, 188–190. [Google Scholar] [CrossRef] [PubMed]
- Verbalis, J.G.; Stricker, E.M. Neuroendocrine Regulation of Fluid Intake and Homeostasis. In Neuroendocrinology in Physiology and Medicine; Conn, P.M., Freeman, M.E., Eds.; Humana Press: Totowa, NJ, USA, 2000; pp. 317–334. ISBN 978-1-61737-153-0. [Google Scholar]
- Mahía, J.; Bernal, A. Animal Models for Diabetes Insipidus. Handb. Clin. Neurol. 2021, 181, 275–288. [Google Scholar] [CrossRef]
- Noda, M.; Matsuda, T. Central Regulation of Body Fluid Homeostasis. Proc. Jpn. Acad. Ser. B Phys. Biol. Sci. 2022, 98, 283–324. [Google Scholar] [CrossRef]
- Lundy, R.F. Potential Mechanisms for Functional Changes in Taste Receptor Cells Following Sodium Deficiency in Mammals. Neurosci. Biobehav. Rev. 1998, 23, 103–109. [Google Scholar] [CrossRef]
- McCaughey, S.A.; Scott, T.R. The Taste of Sodium. Neurosci. Biobehav. Rev. 1998, 22, 663–676. [Google Scholar] [CrossRef]
- Bigiani, A. Does ENaC Work as Sodium Taste Receptor in Humans? Nutrients 2020, 12, 1195. [Google Scholar] [CrossRef]
- Diepeveen, J.; Moerdijk-Poortvliet, T.C.W.; van der Leij, F.R. Molecular Insights into Human Taste Perception and Umami Tastants: A Review. J. Food Sci. 2022, 87, 1449–1465. [Google Scholar] [CrossRef]
- Lindemann, B. Taste Reception. Physiol. Rev. 1996, 76, 719–766. [Google Scholar] [CrossRef]
- Wu, A.; Dvoryanchikov, G.; Pereira, E.; Chaudhari, N.; Roper, S.D. Breadth of Tuning in Taste Afferent Neurons Varies with Stimulus Strength. Nat. Commun. 2015, 6, 8171. [Google Scholar] [CrossRef]
- DeSimone, J.A.; Lyall, V. Taste Receptors in the Gastrointestinal Tract III. Salty and Sour Taste: Sensing of Sodium and Protons by the Tongue. Am. J. Physiol. Gastrointest. Liver Physiol. 2006, 291, G1005–G1010. [Google Scholar] [CrossRef]
- Miyamoto, T.; Fujiyama, R.; Okada, Y.; Sato, T. Acid and Salt Responses in Mouse Taste Cells. Prog. Neurobiol. 2000, 62, 135–157. [Google Scholar] [CrossRef]
- Oakley, B.; Witt, M. Building Sensory Receptors on the Tongue. J. Neurocytol. 2004, 33, 631–646. [Google Scholar] [CrossRef][Green Version]
- Travers, J.B.; Travers, S.P.; Norgren, R. Gustatory Neural Processing in the Hindbrain. Annu. Rev. Neurosci. 1987, 10, 595–632. [Google Scholar] [CrossRef]
- Kinnamon, S.C.; Finger, T.E. Recent Advances in Taste Transduction and Signaling. F1000Res 2019, 8, F1000 Faculty Rev-2117. [Google Scholar] [CrossRef]
- Chandrashekar, J.; Kuhn, C.; Oka, Y.; Yarmolinsky, D.A.; Hummler, E.; Ryba, N.J.P.; Zuker, C.S. The Cells and Peripheral Representation of Sodium Taste in Mice. Nature 2010, 464, 297–301. [Google Scholar] [CrossRef] [PubMed]
- Herness, M.S. Aldosterone Increases the Amiloride-Sensitivity of the Rat Gustatory Neural Response to NaCl. Comp. Biochem. Physiol. Comp. Physiol. 1992, 103, 269–273. [Google Scholar] [CrossRef]
- Shigemura, N.; Iwata, S.; Yasumatsu, K.; Ohkuri, T.; Horio, N.; Sanematsu, K.; Yoshida, R.; Margolskee, R.F.; Ninomiya, Y. Angiotensin II Modulates Salty and Sweet Taste Sensitivities. J. Neurosci. 2013, 33, 6267–6277. [Google Scholar] [CrossRef]
- Frank, M.E.; Contreras, R.J.; Hettinger, T.P. Nerve Fibers Sensitive to Ionic Taste Stimuli in Chorda Tympani of the Rat. J. Neurophysiol. 1983, 50, 941–960. [Google Scholar] [CrossRef] [PubMed]
- McCaughey, S.A. Characterization of Mouse Chorda Tympani Responses Evoked by Stimulation of Anterior or Posterior Fungiform Taste Papillae. Neurosci. Res. 2019, 141, 43–51. [Google Scholar] [CrossRef] [PubMed]
- Spector, A.C.; Grill, H.J. Salt Taste Discrimination after Bilateral Section of the Chorda Tympani or Glossopharyngeal Nerves. Am. J. Physiol. 1992, 263, R169–R176. [Google Scholar] [CrossRef] [PubMed]
- Stricker, E.M.; Gannon, K.S.; Smith, J.C. Thirst and Salt Appetite Induced by Hypovolemia in Rats: Analysis of Drinking Behavior. Physiol. Behav. 1992, 51, 27–37. [Google Scholar] [CrossRef]
- Altschuler, S.M.; Bao, X.; Bieger, D.; Hopkins, D.A.; Miselis, R.R. Viscerotopic Representation of the Upper Alimentary Tract in the Rat: Sensory Ganglia and Nuclei of the Solitary and Spinal Trigeminal Tracts. J. Comp. Neurol. 1989, 283, 248–268. [Google Scholar] [CrossRef]
- Hamilton, R.B.; Norgren, R. Central Projections of Gustatory Nerves in the Rat. J. Comp. Neurol. 1984, 222, 560–577. [Google Scholar] [CrossRef]
- Arnedo, M.; Gallo, M.; Agüero, A.; Puerto, A. Effects of Medullary Afferent Vagal Axotomy and Area Postrema Lesions on Short-Term and Long-Term NaCl-Induced Taste Aversion Learning. Physiol. Behav. 1990, 47, 1067–1074. [Google Scholar] [CrossRef]
- Burman, A.; Kaji, I. Luminal Chemosensory Cells in the Small Intestine. Nutrients 2021, 13, 3712. [Google Scholar] [CrossRef]
- Sbarbati, A.; Osculati, F. The Taste Cell-Related Diffuse Chemosensory System. Prog. Neurobiol. 2005, 75, 295–307. [Google Scholar] [CrossRef]
- Zafra, M.A.; Prados, M.; Molina, F.; Puerto, A. Capsaicin-Sensitive Afferent Vagal Fibers Are Involved in Concurrent Taste Aversion Learning. Neurobiol. Learn. Mem. 2006, 86, 349–352. [Google Scholar] [CrossRef]
- Zimmerman, C.A.; Huey, E.L.; Ahn, J.S.; Beutler, L.R.; Tan, C.L.; Kosar, S.; Bai, L.; Chen, Y.; Corpuz, T.V.; Madisen, L.; et al. A Gut-to-Brain Signal of Fluid Osmolarity Controls Thirst Satiation. Nature 2019, 568, 98–102. [Google Scholar] [CrossRef]
- Johnson, A.K.; Thunhorst, R.L. The Neuroendocrinology of Thirst and Salt Appetite: Visceral Sensory Signals and Mechanisms of Central Integration. Front. Neuroendocr. 1997, 18, 292–353. [Google Scholar] [CrossRef]
- Mediavilla, C.; Bernal, A.; Mahía, J.; Puerto, A. Nucleus of the Solitary Tract and Flavor Aversion Learning: Relevance in Concurrent but Not Sequential Behavioral Test. Behav. Brain Res. 2011, 223, 287–292. [Google Scholar] [CrossRef]
- Mediavilla, C.; Molina, F.; Puerto, A. Concurrent Conditioned Taste Aversion: A Learning Mechanism Based on Rapid Neural versus Flexible Humoral Processing of Visceral Noxious Substances. Neurosci. Biobehav. Rev. 2005, 29, 1107–1118. [Google Scholar] [CrossRef]
- Contreras, R.J.; Kosten, T. Changes in Salt Intake after Abdominal Vagotomy: Evidence for Hepatic Sodium Receptors. Physiol. Behav. 1981, 26, 575–582. [Google Scholar] [CrossRef]
- Chernigovsky, V.N. The Significance of Interoceptive Signals in the Food Behavior in Animals. In The Internal Environment and Alimentary Behavior; Brazier, M.A.B., Ed.; Brain and Behavior; American Institute of Biological Sciences, University of California: Los Angeles, CA, USA, 1963; pp. 319–349. [Google Scholar]
- Blackshaw, L.A.; Grundy, D. Effects of 5-Hydroxytryptamine on Discharge of Vagal Mucosal Afferent Fibres from the Upper Gastrointestinal Tract of the Ferret. J. Auton. Nerv. Syst. 1993, 45, 41–50. [Google Scholar] [CrossRef]
- Mei, N. Intestinal Chemosensitivity. Physiol. Rev. 1985, 65, 211–237. [Google Scholar] [CrossRef]
- Mei, N.; Garnier, L. Osmosensitive Vagal Receptors in the Small Intestine of the Cat. J. Auton. Nerv. Syst. 1986, 16, 159–170. [Google Scholar] [CrossRef]
- Zhu, J.X.; Wu, X.Y.; Owyang, C.; Li, Y. Intestinal Serotonin Acts as a Paracrine Substance to Mediate Vagal Signal Transmission Evoked by Luminal Factors in the Rat. J. Physiol. 2001, 530, 431–442. [Google Scholar] [CrossRef]
- Kahrilas, P.J.; Rogers, R.C. Rat Brainstem Neurons Responsive to Changes in Portal Blood Sodium Concentration. Am. J. Physiol. 1984, 247, R792–R799. [Google Scholar] [CrossRef]
- Morita, H.; Yamashita, Y.; Nishida, Y.; Tokuda, M.; Hatase, O.; Hosomi, H. Fos Induction in Rat Brain Neurons after Stimulation of the Hepatoportal Na-Sensitive Mechanism. Am. J. Physiol. 1997, 272, R913–R923. [Google Scholar] [CrossRef] [PubMed]
- Barraco, R.; El-Ridi, M.; Ergene, E.; Parizon, M.; Bradley, D. An Atlas of the Rat Subpostremal Nucleus Tractus Solitarius. Brain Res. Bull. 1992, 29, 703–765. [Google Scholar] [CrossRef] [PubMed]
- Herbert, H.; Moga, M.M.; Saper, C.B. Connections of the parabrachial nucleus with the nucleus of the solitary tract and the medullary reticular formation in the rat. J. Comp. Neurol. 1990, 293, 540–580. [Google Scholar] [CrossRef] [PubMed]
- Hall, J.E.; Guyton, A.C.; Hall, M.E. Tratado de Fisiología Médica; 14th ed.; Elsevier: Barcelona, Spain, 2021; ISBN 978-84-13-82013-2. [Google Scholar]
- Linz, B.; Saljic, A.; Hohl, M.; Gawałko, M.; Jespersen, T.; Sanders, P.; Böhm, M.; Linz, D. Inhibition of Sodium-Proton-Exchanger Subtype 3-Mediated Sodium Absorption in the Gut: A New Antihypertensive Concept. IJC Heart Vasc. 2020, 29, 100591. [Google Scholar] [CrossRef] [PubMed]
- Spiller, R.C. Intestinal Absorptive Function. Gut 1994, 35, S5–S9. [Google Scholar] [CrossRef]
- Binder, H.J. Movimiento de Fluidos y Electrolitos Intestinales—Boron y Boulpaep.Manual de Fisiología Médica. In Boron & Boulpaep Concise Medical Physiology; Boron, W.F., Boulpaep, E.L., Eds.; Elsevier: Philadelphia, PA, USA, 2021; pp. 476–484. ISBN 978-0-323-65530-9. [Google Scholar]
- Negussie, A.B.; Dell, A.C.; Davis, B.A.; Geibel, J.P. Colonic Fluid and Electrolyte Transport 2022: An Update. Cells 2022, 11, 1712. [Google Scholar] [CrossRef]
- Afsar, B.; Vaziri, N.D.; Aslan, G.; Tarim, K.; Kanbay, M. Gut Hormones and Gut Microbiota: Implications for Kidney Function and Hypertension. J. Am. Soc. Hypertens. 2016, 10, 954–961. [Google Scholar] [CrossRef]
- Coric, T.; Hernandez, N.; de la Rosa, D.A.; Shao, D.; Wang, T.; Canessa, C.M. Expression of ENaC and Serum- and Glucocorticoid-Induced Kinase 1 in the Rat Intestinal Epithelium. Am. J. Physiol. Liver Physiol. 2004, 286, G663–G670. [Google Scholar] [CrossRef]
- Musch, M.W.; Lucioni, A.; Chang, E.B. Aldosterone Regulation of Intestinal Na Absorption Involves SGK-Mediated Changes in NHE3 and Na+ Pump Activity. Am. J. Physiol. Gastrointest. Liver Physiol. 2008, 295, G909–G919. [Google Scholar] [CrossRef]
- Ch’ng, S.S.; Lawrence, A.J. The Subfornical Organ in Sodium Appetite: Recent Insights. Neuropharmacology 2019, 154, 107–113. [Google Scholar] [CrossRef]
- Sladek, C.D.; Armstrong, W.E. Osmotic Control of Vasopressin Release. Trends Neurosci. 1985, 8, 166–169. [Google Scholar] [CrossRef]
- Hiyama, T.Y.; Noda, M. Sodium Sensing in the Subfornical Organ and Body-Fluid Homeostasis. Neurosci. Res. 2016, 113, 1–11. [Google Scholar] [CrossRef]
- Miller, R.L.; Wang, M.H.; Gray, P.A.; Salkoff, L.B.; Loewy, A.D. ENaC-Expressing Neurons in the Sensory Circumventricular Organs Become c-Fos Activated Following Systemic Sodium Changes. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2013, 305, R1141–R1152. [Google Scholar] [CrossRef]
- Solár, P.; Zamani, A.; Kubíčková, L.; Dubový, P.; Joukal, M. Choroid Plexus and the Blood-Cerebrospinal Fluid Barrier in Disease. Fluids Barriers CNS 2020, 17, 35. [Google Scholar] [CrossRef]
- Wang, S.; Liu, J.; Cai, H.; Liu, K.; He, Y.; Liu, S.; Guo, Y.; Guo, L. High Salt Diet Elevates the Mean Arterial Pressure of SLC14α1 Gene Depletion Mice. Life Sci. 2020, 254, 117751. [Google Scholar] [CrossRef]
- Peruzzo, M.; Milani, G.P.; Garzoni, L.; Longoni, L.; Simonetti, G.D.; Bettinelli, A.; Fossali, E.F.; Bianchetti, M.G. Body Fluids and Salt Metabolism—Part II. Ital. J. Pediatr. 2010, 36, 78. [Google Scholar] [CrossRef]
- Geerling, J.C.; Loewy, A.D. Aldosterone in the Brain. Am. J. Physiol. Physiol. 2009, 297, F559–F576. [Google Scholar] [CrossRef]
- Paxinos, G.; Watson, C. The Rat Brain in Stereotaxic Coordinates, 3rd ed.; Academic Press: San Diego, CA, USA, 1997; ISBN 978-0-12-547623-2. [Google Scholar]
- McKinley, M.J.; McAllen, R.M.; Davern, P.; Giles, M.E.; Penschow, J.; Sunn, N.; Uschakov, A.; Oldfield, B.J. The Sensory Circumventricular Organs of the Mammalian Brain. Adv. Anat. Embryol. Cell Biol. 2003, 172, 1–122. [Google Scholar] [CrossRef]
- Matsuda, T.; Hiyama, T.Y.; Niimura, F.; Matsusaka, T.; Fukamizu, A.; Kobayashi, K.; Kobayashi, K.; Noda, M. Distinct Neural Mechanisms for the Control of Thirst and Salt Appetite in the Subfornical Organ. Nat. Neurosci. 2017, 20, 230–241. [Google Scholar] [CrossRef]
- Simpson, F.O.; Waal-Manning, H.J.; Bolli, P.; Phelan, E.L.; Spears, G.F. Relationship of Blood Pressure to Sodium Excretion in a Population Survey. Clin. Sci. Mol. Med. Suppl. 1978, 4, 373s–375s. [Google Scholar] [CrossRef]
- Thrasher, T.N. Osmoreceptor Mediation of Thirst and Vasopressin Secretion in the Dog. Fed. Proc. 1982, 41, 2528–2532. [Google Scholar] [PubMed]
- Shapiro, R.E.; Miselis, R.R. The Central Neural Connections of the Area Postrema of the Rat. J. Comp. Neurol. 1985, 234, 344–364. [Google Scholar] [CrossRef] [PubMed]
- Stein, M.K.; Loewy, A.D. Area Postrema Projects to FoxP2 Neurons of the Pre-Locus Coeruleus and Parabrachial Nuclei: Brainstem Sites Implicated in Sodium Appetite Regulation. Brain Res. 2010, 1359, 116–127. [Google Scholar] [CrossRef] [PubMed]
- Miller, R.L.; Loewy, A.D. ENaC γ-Expressing Astrocytes in the Circumventricular Organs, White Matter, and Ventral Medullary Surface: Sites for Na+ Regulation by Glial Cells. J. Chem. Neuroanat. 2013, 53, 72–80. [Google Scholar] [CrossRef] [PubMed]
- Miller, R.L.; Loewy, A.D. 5-HT Neurons of the Area Postrema Become c-Fos-Activated after Increases in Plasma Sodium Levels and Transmit Interoceptive Information to the Nucleus Accumbens. Am. J. Physiol. Integr. Comp. Physiol. 2014, 306, R663–R673. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Sunn, N.; McKinley, M.J.; Oldfield, B.J. Circulating Angiotensin II Activates Neurones in Circumventricular Organs of the Lamina Terminalis That Project to the Bed Nucleus of the Stria Terminalis. J. Neuroendocrinol. 2003, 15, 725–731. [Google Scholar] [CrossRef]
- Jarvie, B.C.; Palmiter, R.D. HSD2 Neurons in the Hindbrain Drive Sodium Appetite. Nat. Neurosci. 2017, 20, 167–169. [Google Scholar] [CrossRef]
- Han, W.; Tellez, L.A.; Perkins, M.H.; Perez, I.O.; Qu, T.; Ferreira, J.; Ferreira, T.L.; Quinn, D.; Liu, Z.-W.; Gao, X.-B.; et al. A Neural Circuit for Gut-Induced Reward. Cell 2018, 175, 665–678.e23. [Google Scholar] [CrossRef]
- Hsu, T.M.; McCutcheon, J.E.; Roitman, M.F. Parallels and Overlap: The Integration of Homeostatic Signals by Mesolimbic Dopamine Neurons. Front. Psychiatry 2018, 9, 410. [Google Scholar] [CrossRef]
- Sandhu, E.C.; Fernando, A.B.P.; Irvine, E.E.; Tossell, K.; Kokkinou, M.; Glegola, J.; Smith, M.A.; Howes, O.D.; Withers, D.J.; Ungless, M.A. Phasic Stimulation of Midbrain Dopamine Neuron Activity Reduces Salt Consumption. eNeuro 2018, 5, e0064-18.2018 1–15. [Google Scholar] [CrossRef]
- Lee, S.; Augustine, V.; Zhao, Y.; Ebisu, H.; Ho, B.; Kong, D.; Oka, Y. Chemosensory Modulation of Neural Circuits for Sodium Appetite. Nature 2019, 568, 93–97. [Google Scholar] [CrossRef] [PubMed]
- Edwards, G.L.; Beltz, T.G.; Power, J.D.; Johnson, A.K. Rapid-Onset “Need-Free” Sodium Appetite after Lesions of the Dorsomedial Medulla. Am. J. Physiol. Integr. Comp. Physiol. 1993, 264, R1242–R1247. [Google Scholar] [CrossRef]
- Menani, J.V.; Thunhorst, R.L.; Johnson, A.K. Lateral Parabrachial Nucleus and Serotonergic Mechanisms in the Control of Salt Appetite in Rats. J. Physiol. Integr. Comp. Physiol. 1996, 270, R162–R168. [Google Scholar] [CrossRef]
- De Gobbi, J.I.F.; De Luca, L.A.; Menani, J.V. Serotonergic Mechanisms of the Lateral Parabrachial Nucleus on DOCA-Induced Sodium Intake. Brain Res. 2000, 880, 131–138. [Google Scholar] [CrossRef]
- De luca, L.A.; Barbosa, S.P.; Menani, J.V. Brain Serotonin Blockade and Paradoxical Salt Intake in Rats. Neuroscience 2003, 121, 1055–1061. [Google Scholar] [CrossRef]
- Andrade-Franzé, G.M.F.; Andrade, C.A.F.; De Luca, L.A.; De Paula, P.M.; Menani, J.V. Lateral Parabrachial Nucleus and Central Amygdala in the Control of Sodium Intake. Neuroscience 2010, 165, 633–641. [Google Scholar] [CrossRef]
- Godino, A.; Margatho, L.O.; Caeiro, X.E.; Antunes-Rodrigues, J.; Vivas, L. Activation of Lateral Parabrachial Afferent Pathways and Endocrine Responses during Sodium Appetite Regulation. Exp. Neurol. 2010, 221, 275–284. [Google Scholar] [CrossRef]
- Margatho, L.O.; Porcari, C.Y.; Macchione, A.F.; da Silva Souza, G.D.; Caeiro, X.E.; Antunes-Rodrigues, J.; Vivas, L.; Godino, A. Temporal Dissociation between Sodium Depletion and Sodium Appetite Appearance: Involvement of Inhibitory and Stimulatory Signals. Neuroscience 2015, 297, 78–88. [Google Scholar] [CrossRef]
- Andrade-Franzé, G.M.F.; Andrade, C.A.F.; De Luca, L.A.; De Paula, P.M.; Colombari, D.S.A.; Menani, J.V. Lesions in the Central Amygdala Impair Sodium Intake Induced by the Blockade of the Lateral Parabrachial Nucleus. Brain Res. 2010, 1332, 57–64. [Google Scholar] [CrossRef]
- David, R.B.; Roncari, C.F.; Lauar, M.R.; Vendramini, R.C.; Antunes-Rodrigues, J.; Menani, J.V.; De Luca, L.A. Sodium Intake, Brain c-Fos Protein and Gastric Emptying in Cell-Dehydrated Rats Treated with Methysergide into the Lateral Parabrachial Nucleus. Physiol. Behav. 2015, 151, 111–120. [Google Scholar] [CrossRef] [PubMed]
- Fulwiler, C.E.; Saper, C.B. Subnuclear Organization of the Efferent Connections of the Parabrachial Nucleus in the Rat. Brain Res. 1984, 319, 229–259. [Google Scholar] [CrossRef] [PubMed]
- Menani, J.V.; De Luca, L.A.; Johnson, A.K. Role of the Lateral Parabrachial Nucleus in the Control of Sodium Appetite. American Am. J. Physiol. Integr. Comp. Physiol. 2014, 306, R201–R210. [Google Scholar] [CrossRef] [PubMed]
- Ryan, P.J.; Ross, S.I.; Campos, C.A.; Derkach, V.A.; Palmiter, R.D. Oxytocin-Receptor-Expressing Neurons in the Parabrachial Nucleus Regulate Fluid Intake. Nat. Neurosci. 2017, 20, 1722–1733. [Google Scholar] [CrossRef] [PubMed]
- Cort, J.H. Spontaneous Salt Intake in the Rat Following Lesions in the Posterior Hypothalamus. Physiol. Bohemoslov. 1963, 12, 502–505. [Google Scholar] [PubMed]
- Natcheff, N.; Piryova, B.; Garchev, R.; Kirkova, L. Influence of the Hypothalamic Mammillary Area on the Kidney Function in Rats. Agressologie 1975, 16, 367–372. [Google Scholar] [PubMed]
- Mahía, J.; Bernal, A.; Puerto, A. Inhibition of Natriuresis in Median Eminence Polydipsia: Effects after Intake of Diets with Different Osmolalities and after Hypertonic NaCl Administration. Acta Neurobiol. Exp. 2013, 73, 326–337. [Google Scholar]
- Mahía, J.; Bernal, A.; García Del Rio, C.; Puerto, A. The Natriuretic Effect of Oxytocin Blocks Medial Tuberomammillary Polydipsia and Polyuria in Male Rats. Eur. J. Neurosci. 2009, 29, 1440–1446. [Google Scholar] [CrossRef]
- Bacić, A.; Gluncić, I.; Gluncić, V. Disturbances in Plasma Sodium in Patients with War Head Injuries. Mil. Med. 1999, 164, 214–217. [Google Scholar] [CrossRef]
- Cerdà-Esteve, M.; Cuadrado-Godia, E.; Chillaron, J.J.; Pont-Sunyer, C.; Cucurella, G.; Fernández, M.; Goday, A.; Cano-Pérez, J.F.; Rodríguez-Campello, A.; Roquer, J. Cerebral Salt Wasting Syndrome: Review. Eur. J. Intern. Med. 2008, 19, 249–254. [Google Scholar] [CrossRef]
- Yee, A.H.; Burns, J.D.; Wijdicks, E.F.M. Cerebral Salt Wasting: Pathophysiology, Diagnosis, and Treatment. Neurosurg. Clin. N. Am. 2010, 21, 339–352. [Google Scholar] [CrossRef]
- Verbalis, J.G. Disorders of Body Water Homeostasis. Best Pr. Res. Clin. Endocrinol. Metab. 2003, 17, 471–503. [Google Scholar] [CrossRef]
- Antunes-Rodrigues, J.; Turrin, M.Q.; Gutkowska, J.; McCann, S.M. Blockade of Volume Expansion-Induced Release of Atrial Natriuretic Peptide by Median Eminence Lesions in the Rat. Braz. J. Med. Biol. Res. 1990, 23, 355–359. [Google Scholar]
- Hennessy, J.W.; Grossman, S.P.; Kanner, M. A Study of the Etiology of the Hyperdipsia Produced by Coronal Knife Cuts in the Posterior Hypothalamus. Physiol. Behav. 1977, 18, 73–80. [Google Scholar] [CrossRef]
- Mahía, J.; Bernal, A.; Puerto, A. Hyperphagia and Increased Body Weight Induced by Lesions of the Ventral Tuberomammillary System. Behav. Brain Res. 2007, 181, 147–152. [Google Scholar] [CrossRef]
- Mahía, J.; Bernal, A.; Puerto, A. Dipsogenic Potentiation by Sodium Chloride but Not by Sucrose or Polyethylene Glycol in Tuberomammillary-Mediated Polydipsia. Exp. Brain Res. 2007, 183, 27–39. [Google Scholar] [CrossRef]
- Mahía, J.; Bernal, A.; Puerto, A. NaCl Preference and Water Intake Effects of Food Availability in Median Eminence Polydipsia. Neurosci. Lett. 2008, 447, 7–11. [Google Scholar] [CrossRef]
- Mahía, J.; Puerto, A. Lesions of Tuberomammillary Nuclei Induce Differential Polydipsic and Hyperphagic Effects. Eur. J. Neurosci. 2006, 23, 1321–1331. [Google Scholar] [CrossRef]
- Blessing, W.W.; Sved, A.F.; Reis, D.J. Destruction of Noradrenergic Neurons in Rabbit Brainstem Elevates Plasma Vasopressin, Causing Hypertension. Science 1982, 217, 661–663. [Google Scholar] [CrossRef]
- Ramos, J.M.; Castillo, M.E.; Puerto, A. Submandibular and Parotid Salivary Secretion after Electrolytic Lesioning of the Brainstem Nucleus Parvocellularis in the Rat. Physiol. Behav. 1988, 44, 173–180. [Google Scholar] [CrossRef]
- von Bismarck, P.; Ankermann, T.; Eggert, P.; Claviez, A.; Fritsch, M.J.; Krause, M.F. Diagnosis and Management of Cerebral Salt Wasting (CSW) in Children: The Role of Atrial Natriuretic Peptide (ANP) and Brain Natriuretic Peptide (BNP). Childs Nerv. Syst. 2006, 22, 1275–1281. [Google Scholar] [CrossRef]
- Sclafani, A.; Grossman, S.P. Hyperphagia Produced by Knife Cuts between the Medial and Lateral Hypothalamus in the Rat. Physiol. Behav. 1969, 4, 533–537. [Google Scholar] [CrossRef]
- Fried, L.F.; Palevsky, P.M. Hyponatremia and Hypernatremia. Med. Clin. N. Am. 1997, 81, 585–609. [Google Scholar] [CrossRef] [PubMed]
- Verghese, C.; De Leon, J.; Simpson, G.M. Neuroendocrine Factors Influencing Polydipsia in Psychiatric Patients: An Hypothesis. Neuropsychopharmacology 1993, 9, 157–166. [Google Scholar] [CrossRef] [PubMed]
- Goldman, M.B. Brain Circuit Dysfunction in a Distinct Subset of Chronic Psychotic Patients. Schizophr. Res. 2014, 157, 204–213. [Google Scholar] [CrossRef] [PubMed]
- De Berardis, D.; Marini, S.; Carano, A.; Lang, A.P.; Cavuto, M.; Piersanti, M.; Fornaro, M.; Perna, G.; Valchera, A.; Mazza, M.; et al. Efficacy and Safety of Long Acting Injectable Atypical Antipsychotics: A Review. Curr. Clin. Pharmacol. 2013, 8, 256–264. [Google Scholar] [CrossRef]
- Briess, D.; Cotter, D.; Doshi, R.; Everall, I. Mamillary Body Abnormalities in Schizophrenia. Lancet 1998, 352, 789–790. [Google Scholar] [CrossRef]
- Loh, J.A.; Verbalis, J.G. Diabetes Insipidus as a Complication after Pituitary Surgery. Nat. Clin. Pract. Endocrinol. Metab. 2007, 3, 489–494. [Google Scholar] [CrossRef]
- Ericson, H.; Blomqvist, A.; Köhler, C. Origin of Neuronal Inputs to the Region of the Tuberomammillary Nucleus of the Rat Brain. J. Comp. Neurol. 1991, 311, 45–64. [Google Scholar] [CrossRef]
- Inagaki, N.; Toda, K.; Taniuchi, I.; Panula, P.; Yamatodani, A.; Tohyama, M.; Watanabe, T.; Wada, H. An Analysis of Histaminergic Efferents of the Tuberomammillary Nucleus to the Medial Preoptic Area and Inferior Colliculus of the Rat. Exp. Brain Res. 1990, 80, 374–380. [Google Scholar] [CrossRef]
- Köhler, C.; Ericson, H.; Watanabe, T.; Polak, J.; Palay, S.L.; Palay, V.; Chan-Palay, V. Galanin Immunoreactivity in Hypothalamic Neurons: Further Evidence for Multiple Chemical Messengers in the Tuberomammillary Nucleus. J. Comp. Neurol. 1986, 250, 58–64. [Google Scholar] [CrossRef]
- Panula, P.; Karlstedt, K.; Sallmen, T.; Peitsaro, N.; Kaslin, J.; Michelsen, K.A.; Anichtchik, O.; Kukko-Lukjanov, T.; Lintunen, M. The Histaminergic System in the Brain: Structural Characteristics and Changes in Hibernation. J. Chem. Neuroanat. 2000, 18, 65–74. [Google Scholar] [CrossRef]
- Ericson, H.; Watanabe, T.; Köhler, C. Morphological Analysis of the Tuberomammillary Nucleus in the Rat Brain: Delineation of Subgroups with Antibody against L-Histidine Decarboxylase as a Marker. J. Comp. Neurol. 1987, 263, 1–24. [Google Scholar] [CrossRef]
- Reiner, P.B.; Semba, K.; Watanabe, T.; Wada, H. En Bloc Immunohistochemistry Reveals Extensive Distribution of Histidine Decarboxylase-Immunoreactive Neurons on the Ventral Surface of the Rat Hypothalamus. Neurosci. Lett. 1987, 77, 137–142. [Google Scholar] [CrossRef]
- Watanabe, T.; Taguchi, Y.; Shiosaka, S.; Tanaka, J.; Kubota, H.; Terano, Y.; Tohyama, M.; Wada, H. Distribution of the Histaminergic Neuron System in the Central Nervous System of Rats; a Fluorescent Immunohistochemical Analysis with Histidine Decarboxylase as a Marker. Brain Res. 1984, 295, 13–25. [Google Scholar] [CrossRef]
- Schwartz, J.C.; Arrang, J.M.; Garbarg, M.; Pollard, H.; Ruat, M. Histaminergic Transmission in the Mammalian Brain. Physiol. Rev. 1991, 71, 1–51. [Google Scholar] [CrossRef]
- Hatton, G.I.; Li, Z.H. Neurophysiology of Magnocellular Neuroendocrine Cells: Recent Advances. Prog. Brain Res. 1998, 119, 77–99. [Google Scholar] [CrossRef]
- Kjaer, A.; Larsen, P.J.; Knigge, U.; Warberg, J. Dehydration Stimulates Hypothalamic Gene Expression of Histamine Synthesis Enzyme: Importance for Neuroendocrine Regulation of Vasopressin and Oxytocin Secretion. Endocrinology 1995, 136, 2189–2197. [Google Scholar] [CrossRef]
- Kjaer, A.; Knigge, U.; Rouleau, A.; Garbarg, M.; Warberg, J. Dehydration-Induced Release of Vasopressin Involves Activation of Hypothalamic Histaminergic Neurons. Endocrinology 1994, 135, 675–681. [Google Scholar] [CrossRef]
- Pollard, H.; Bischoff, S.; Llorens-Cortes, C.; Schwartz, J.C. Histidine Decarboxylase and Histamine in Discrete Nuclei of Rat Hypothalamus and the Evidence for Mast-Cells in the Median Eminence. Brain Res. 1976, 118, 509–513. [Google Scholar] [CrossRef]
- Weiss, M.L.; Yang, Q.Z.; Hatton, G.I. Magnocellular Tuberomammillary Nucleus Input to the Supraoptic Nucleus in the Rat: Anatomical and in Vitro Electrophysiological Investigations. Neuroscience 1989, 31, 299–311. [Google Scholar] [CrossRef]
- Akins, V.F.; Bealer, S.L. Brain Histamine Regulates Pressor Responses to Peripheral Hyperosmolality. Am. J. Physiol. 1990, 259, R507–R513. [Google Scholar] [CrossRef] [PubMed]
- Knigge, U.; Willems, E.; Kjaer, A.; Jørgensen, H.; Warberg, J. Histaminergic and Catecholaminergic Interactions in the Central Regulation of Vasopressin and Oxytocin Secretion. Endocrinology 1999, 140, 3713–3719. [Google Scholar] [CrossRef] [PubMed]
- Swaab, D.F.; Nijveldt, F.; Pool, C.W. Distribution of Oxytocin and Vasopressin in the Rat Supraoptic and Paraventricular Nucleus. J. Endocrinol. 1975, 67, 461–462. [Google Scholar] [CrossRef] [PubMed]
- Bealer, S.L.; Crowley, W.R. Stimulation of Central and Systemic Oxytocin Release by Histamine in the Paraventricular Hypothalamic Nucleus: Evidence for an Interaction with Norepinephrine*. Endocrinology 1999, 140, 1158–1164. [Google Scholar] [CrossRef] [PubMed]
- Johns, E.J. The Autonomic Nervous System and Pressure-Natriuresis in Cardiovascular-Renal Interactions in Response to Salt. Clin. Auton. Res. 2002, 12, 256–263. [Google Scholar] [CrossRef]
- van Westing, A.C.; Küpers, L.K.; Geleijnse, J.M. Diet and Kidney Function: A Literature Review. Curr. Hypertens. Rep. 2020, 22, 14. [Google Scholar] [CrossRef]
- Guthrie, D.; Yucha, C. Urinary Concentration and Dilution. Nephrol. Nurs. J. 2004, 31, 297–301; quiz 302–303. [Google Scholar]
- Natochin, Y.V.; Golosova, D.V. Vasopressin Receptor Subtypes and Renal Sodium Transport. Vitam. Horm. 2020, 113, 239–258. [Google Scholar] [CrossRef]
- Rose, B.D.; Post, T.W. Clinical Physiology of Acid-Base and Electrolyte Disorders, 5th ed.; Medical Pub. Division, McGraw-Hill: New York, NY, USA, 2001; ISBN 978-0-07-134682-5. [Google Scholar]
- Tigerstedt, R.; Bergman, P.G. Niere and Kreislauf. Skand. Archly Physiol. 1898, 8, 223–271. [Google Scholar] [CrossRef]
- Dzau, V.J.; Ingelfinger, J.; Pratt, R.E.; Ellison, K.E. Identification of Renin and Angiotensinogen Messenger RNA Sequences in Mouse and Rat Brains. Hypertension 1986, 8, 544–548. [Google Scholar] [CrossRef]
- Healy, D.P.; Printz, M.P. Distribution of Immunoreactive Angiotensin II, Angiotensin I, Angiotensinogen and Renin in the Central Nervous System of Intact and Nephrectomized Rats. Hypertension 1984, 6, I130–I136. [Google Scholar] [CrossRef]
- Lynch, K.R.; Hawelu-Johnson, C.L.; Guyenet, P.G. Localization of Brain Angiotensinogen MRNA by Hybridization Histochemistry. Brain Res. 1987, 388, 149–158. [Google Scholar] [CrossRef]
- DiBartola, S.P. Applied Renal Physiology. In Fluid, Electrolyte, and Acid-Base Disorders in Small Animal Practice; Elsevier: Amsterdam, The Netherlands, 2012; pp. 26–43. ISBN 978-1-4377-0654-3. [Google Scholar]
- Hall, J.E.; Guyton, A.C.; Mizelle, H.L. Role of the Renin-Angiotensin System in Control of Sodium Excretion and Arterial Pressure. Acta Physiol. Scand. Suppl. 1990, 591, 48–62. [Google Scholar]
- Harrison-Bernard, L.M. The Renal Renin-Angiotensin System. Adv. Physiol. Educ. 2009, 33, 270–274. [Google Scholar] [CrossRef]
- Nishimura, H. Renin-Angiotensin System in Vertebrates: Phylogenetic View of Structure and Function. Anat. Sci. Int. 2017, 92, 215–247. [Google Scholar] [CrossRef]
- Stroth, U.; Unger, T. The Renin-Angiotensin System and Its Receptors. J. Cardiovasc. Pharmacol. 1999, 33 (Suppl. 1), S21–S28; discussion S41–S43. [Google Scholar] [CrossRef]
- Cusi, D. Renal Hypertension. In Encyclopedia of Endocrine Diseases; Elsevier: Amsterdam, The Netherlands, 2019; pp. 383–389. ISBN 978-0-12-812200-6. [Google Scholar]
- Hanukoglu, I.; Hanukoglu, A. Epithelial Sodium Channel (ENaC) Family: Phylogeny, Structure-Function, Tissue Distribution, and Associated Inherited Diseases. Gene 2016, 579, 95–132. [Google Scholar] [CrossRef]
- Song, C.; Ma, H.-P.; Eaton, D.C. Epithelial Sodium Channels (ENaC). In Studies of Epithelial Transporters and Ion Channels; Hamilton, K.L., Devor, D.C., Eds.; Physiology in Health and Disease; Springer International Publishing: Cham, Switzerland, 2020; pp. 697–803. ISBN 978-3-030-55453-8. [Google Scholar]
- Beutler, K.T.; Masilamani, S.; Turban, S.; Nielsen, J.; Brooks, H.L.; Ageloff, S.; Fenton, R.A.; Packer, R.K.; Knepper, M.A. Long-Term Regulation of ENaC Expression in Kidney by Angiotensin II. Hypertension 2003, 41, 1143–1150. [Google Scholar] [CrossRef]
- Ferrão, F.M.; Lara, L.S.; Lowe, J. Renin-Angiotensin System in the Kidney: What Is New? World J. Nephrol. 2014, 3, 64–76. [Google Scholar] [CrossRef]
- Baek, E.J.; Kim, S. Current Understanding of Pressure Natriuresis. Electrolyte Blood Press. 2021, 19, 38–45. [Google Scholar] [CrossRef]
- Fitzsimons, J.T. Angiotensin, Thirst, and Sodium Appetite. Physiol. Rev. 1998, 78, 583–686. [Google Scholar] [CrossRef] [PubMed]
- Richter, C.P. Increased Salt Appetite in Adrenalectomized Rats. Am. J. Physiol. 1936, 115, 155–161. [Google Scholar] [CrossRef]
- Fuller, P.J.; Young, M.J. Aldosterone Secretion and Action. In Endocrinology: Adult and Pediatric; Elsevier: Amsterdam, The Netherlands, 2016; pp. 1756–1762.e3. ISBN 978-0-323-18907-1. [Google Scholar]
- MacKenzie, S.M.; van Kralingen, J.C.; Davies, E. Regulation of Aldosterone Secretion. Vitam. Horm. 2019, 109, 241–263. [Google Scholar] [CrossRef] [PubMed]
- Aquitlera, G.; Marusic, E.T. Role of the Renin Angiotensin System on the Biosynthesis of Aldosterone. Endocrinology 1971, 89, 1524–1529. [Google Scholar] [CrossRef] [PubMed]
- Catt, K.J.; Carson, M.C.; Hausdorff, W.P.; Leach-Harper, C.M.; Baukal, A.J.; Guillemette, G.; Balla, T.; Aguilera, G. Angiotensin II Receptors and Mechanisms of Action in Adrenal Glomerulosa Cells. J. Steroid Biochem. 1987, 27, 915–927. [Google Scholar] [CrossRef]
- Merrill, D.C.; Ebert, T.J.; Skelton, M.M.; Cowley, A.W. Effect of Plasma Sodium on Aldosterone Secretion during Angiotensin II Stimulation in Normal Humans. Hypertension 1989, 14, 164–169. [Google Scholar] [CrossRef]
- Merrill, D.C.; Skelton, M.M.; Cowley, A.W. Angiotensin II Sensitization of Aldosterone Responsiveness to Plasma Sodium in Conscious Dogs. Am. J. Physiol. 1987, 253, R832–R837. [Google Scholar] [CrossRef]
- Blair West, J.R.; Coghlan, J.P.; Denton, D.A.; Goding, J.R.; Wintour, M.; Wright, R.D. The Control of Aldosterone Secretion. Recent Prog. Horm. Res. 1963, 19, 311–383. [Google Scholar]
- Cade, R.; Perenich, T. Secretion of Aldosterone by Rats. Am. J. Physiol. 1965, 208, 1026–1030. [Google Scholar] [CrossRef]
- Eisen, L.P.; Harmon, J.M. Activation of the Rat Kidney Mineralocorticoid Receptor. Endocrinology 1986, 119, 1419–1426. [Google Scholar] [CrossRef]
- Katz, A.I. Corticosteroid Regulation of NaK-ATPase along the Mammalian Nephron. Semin. Nephrol. 1990, 10, 388–399. [Google Scholar]
- Sheppard, K.E.; Funder, J.W. Equivalent Affinity of Aldosterone and Corticosterone for Type I Receptors in Kidney and Hippocampus: Direct Binding Studies. J. Steroid Biochem. 1987, 28, 737–742. [Google Scholar] [CrossRef]
- Soundararajan, R.; Pearce, D.; Ziera, T. The Role of the ENaC-Regulatory Complex in Aldosterone-Mediated Sodium Transport. Mol. Cell Endocrinol. 2012, 350, 242–247. [Google Scholar] [CrossRef]
- Verrey, F. Regulation of Gene Expression by Aldosterone in Tight Epithelia. Semin. Nephrol. 1990, 10, 410–420. [Google Scholar]
- White, P.C. Disorders of Aldosterone Biosynthesis and Action. N. Engl. J. Med. 1994, 331, 250–258. [Google Scholar] [CrossRef]
- Hall, J.E. The Kidney, Hypertension, and Obesity. Hypertension 2003, 41, 625–633. [Google Scholar] [CrossRef]
- Haas, J.A.; Granger, J.P.; Knox, F.G. Effect of Renal Perfusion Pressure on Sodium Reabsorption from Proximal Tubules of Superficial and Deep Nephrons. Am. J. Physiol. 1986, 250, F425–F429. [Google Scholar] [CrossRef]
- Roman, R.J. Pressure-Diuresis in Volume-Expanded Rats. Tubular Reabsorption in Superficial and Deep Nephrons. Hypertension 1988, 12, 177–183. [Google Scholar] [CrossRef]
- Granger, J.P.; Alexander, B.T.; Llinas, M. Mechanisms of Pressure Natriuresis. Curr. Hypertens. Rep. 2002, 4, 152–159. [Google Scholar] [CrossRef]
- Johns, E.J.; Kopp, U.C.; DiBona, G.F. Neural Control of Renal Function. Compr. Physiol. 2011, 1, 731–767. [Google Scholar] [CrossRef]
- DiBona, G.F. Physiology in Perspective: The Wisdom of the Body. Neural Control of the Kidney. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2005, 289, R633–R641. [Google Scholar] [CrossRef] [PubMed]
- DiBona, G.F.; Kopp, U.C. Neural Control of Renal Function. Physiol. Rev. 1997, 77, 75–197. [Google Scholar] [CrossRef] [PubMed]
- Guild, S.-J.; Eppel, G.A.; Malpas, S.C.; Rajapakse, N.W.; Stewart, A.; Evans, R.G. Regional Responsiveness of Renal Perfusion to Activation of the Renal Nerves. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2002, 283, R1177–R1186. [Google Scholar] [CrossRef] [PubMed]
- Barajas, L.; Liu, L.; Powers, K. Anatomy of the Renal Innervation: Intrarenal Aspects and Ganglia of Origin. Can. J. Physiol. Pharmacol. 1992, 70, 735–749. [Google Scholar] [CrossRef] [PubMed]
- Johns, E.J. An Investigation into the Type of Beta-Adrenoceptor Mediating Sympathetically Activated Renin Release in the Cat. Br. J. Pharmacol. 1981, 73, 749–754. [Google Scholar] [CrossRef]
- Handa, R.K.; Johns, E.J. Interaction of the Renin-Angiotensin System and the Renal Nerves in the Regulation of Rat Kidney Function. J. Physiol. 1985, 369, 311–321. [Google Scholar] [CrossRef]
- Aperia, A.; Ibarra, F.; Svensson, L.B.; Klee, C.; Greengard, P. Calcineurin Mediates Alpha-Adrenergic Stimulation of Na+,K(+)-ATPase Activity in Renal Tubule Cells. Proc. Natl. Acad. Sci. USA 1992, 89, 7394–7397. [Google Scholar] [CrossRef]
- Pettinger, W.A.; Umemura, S.; Smyth, D.D.; Jeffries, W.B. Renal Alpha 2-Adrenoceptors and the Adenylate Cyclase-CAMP System: Biochemical and Physiological Interactions. Am. J. Physiol. 1987, 252, F199–F208. [Google Scholar] [CrossRef]
- Luchner, A.; Schunkert, H. Interactions between the Sympathetic Nervous System and the Cardiac Natriuretic Peptide System. Cardiovasc. Res. 2004, 63, 443–449. [Google Scholar] [CrossRef]
- van den Meiracker, A.H.; Boomsma, F. The Angiotensin II-Sympathetic Nervous System Connection. J. Hypertens. 2003, 21, 1453–1454. [Google Scholar] [CrossRef]
- Aileru, A.A.; Logan, E.; Callahan, M.; Ferrario, C.M.; Ganten, D.; Diz, D.I. Alterations in Sympathetic Ganglionic Transmission in Response to Angiotensin II in (MRen2)27 Transgenic Rats. Hypertension 2004, 43, 270–275. [Google Scholar] [CrossRef]
- Veelken, R.; Hilgers, K.F.; Stetter, A.; Siebert, H.G.; Schmieder, R.E.; Mann, J.F. Nerve-Mediated Antidiuresis and Antinatriuresis after Air-Jet Stress Is Modulated by Angiotensin II. Hypertension 1996, 28, 825–832. [Google Scholar] [CrossRef]
- Cao, L.-H.; Yang, X.-L. Natriuretic Peptides and Their Receptors in the Central Nervous System. Prog. Neurobiol. 2008, 84, 234–248. [Google Scholar] [CrossRef]
- de Bold, A.J.; Borenstein, H.B.; Veress, A.T.; Sonnenberg, H. A Rapid and Potent Natriuretic Response to Intravenous Injection of Atrial Myocardial Extract in Rats. Life Sci. 1981, 28, 89–94. [Google Scholar] [CrossRef]
- Flynn, T.G.; de Bold, M.L.; de Bold, A.J. The Amino Acid Sequence of an Atrial Peptide with Potent Diuretic and Natriuretic Properties. Biochem. Biophys. Res. Commun. 1983, 117, 859–865. [Google Scholar] [CrossRef]
- Kangawa, K.; Matsuo, H. Purification and Complete Amino Acid Sequence of Alpha-Human Atrial Natriuretic Polypeptide (Alpha-HANP). Biochem. Biophys. Res. Commun. 1984, 118, 131–139. [Google Scholar] [CrossRef]
- Goetze, J.P.; Hansen, L.H.; Terzic, D.; Zois, N.E.; Albrethsen, J.; Timm, A.; Smith, J.; Soltysinska, E.; Lippert, S.K.; Hunter, I. Atrial Natriuretic Peptides in Plasma. Clin. Chim. Acta 2015, 443, 25–28. [Google Scholar] [CrossRef]
- Rao, S.; Pena, C.; Shurmur, S.; Nugent, K. Atrial Natriuretic Peptide: Structure, Function, and Physiological Effects: A Narrative Review. Curr. Cardiol. Rev. 2021, 17, e051121191003. [Google Scholar] [CrossRef]
- Saito, Y.; Nakao, K.; Itoh, H.; Yamada, T.; Mukoyama, M.; Arai, H.; Hosoda, K.; Shirakami, G.; Suga, S.; Minamino, N. Brain Natriuretic Peptide Is a Novel Cardiac Hormone. Biochem. Biophys. Res. Commun. 1989, 158, 360–368. [Google Scholar] [CrossRef]
- Nishikimi, T.; Maeda, N.; Matsuoka, H. The Role of Natriuretic Peptides in Cardioprotection. Cardiovasc. Res. 2006, 69, 318–328. [Google Scholar] [CrossRef]
- Edwards, B.S.; Zimmerman, R.S.; Schwab, T.R.; Heublein, D.M.; Burnett, J.C. Atrial Stretch, Not Pressure, Is the Principal Determinant Controlling the Acute Release of Atrial Natriuretic Factor. Circ. Res. 1988, 62, 191–195. [Google Scholar] [CrossRef] [PubMed]
- Ichai, C.; Bichet, D.G. Water and Sodium Balance. In Metabolic Disorders and Critically Ill Patients; Ichai, C., Quintard, H., Orban, J.-C., Eds.; Springer International Publishing: Cham, Switzerland, 2018; pp. 3–31. ISBN 978-3-319-64008-2. [Google Scholar]
- Patel, S. Sodium Balance-an Integrated Physiological Model and Novel Approach. Saudi J. Kidney Dis. Transpl. 2009, 20, 560–569. [Google Scholar] [PubMed]
- Goetze, J.P.; Bruneau, B.G.; Ramos, H.R.; Ogawa, T.; de Bold, M.K.; de Bold, A.J. Cardiac Natriuretic Peptides. Nat. Rev. Cardiol. 2020, 17, 698–717. [Google Scholar] [CrossRef] [PubMed]
- Misono, K.S.; Philo, J.S.; Arakawa, T.; Ogata, C.M.; Qiu, Y.; Ogawa, H.; Young, H.S. Structure, Signaling Mechanism and Regulation of the Natriuretic Peptide Receptor Guanylate Cyclase. FEBS J. 2011, 278, 1818–1829. [Google Scholar] [CrossRef] [PubMed]
- Ogawa, Y.; Mukoyama, M.; Yokoi, H.; Kasahara, M.; Mori, K.; Kato, Y.; Kuwabara, T.; Imamaki, H.; Kawanishi, T.; Koga, K.; et al. Natriuretic Peptide Receptor Guanylyl Cyclase-A Protects Podocytes from Aldosterone-Induced Glomerular Injury. J. Am. Soc. Nephrol. 2012, 23, 1198–1209. [Google Scholar] [CrossRef]
- Staffel, J.; Valletta, D.; Federlein, A.; Ehm, K.; Volkmann, R.; Füchsl, A.M.; Witzgall, R.; Kuhn, M.; Schweda, F. Natriuretic Peptide Receptor Guanylyl Cyclase-A in Podocytes Is Renoprotective but Dispensable for Physiologic Renal Function. J. Am. Soc. Nephrol. 2017, 28, 260–277. [Google Scholar] [CrossRef]
- Suga, S.; Nakao, K.; Hosoda, K.; Mukoyama, M.; Ogawa, Y.; Shirakami, G.; Arai, H.; Saito, Y.; Kambayashi, Y.; Inouye, K. Receptor Selectivity of Natriuretic Peptide Family, Atrial Natriuretic Peptide, Brain Natriuretic Peptide, and C-Type Natriuretic Peptide. Endocrinology 1992, 130, 229–239. [Google Scholar] [CrossRef]
- Dunn, B.R.; Ichikawa, I.; Pfeffer, J.M.; Troy, J.L.; Brenner, B.M. Renal and Systemic Hemodynamic Effects of Synthetic Atrial Natriuretic Peptide in the Anesthetized Rat. Circ. Res. 1986, 59, 237–246. [Google Scholar] [CrossRef]
- McCoy, D.E.; Guggino, S.E.; Stanton, B.A. The Renal CGMP-Gated Cation Channel: Its Molecular Structure and Physiological Role. Kidney Int. 1995, 48, 1125–1133. [Google Scholar] [CrossRef][Green Version]
- Guo, L.-J.; Alli, A.A.; Eaton, D.C.; Bao, H.-F. ENaC Is Regulated by Natriuretic Peptide Receptor-Dependent CGMP Signaling. Am. J. Physiol. Renal. Physiol. 2013, 304, F930–F937. [Google Scholar] [CrossRef]
- Klokkers, J.; Langehanenberg, P.; Kemper, B.; Kosmeier, S.; von Bally, G.; Riethmüller, C.; Wunder, F.; Sindic, A.; Pavenstädt, H.; Schlatter, E.; et al. Atrial Natriuretic Peptide and Nitric Oxide Signaling Antagonizes Vasopressin-Mediated Water Permeability in Inner Medullary Collecting Duct Cells. Am. J. Physiol. Renal. Physiol. 2009, 297, F693–F703. [Google Scholar] [CrossRef]
- McGrath, M.F.; de Bold, M.L.K.; de Bold, A.J. The Endocrine Function of the Heart. Trends Endocrinol. Metab. 2005, 16, 469–477. [Google Scholar] [CrossRef]
- Theilig, F.; Wu, Q. ANP-Induced Signaling Cascade and Its Implications in Renal Pathophysiology. Am. J. Physiol. Renal. Physiol. 2015, 308, F1047–F1055. [Google Scholar] [CrossRef]
- Choi, M.R.; Fernández, B.E. Protective Renal Effects of Atrial Natriuretic Peptide: Where Are We Now? Front. Physiol. 2021, 12, 680213. [Google Scholar] [CrossRef]
- Nakagawa, Y.; Nishikimi, T.; Kuwahara, K. Atrial and Brain Natriuretic Peptides: Hormones Secreted from the Heart. Peptides 2019, 111, 18–25. [Google Scholar] [CrossRef]
- Kurtz, A.; Della Bruna, R.; Pfeilschifter, J.; Taugner, R.; Bauer, C. Atrial Natriuretic Peptide Inhibits Renin Release from Juxtaglomerular Cells by a CGMP-Mediated Process. Proc. Natl. Acad. Sci. USA 1986, 83, 4769–4773. [Google Scholar] [CrossRef]
- Brenner, B.M.; Ballermann, B.J.; Gunning, M.E.; Zeidel, M.L. Diverse Biological Actions of Atrial Natriuretic Peptide. Physiol. Rev. 1990, 70, 665–699. [Google Scholar] [CrossRef]
- Volpe, M.; Rubattu, S.; Battistoni, A. ARNi: A Novel Approach to Counteract Cardiovascular Diseases. Int. J. Mol. Sci. 2019, 20, 2092. [Google Scholar] [CrossRef]
- Motzfeldt, K. Experimental Studies on the Relation of the Pituitary Body To Renal Function. J. Exp. Med. 1917, 25, 153–188. [Google Scholar] [CrossRef][Green Version]
- Bernal, A.; Mahía, J.; Puerto, A. Animal Models of Central Diabetes Insipidus: Human Relevance of Acquired beyond Hereditary Syndromes and the Role of Oxytocin. Neurosci. Biobehav. Rev. 2016, 66, 1–14. [Google Scholar] [CrossRef]
- Bankir, L.; Bichet, D.G.; Morgenthaler, N.G. Vasopressin: Physiology, Assessment and Osmosensation. J. Intern. Med. 2017, 282, 284–297. [Google Scholar] [CrossRef] [PubMed]
- Stockand, J.D. Vasopressin Regulation of Renal Sodium Excretion. Kidney Int. 2010, 78, 849–856. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Wang, W.; Jiang, T.; Yang, B. Aquaporins in Urinary System. Adv. Exp. Med. Biol. 2017, 969, 131–148. [Google Scholar] [CrossRef] [PubMed]
- Preston, G.M.; Carroll, T.P.; Guggino, W.B.; Agre, P. Appearance of Water Channels in Xenopus Oocytes Expressing Red Cell CHIP28 Protein. Science 1992, 256, 385–387. [Google Scholar] [CrossRef] [PubMed]
- Perucca, J.; Bichet, D.G.; Bardoux, P.; Bouby, N.; Bankir, L. Sodium Excretion in Response to Vasopressin and Selective Vasopressin Receptor Antagonists. J. Am. Soc. Nephrol. 2008, 19, 1721–1731. [Google Scholar] [CrossRef]
- Kortenoeven, M.L.A.; Pedersen, N.B.; Rosenbaek, L.L.; Fenton, R.A. Vasopressin Regulation of Sodium Transport in the Distal Nephron and Collecting Duct. Am. J. Physiol. Renal. Physiol. 2015, 309, F280–F299. [Google Scholar] [CrossRef]
- Marunaka, Y.; Eaton, D.C. Effects of Vasopressin and CAMP on Single Amiloride-Blockable Na Channels. Am. J. Physiol. 1991, 260, C1071–C1084. [Google Scholar] [CrossRef]
- Snyder, P.M. Minireview: Regulation of Epithelial Na+ Channel Trafficking. Endocrinology 2005, 146, 5079–5085. [Google Scholar] [CrossRef]
- Knepper, M.A.; Kwon, T.-H.; Nielsen, S. Molecular Physiology of Water Balance. N. Engl. J. Med. 2015, 373, 196. [Google Scholar] [CrossRef]
- Mironova, E.; Bugaj, V.; Roos, K.P.; Kohan, D.E.; Stockand, J.D. Aldosterone-Independent Regulation of the Epithelial Na+ Channel (ENaC) by Vasopressin in Adrenalectomized Mice. Proc. Natl. Acad. Sci. USA 2012, 109, 10095–10100. [Google Scholar] [CrossRef]
- Snyder, P.M.; Olson, D.R.; Kabra, R.; Zhou, R.; Steines, J.C. CAMP and Serum and Glucocorticoid-Inducible Kinase (SGK) Regulate the Epithelial Na(+) Channel through Convergent Phosphorylation of Nedd4-2. J. Biol. Chem. 2004, 279, 45753–45758. [Google Scholar] [CrossRef]
- Staruschenko, A. Regulation of Transport in the Connecting Tubule and Cortical Collecting Duct. Compr. Physiol. 2012, 2, 1541–1584. [Google Scholar] [CrossRef]
- Choukroun, G.; Schmitt, F.; Martinez, F.; Drüeke, T.B.; Bankir, L. Low Urine Flow Reduces the Capacity to Excrete a Sodium Load in Humans. Am. J. Physiol. 1997, 273, R1726–R1733. [Google Scholar] [CrossRef]
- Bankir, L.; Fernandes, S.; Bardoux, P.; Bouby, N.; Bichet, D.G. Vasopressin-V2 Receptor Stimulation Reduces Sodium Excretion in Healthy Humans. J. Am. Soc. Nephrol. 2005, 16, 1920–1928. [Google Scholar] [CrossRef]
- Maybauer, M.O.; Maybauer, D.M.; Enkhbaatar, P.; Traber, D.L. Physiology of the Vasopressin Receptors. Best Pract. Res. Clin. Anaesthesiol. 2008, 22, 253–263. [Google Scholar] [CrossRef]
- Giesecke, T.; Himmerkus, N.; Leipziger, J.; Bleich, M.; Koshimizu, T.-A.; Fähling, M.; Smorodchenko, A.; Shpak, J.; Knappe, C.; Isermann, J.; et al. Vasopressin Increases Urinary Acidification via V1a Receptors in Collecting Duct Intercalated Cells. J. Am. Soc. Nephrol. 2019, 30, 946–961. [Google Scholar] [CrossRef]
- Bernal, A.; Mahía, J.; Puerto, A. Oxytocin, Water Intake, and Food Sodium Availability in Male Rats. Horm. Behav. 2007, 52, 289–296. [Google Scholar] [CrossRef]
- Christ-Crain, M.; Bichet, D.G.; Fenske, W.K.; Goldman, M.B.; Rittig, S.; Verbalis, J.G.; Verkman, A.S. Diabetes Insipidus. Nat. Rev. Dis. Primers 2019, 5, 54. [Google Scholar] [CrossRef]
- Kavanagh, C.; Uy, N.S. Nephrogenic Diabetes Insipidus. Pediatr. Clin. N. Am. 2019, 66, 227–234. [Google Scholar] [CrossRef]
- Qureshi, S.; Galiveeti, S.; Bichet, D.G.; Roth, J. Diabetes Insipidus: Celebrating a Century of Vasopressin Therapy. Endocrinology 2014, 155, 4605–4621. [Google Scholar] [CrossRef]
- Di Iorgi, N.; Napoli, F.; Allegri, A.E.M.; Olivieri, I.; Bertelli, E.; Gallizia, A.; Rossi, A.; Maghnie, M. Diabetes Insipidus—Diagnosis and Management. Horm. Res. Paediatr. 2012, 77, 69–84. [Google Scholar] [CrossRef] [PubMed]
- Antunes-Rodrigues, J.; de Castro, M.; Elias, L.L.K.; Valença, M.M.; McCann, S.M. Neuroendocrine Control of Body Fluid Metabolism. Physiol. Rev. 2004, 84, 169–208. [Google Scholar] [CrossRef] [PubMed]
- Makaryus, A.N.; McFarlane, S.I. Diabetes Insipidus: Diagnosis and Treatment of a Complex Disease. Clevel. Clin. J. Med. 2006, 73, 65–71. [Google Scholar] [CrossRef] [PubMed]
- Aulinas, A.; Plessow, F.; Asanza, E.; Silva, L.; Marengi, D.A.; Fan, W.; Abedi, P.; Verbalis, J.; Tritos, N.A.; Nachtigall, L.; et al. Low Plasma Oxytocin Levels and Increased Psychopathology in Hypopituitary Men With Diabetes Insipidus. J. Clin. Endocrinol. Metab. 2019, 104, 3181–3191. [Google Scholar] [CrossRef] [PubMed]
- Armstrong, W.E. Hypothalamic Supraoptic and Paraventricular Nuclei. In The Rat Nervous System; Elsevier: Amsterdam, The Netherlands, 2015; pp. 295–314. ISBN 978-0-12-374245-2. [Google Scholar]
- Balment, R.J.; Brimble, M.J.; Forsling, M.L. Release of Oxytocin Induced by Salt Loading and Its Influence on Renal Excretion in the Male Rat. J. Physiol. 1980, 308, 439–449. [Google Scholar] [CrossRef]
- Bernal, A.; Mahía, J.; García Del Rio, C.; Puerto, A. Oxytocin Polyuria and Polydipsia Is Blocked by NaCl Administration in Food-Deprived Male Rats. J. Neuroendocrinol. 2010, 22, 1043–1051. [Google Scholar] [CrossRef]
- Bernal, A.; Mahía, J.; Puerto, A. Potentiated Effect of Systemic Administration of Oxytocin on Hypertonic NaCl Intake in Food-Deprived Male Rats. Horm. Behav. 2010, 57, 284–290. [Google Scholar] [CrossRef]
- Bernal, A.; Mahía, J.; Mediavilla, C.; Puerto, A. Opposite Effects of Oxytocin on Water Intake Induced by Hypertonic NaCl or Polyethylene Glycol Administration. Physiol. Behav. 2015, 141, 135–142. [Google Scholar] [CrossRef]
- Mahía, J.; Bernal, A.; Puerto, A. Effects of Oxytocin Administration on the Hydromineral Balance of Median Eminence-Lesioned Rats. J. Neuroendocrinol. 2019, 31, e12778. [Google Scholar] [CrossRef]
- Verbalis, J.G.; Mangione, M.P.; Stricker, E.M. Oxytocin Produces Natriuresis in Rats at Physiological Plasma Concentrations. Endocrinology 1991, 128, 1317–1322. [Google Scholar] [CrossRef]
- Conrad, K.P.; Gellai, M.; North, W.G.; Valtin, H. Influence of Oxytocin on Renal Hemodynamics and Electrolyte and Water Excretion. Am. J. Physiol. 1986, 251, F290–F296. [Google Scholar] [CrossRef]
- Lippert, T.H.; Mueck, A.O.; Seeger, H.; Pfaff, A. Effects of Oxytocin Outside Pregnancy. Horm. Res. 2003, 60, 262–271. [Google Scholar] [CrossRef]
- Haanwinckel, M.A.; Elias, L.K.; Favaretto, A.L.; Gutkowska, J.; McCann, S.M.; Antunes-Rodrigues, J. Oxytocin Mediates Atrial Natriuretic Peptide Release and Natriuresis after Volume Expansion in the Rat. Proc. Natl. Acad. Sci. USA 1995, 92, 7902–7906. [Google Scholar] [CrossRef]
- Andersen, S.E.; Engstrøm, T.; Bie, P. Effects on Renal Sodium and Potassium Excretion of Vasopressin and Oxytocin in Conscious Dogs. Acta Physiol. Scand. 1992, 145, 267–274. [Google Scholar] [CrossRef]
- Windle, R.J.; Judah, J.M.; Forsling, M.L. Do Vasopressin and Oxytocin Have Synergistic Renal Effects in the Conscious Rat? J. Endocrinol. 1995, 144, 441–448. [Google Scholar] [CrossRef]
- Balment, R.J.; Brimble, M.J.; Forsling, M.L.; Kelly, L.P.; Musabayane, C.T. A Synergistic Effect of Oxytocin and Vasopressin on Sodium Excretion in the Neurohypophysectomized Rat. J. Physiol. 1986, 381, 453–464. [Google Scholar] [CrossRef]
- Balment, R.J.; Brimble, M.J.; Forsling, M.L.; Musabayane, C.T. The Influence of Neurohypophysial Hormones on Renal Function in the Acutely Hypophysectomized Rat. J. Physiol. 1986, 381, 439–452. [Google Scholar] [CrossRef]
- Bernal, A.; Mahía, J.; Puerto, A. Differential Lasting Inhibitory Effects of Oxytocin and Food-Deprivation on Mediobasal Hypothalamic Polydipsia. Brain Res. Bull. 2013, 94, 40–48. [Google Scholar] [CrossRef]
- Holcomb, S.S. Diabetes Insipidus. Dimens. Crit. Care Nurs. 2002, 21, 94–97. [Google Scholar] [CrossRef]
- Joo, K.W.; Jeon, U.S.; Kim, G.-H.; Park, J.; Oh, Y.K.; Kim, Y.S.; Ahn, C.; Kim, S.; Kim, S.Y.; Lee, J.S.; et al. Antidiuretic Action of Oxytocin Is Associated with Increased Urinary Excretion of Aquaporin-2. Nephrol. Dial. Transplant. 2004, 19, 2480–2486. [Google Scholar] [CrossRef]
- Rivkees, S.A.; Dunbar, N.; Wilson, T.A. The Management of Central Diabetes Insipidus in Infancy: Desmopressin, Low Renal Solute Load Formula, Thiazide Diuretics. J. Pediatr. Endocrinol. Metab. 2007, 20, 459–469. [Google Scholar] [CrossRef]
- Adrogué, H.J.; Madias, N.E. Hyponatremia. N. Engl. J. Med. 2000, 342, 1581–1589. [Google Scholar] [CrossRef] [PubMed]
- Sasaki, S. Is Oxytocin a Player in Antidiuresis? J. Am. Soc. Nephrol. 2008, 19, 189–190. [Google Scholar] [CrossRef] [PubMed]
- Kanbay, M.; Chen, Y.; Solak, Y.; Sanders, P.W. Mechanisms and Consequences of Salt Sensitivity and Dietary Salt Intake. Curr. Opin. Nephrol. Hypertens. 2011, 20, 37–43. [Google Scholar] [CrossRef]
- Balafa, O.; Kalaitzidis, R.G. Salt Sensitivity and Hypertension. J. Hum. Hypertens. 2021, 35, 184–192. [Google Scholar] [CrossRef] [PubMed]
- Dahl, L.K.; Heine, M. Primary Role of Renal Homografts in Setting Chronic Blood Pressure Levels in Rats. Circ. Res. 1975, 36, 692–696. [Google Scholar] [CrossRef]
- Mishra, S.; Ingole, S.; Jain, R. Salt Sensitivity and Its Implication in Clinical Practice. Indian Hear. J. 2018, 70, 556–564. [Google Scholar] [CrossRef]
- Bier, A.; Braun, T.; Khasbab, R.; Di Segni, A.; Grossman, E.; Haberman, Y.; Leibowitz, A. A High Salt Diet Modulates the Gut Microbiota and Short Chain Fatty Acids Production in a Salt-Sensitive Hypertension Rat Model. Nutrients 2018, 10, 1154. [Google Scholar] [CrossRef]
- Elijovich, F.; Laffer, C.L.; Sahinoz, M.; Pitzer, A.; Ferguson, J.F.; Kirabo, A. The Gut Microbiome, Inflammation, and Salt-Sensitive Hypertension. Curr. Hypertens. Rep. 2020, 22, 79. [Google Scholar] [CrossRef]
- Long-Smith, C.; O’Riordan, K.J.; Clarke, G.; Stanton, C.; Dinan, T.G.; Cryan, J.F. Microbiota-Gut-Brain Axis: New Therapeutic Opportunities. Annu. Rev. Pharmacol. Toxicol. 2020, 60, 477–502. [Google Scholar] [CrossRef]
- Kim, Y.S.; Unno, T.; Kim, B.-Y.; Park, M.-S. Sex Differences in Gut Microbiota. World J. Men’s Health 2020, 38, 48. [Google Scholar] [CrossRef]
- Mishima, E.; Abe, T. Role of the Microbiota in Hypertension and Antihypertensive Drug Metabolism. Hypertens. Res. 2022, 45, 246–253. [Google Scholar] [CrossRef]
- Yang, T.; Santisteban, M.M.; Rodriguez, V.; Li, E.; Ahmari, N.; Carvajal, J.M.; Zadeh, M.; Gong, M.; Qi, Y.; Zubcevic, J.; et al. Gut Dysbiosis Is Linked to Hypertension. Hypertension 2015, 65, 1331–1340. [Google Scholar] [CrossRef]





Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).