
Metformin Inefficiency to Lower Lipids in Vitamin B12 Deficient HepG2 Cells Is Alleviated via Adiponectin-AMPK Axis
 and
and {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Experimental Procedures
3. Results
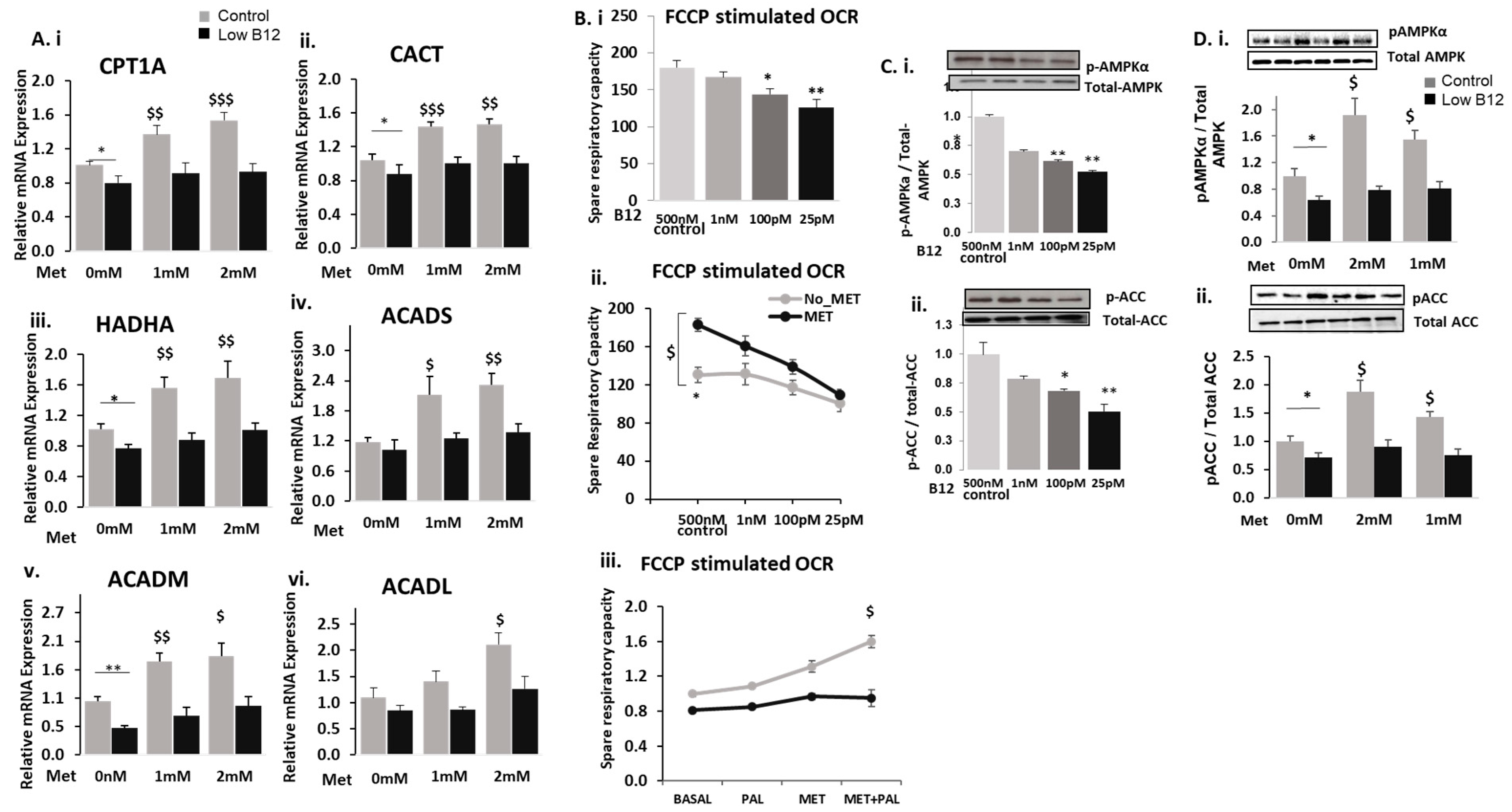
3.1. Low B12 Impedes Metformin Action on Intracellular TG and Expression of Genes in Fatty Acid Synthesis
3.2. Low B12 Impedes Metformin Action on Fatty Acid Oxidation (FAO) and Mitochondria Function via Impaired AMPK
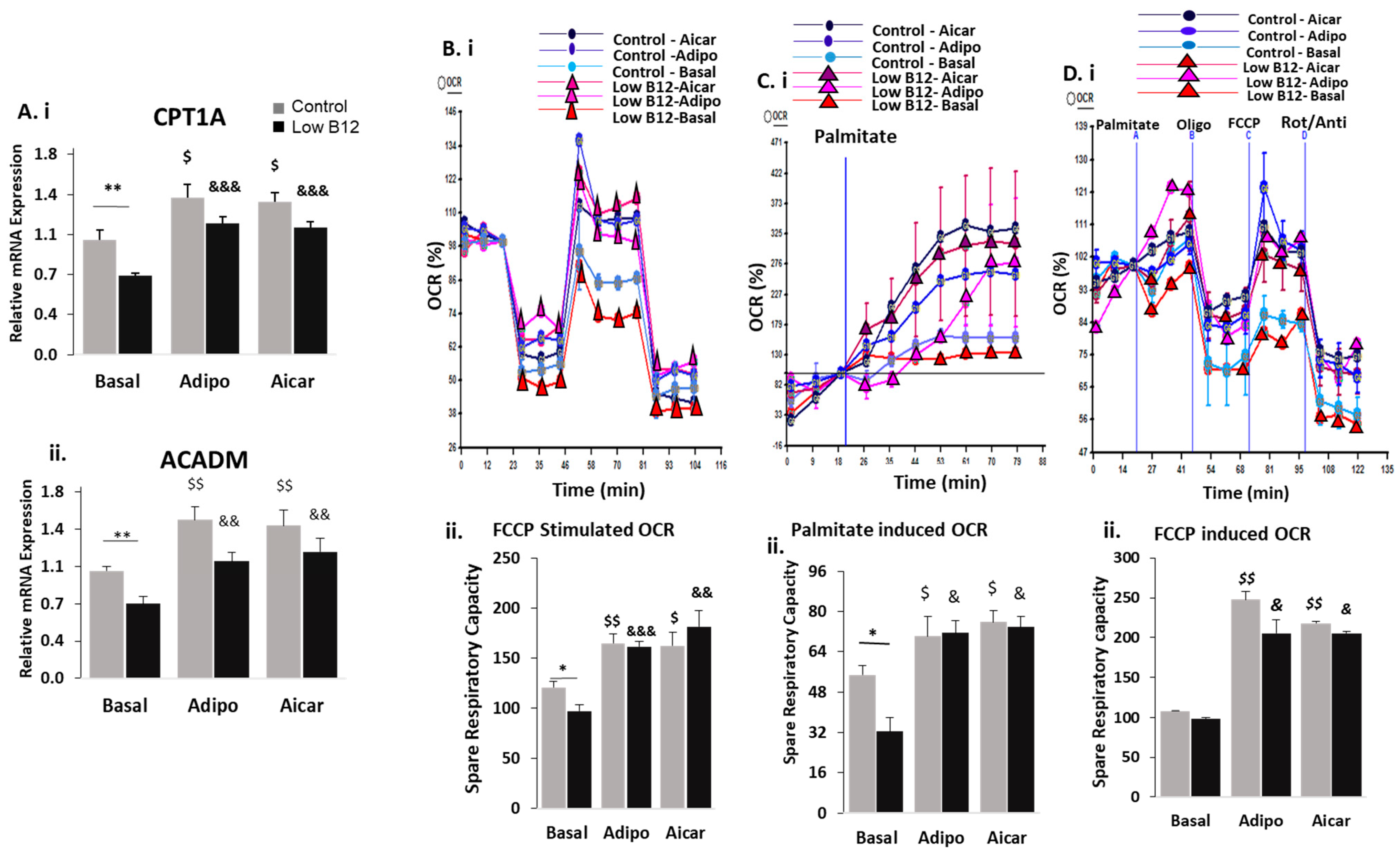
3.3. Adiponectin Ameliorates the Regulation of FA Synthesis in Low B12 Hepatocytes via Increased AMPK Activation and AdipoR1/R2
3.4. Adiponectin Ameliorates the Regulation of FAO in Low B12 Hepatocytes via Increased AMPK Activation
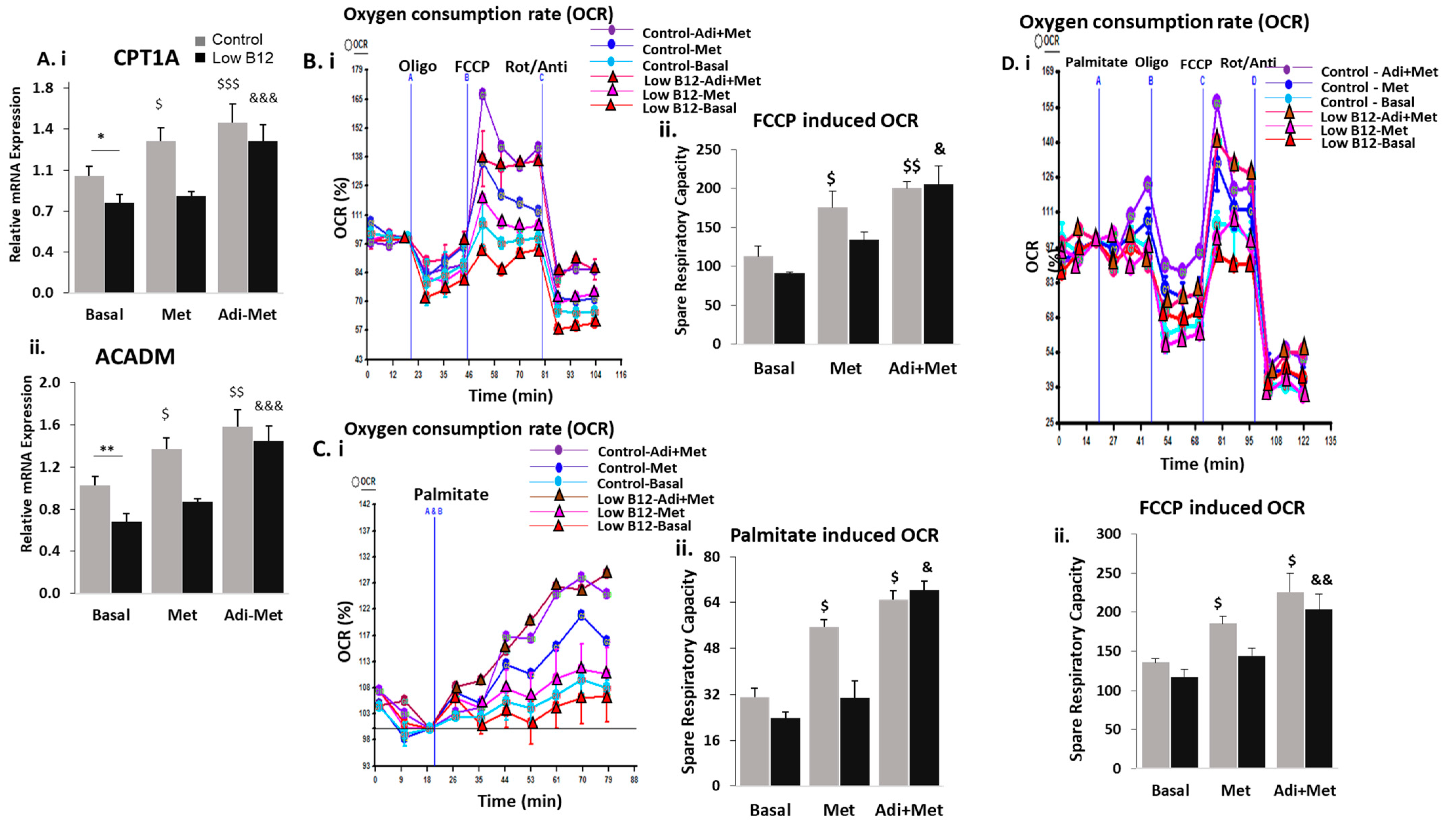
3.5. Combined Treatment of Adiponectin and Metformin Ameliorates the Regulation of FA Synthesis and FAO in Low B12 Hepatocytes via Increased AMPK Activation
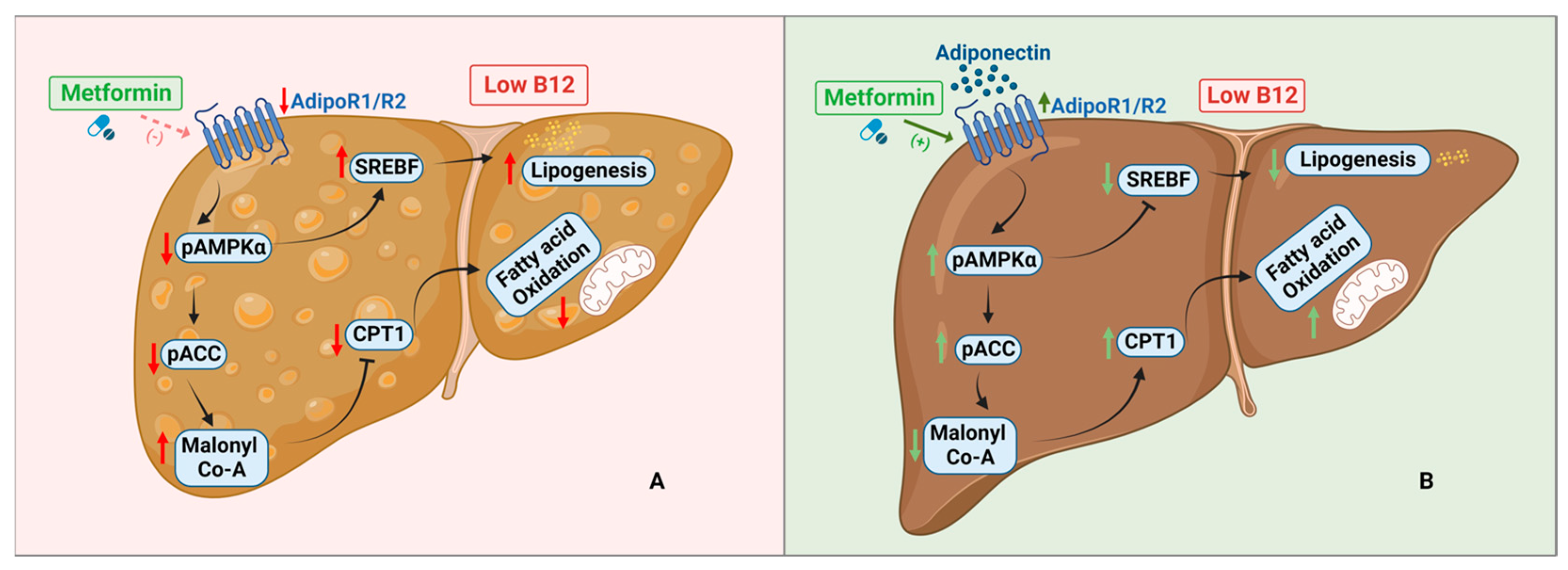
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Townsend, M.J.; Reddy, N.; Stanford, F.C. Geography and equity: Expanding access to obesity medicine diplomate care. Int. J. Obes. 2022, 46, 447–448. [Google Scholar] [CrossRef] [PubMed]
- Riazi, K.; Azhari, H.; Charette, J.H.; Underwood, F.E.; King, J.A.; Afshar, E.E.; Swain, M.G.; Congly, S.E.; Kaplan, G.G.; Shaheen, A.-A. The prevalence and incidence of NAFLD worldwide: A systematic review and meta-analysis. Lancet Gastroenterol. Hepatol. 2022, 7, 851–861. [Google Scholar] [CrossRef] [PubMed]
- Karlsen, T.H.; Sheron, N.; Zelber-Sagi, S.; Carrieri, P.; Dusheiko, G.; Bugianesi, E.; Pryke, R.; Hutchinson, S.J.; Sangro, B.; Martin, N.K.; et al. The EASL–Lancet Liver Commission: Protecting the next generation of Europeans against liver disease complications and premature mortality. Lancet 2022, 399, 61–116. [Google Scholar] [CrossRef] [PubMed]
- Hannah, W.; Bhavadharini, B.; Beks, H.; Deepa, M.; Anjana, R.M.; Uma, R.; Martin, E.; McNamara, K.; Versace, V.; Saravanan, P.; et al. Global burden of early pregnancy gestational diabetes mellitus (eGDM): A systematic review. Acta Diabetol. 2021, 59, 403–427. [Google Scholar] [CrossRef]
- Adaikalakoteswari, A.; Vatish, M.; Lawson, A.; Wood, C.; Sivakumar, K.; McTernan, P.G.; Webster, C.; Anderson, N.; Yajnik, C.S.; Tripathi, G.; et al. Low Maternal Vitamin B12 Status Is Associated with Lower Cord Blood HDL Cholesterol in White Caucasians Living in the UK. Nutrients 2015, 7, 2401–2414. [Google Scholar] [CrossRef] [PubMed]
- Adaikalakoteswari, A.; Jayashri, R.; Sukumar, N.; Venkataraman, H.; Pradeepa, R.; Gokulakrishnan, K.; Anjana, R.M.; McTernan, P.G.; Tripathi, G.; Patel, V.; et al. Vitamin B12 deficiency is associated with adverse lipid profile in Europeans and Indians with type 2 diabetes. Cardiovasc. Diabetol. 2014, 13, 129. [Google Scholar] [CrossRef] [PubMed]
- Ghosh, S.; Sinha, J.K.; Putcha, U.K.; Raghunath, M. Severe but Not Moderate Vitamin B12 Deficiency Impairs Lipid Profile, Induces Adiposity, and Leads to Adverse Gestational Outcome in Female C57BL/6 Mice. Front. Nutr. 2016, 3, 1. [Google Scholar] [CrossRef]
- Adaikalakoteswari, A.; Finer, S.; Voyias, P.D.; McCarthy, C.M.; Vatish, M.; Moore, J.; Smart-Halajko, M.; Bawazeer, N.; Al-Daghri, N.M.; McTernan, P.G.; et al. Vitamin B12 insufficiency induces cholesterol biosynthesis by limiting s-adenosylmethionine and modulating the methylation of SREBF1 and LDLR genes. Clin. Epigenet. 2015, 7, 14. [Google Scholar] [CrossRef]
- Boachie, J.; Adaikalakoteswari, A.; Gázquez, A.; Zammit, V.; Larqué, E.; Saravanan, P. Vitamin B12 Induces Hepatic Fatty Infiltration through Altered Fatty Acid Metabolism. Cell. Physiol. Biochem. 2021, 55, 241–255. [Google Scholar] [CrossRef]
- Infante, M.; Leoni, M.; Caprio, M.; Fabbri, A. Long-term metformin therapy and vitamin B12 deficiency: An association to bear in mind. World J. Diabetes 2021, 12, 916–931. [Google Scholar] [CrossRef]
- Zhu, X.; Yan, H.; Xia, M.; Chang, X.; Xu, X.; Wang, L.; Sun, X.; Lu, Y.; Bian, H.; Li, X.; et al. Metformin attenuates triglyceride accumulation in HepG2 cells through decreasing stearyl-coenzyme A desaturase 1 expression. Lipids Health Dis. 2018, 17, 114. [Google Scholar] [CrossRef] [PubMed]
- Szymczak-Pajor, I.; Wenclewska, S.; Śliwińska, A. Metabolic Action of Metformin. Pharmaceuticals 2022, 15, 810. [Google Scholar] [CrossRef] [PubMed]
- Tokubuchi, I.; Tajiri, Y.; Iwata, S.; Hara, K.; Wada, N.; Hashinaga, T.; Nakayama, H.; Mifune, H.; Yamada, K. Beneficial effects of metformin on energy metabolism and visceral fat volume through a possible mechanism of fatty acid oxidation in human subjects and rats. PLoS ONE 2017, 12, e0171293. [Google Scholar] [CrossRef] [PubMed]
- Green, C.J.; Marjot, T.; Walsby-Tickle, J.; Charlton, C.; Cornfield, T.; Westcott, F.; Pinnick, E.K.; Moolla, A.; Hazlehurst, J.M.; McCullagh, J.; et al. Metformin maintains intrahepatic triglyceride content through increased hepatic de novo lipogenesis. Eur. J. Endocrinol. 2022, 186, 367–377. [Google Scholar] [CrossRef] [PubMed]
- Kumagai, A.; Itakura, A.; Koya, D.; Kanasaki, K. AMP-Activated Protein (AMPK) in Pathophysiology of Pregnancy Complications. Int. J. Mol. Sci. 2018, 19, 3076. [Google Scholar] [CrossRef] [PubMed]
- Senkus, K.E.; Crowe-White, K.M.; Bolland, A.C.; Locher, J.L.; Ard, J.D. Changes in adiponectin:leptin ratio among older adults with obesity following a 12-month exercise and diet intervention. Nutr. Diabetes 2022, 12, 30. [Google Scholar] [CrossRef] [PubMed]
- Achari, A.E.; Jain, S.K. Adiponectin, a therapeutic target for obesity, diabetes, and endothelial dysfunction. Int. J. Mol. Sci. 2017, 18, 1321. [Google Scholar] [CrossRef] [PubMed]
- Xu, A.; Wang, Y.; Keshaw, H.; Xu, L.Y.; Lam, K.S.; Cooper, G.J. The fat-derived hormone adiponectin alleviates alcoholic and nonalcoholic fatty liver diseases in mice. J. Clin. Investig. 2003, 112, 91–100. [Google Scholar] [CrossRef]
- Koh, E.H.; Park, J.-Y.; Park, H.-S.; Jeon, M.J.; Ryu, J.W.; Kim, M.; Kim, S.Y.; Kim, M.-S.; Kim, S.-W.; Park, I.S. Essential role of mitochondrial function in adiponectin synthesis in adipocytes. Diabetes 2007, 56, 2973–2981. [Google Scholar] [CrossRef]
- Kadowaki, T.; Yamauchi, T.; Kubota, N.; Hara, K.; Ueki, K.; Tobe, K. Adiponectin and adiponectin receptors in insulin resistance, diabetes, and the metabolic syndrome. J. Clin. Investig. 2006, 116, 1784–1792. [Google Scholar] [CrossRef]
- Park, D.; Lee, S.; Boo, H. Metformin Induces Lipogenesis and Apoptosis in H4IIE Hepatocellular Carcinoma Cells. Dev. Reprod. 2023, 27, 77–89. [Google Scholar] [CrossRef] [PubMed]
- Sui, Y.; Kong, X.; Fan, R.; Ye, Y.; Mai, H.; Zhuo, S.; Lu, W.; Ruan, P.; Fang, S.; Yang, T. Long-term treatment with metformin in the prevention of fatty liver in Zucker diabetic fatty rats. Diabetol. Metab. Syndr. 2019, 11, 94. [Google Scholar] [CrossRef]
- Jackisch, L.; Kumsaiyai, W.; Moore, J.D.; Al-Daghri, N.; Kyrou, I.; Barber, T.M.; Randeva, H.; Kumar, S.; Tripathi, G.; McTernan, P.G. Differential expression of Lp-PLA2 in obesity and type 2 diabetes and the influence of lipids. Diabetologia 2018, 61, 1155–1166. [Google Scholar] [CrossRef] [PubMed]
- Schisano, B.; Tripathi, G.; McGee, K.; McTernan, P.G.; Ceriello, A. Glucose oscillations, more than constant high glucose, induce p53 activation and a metabolic memory in human endothelial cells. Diabetologia 2011, 54, 1219–1226. [Google Scholar] [CrossRef] [PubMed]
- van der Windt, G.J.W.; Chang, C.-H.; Pearce, E.L. Measuring bioenergetics in T cells using a Seahorse Extracellular Flux Analyzer. Curr. Protoc. Immunol. 2016, 113, 3–16. [Google Scholar] [CrossRef] [PubMed]
- Gluais-Dagorn, P.; Foretz, M.; Steinberg, G.R.; Batchuluun, B.; Zawistowska-Deniziak, A.; Lambooij, J.M.; Guigas, B.; Carling, D.; Monternier, P.; Moller, D.E.; et al. Direct AMPK Activation Corrects NASH in Rodents Through Metabolic Effects and Direct Action on Inflammation and Fibrogenesis. Hepatol. Commun. 2022, 6, 101–119. [Google Scholar] [CrossRef] [PubMed]
- Peng, H.; Xu, H.; Wu, J.; Li, J.; Wang, X.; Liu, Z.; Kim, M.; Jeon, M.S.; Zhang, K.K.; Xie, L. Maternal One-Carbon Supplement Reduced the Risk of Non-Alcoholic Fatty Liver Disease in Male Offspring. Nutrients 2022, 14, 2545. [Google Scholar] [CrossRef] [PubMed]
- Boudaba, N.; Marion, A.; Huet, C.; Pierre, R.; Viollet, B.; Foretz, M. AMPK Re-Activation Suppresses Hepatic Steatosis but its Downregulation Does Not Promote Fatty Liver Development. EBioMedicine 2018, 28, 194–209. [Google Scholar] [CrossRef]
- Greibe, E.; Miller, J.; Foutouhi, S.; Green, R.; Nexo, E. Metformin increases liver accumulation of vitamin B12—An experimental study in rats. Biochimie 2013, 95, 1062–1065. [Google Scholar] [CrossRef]
- Hasanvand, A. The role of AMPK-dependent pathways in cellular and molecular mechanisms of metformin: A new perspective for treatment and prevention of diseases. Inflammopharmacology 2022, 30, 775–788. [Google Scholar] [CrossRef]
- Rena, G.; Pearson, E.R.; Sakamoto, K. Molecular mechanism of action of metformin: Old or new insights? Diabetologia 2013, 56, 1898–18906. [Google Scholar] [CrossRef] [PubMed]
- Henry, C.J.; Nemkov, T.; Casás-Selves, M.; Bilousova, G.; Zaberezhnyy, V.; Higa, K.C.; Serkova, N.J.; Hansen, K.C.; D’alessandro, A.; DeGregori, J. Folate dietary insufficiency and folic acid supplementation similarly impair metabolism and compromise hematopoiesis. Haematologica 2017, 102, 1985–1994. [Google Scholar] [CrossRef] [PubMed]
- Madsen, A.; Bozickovic, O.; Bjune, J.-I.; Mellgren, G.; Sagen, J.V. Metformin inhibits hepatocellular glucose, lipid and cholesterol biosynthetic pathways by transcriptionally suppressing steroid receptor coactivator 2 (SRC-2). Sci. Rep. 2015, 5, 16430. [Google Scholar] [CrossRef] [PubMed]
- Green, C.J.; Marjot, T.; Tomlinson, J.W.; Hodson, L. Of mice and men: Is there a future for metformin in the treatment of hepatic steatosis? Diabetes Obes. Metab. 2019, 21, 749–760. [Google Scholar] [CrossRef] [PubMed]
- Aroda, V.R.; Edelstein, S.L.; Goldberg, R.B.; Knowler, W.C.; Marcovina, S.M.; Orchard, T.J.; Bray, G.A.; Schade, D.S.; Temprosa, M.G.; White, N.H.; et al. Long-term Metformin Use and Vitamin B12 Deficiency in the Diabetes Prevention Program Outcomes Study. J. Clin. Endocrinol. Metab. 2016, 101, 1754–1761. [Google Scholar] [CrossRef] [PubMed]
- Andrès, E.; Serraj, K.; Zhu, J.; Vermorken, A. The pathophysiology of elevated vitamin B12 in clinical practice. QJM Int. J. Med. 2013, 106, 505–515. [Google Scholar] [CrossRef]
- Zabielski, P.; Hady, H.R.; Chacinska, M.; Roszczyc, K.; Gorski, J.; Blachnio-Zabielska, A.U. The effect of high fat diet and metformin treatment on liver lipids accumulation and their impact on insulin action. Sci. Rep. 2018, 8, 7249. [Google Scholar] [CrossRef]
- Fullerton, M.D.; Galic, S.; Marcinko, K.; Sikkema, S.; Pulinilkunnil, T.; Chen, Z.-P.; O’Neill, H.M.; Ford, R.J.; Palanivel, R.; O’Brien, M.; et al. Single phosphorylation sites in Acc1 and Acc2 regulate lipid homeostasis and the insulin-sensitizing effects of metformin. Nat. Med. 2013, 19, 1649–1654. [Google Scholar] [CrossRef]
- Fulgencio, J.-P.; Kohl, C.; Girard, J.; Pégorier, J.-P. Effect of metformin on fatty acid and glucose metabolism in freshly isolated hepatocytes and on specific gene expression in cultured hepatocytes. Biochem. Pharmacol. 2001, 62, 439–446. [Google Scholar] [CrossRef]
- Gormsen, L.C.; Søndergaard, E.; Christensen, N.L.; Jakobsen, S.; Nielsen, E.H.T.; Munk, O.L.; Tolbod, L.P.; Jessen, N.; Nielsen, S. Metformin does not affect postabsorptive hepatic free fatty acid uptake, oxidation or resecretion in humans: A 3-month placebo-controlled clinical trial in patients with type 2 diabetes and healthy controls. Diabetes Obes. Metab. 2018, 20, 1435–1444. [Google Scholar] [CrossRef]
- Li, X.; Zhang, D.; Vatner, D.F.; Goedeke, L.; Hirabara, S.M.; Zhang, Y.; Perry, R.J.; Shulman, G.I. Mechanisms by which adiponectin reverses high fat diet-induced insulin resistance in mice. Proc. Natl. Acad. Sci. USA 2020, 117, 32584–32593. [Google Scholar] [CrossRef] [PubMed]
- Fukui, H.; Uemura, M.; Akahane, T.; Tsujimoto, T.; Yanase, K.; Namisaki, T.; Yamazaki, M.; Yoshii, J.; Kaji, K.; Kitade, M.; et al. Crosstalk between high-molecular-weight adiponectin and T-cadherin during liver fibrosis development in rats. Int. J. Mol. Med. 2007, 20, 725–729. [Google Scholar] [CrossRef]
- Kadowaki, T.; Yamauchi, T. Adiponectin and adiponectin receptors. Endocr. Rev. 2005, 26, 439–451. [Google Scholar] [CrossRef] [PubMed]
- Gamberi, T.; Magherini, F.; Modesti, A.; Fiaschi, T. Adiponectin Signaling Pathways in Liver Diseases. Biomedicines 2018, 6, 52. [Google Scholar] [CrossRef] [PubMed]
- Awazawa, M.; Ueki, K.; Inabe, K.; Yamauchi, T.; Kaneko, K.; Okazaki, Y.; Bardeesy, N.; Ohnishi, S.; Nagai, R.; Kadowaki, T. Adiponectin suppresses hepatic SREBP1c expression in an AdipoR1/LKB1/AMPK dependent pathway. Biochem. Biophys. Res. Commun. 2009, 382, 51–56. [Google Scholar] [CrossRef] [PubMed]
- Liu, Q.; Yuan, B.; Lo, K.A.; Patterson, H.C.; Sun, Y.; Lodish, H.F. Adiponectin regulates expression of hepatic genes critical for glucose and lipid metabolism. Proc. Natl. Acad. Sci. USA 2012, 109, 14568–14573. [Google Scholar] [CrossRef]
- Wang, Y.; Zidichouski, J.A. Update on the Benefits and Mechanisms of Action of the Bioactive Vegetal Alkaloid Berberine on Lipid Metabolism and Homeostasis. Cholesterol 2018, 2018, 7173920. [Google Scholar] [CrossRef]
- Yoon, M.J.; Lee, G.Y.; Chung, J.-J.; Ahn, Y.H.; Hong, S.H.; Kim, J.B. Adiponectin increases fatty acid oxidation in skeletal muscle cells by sequential activation of AMPK, p38 MAPK, and PPARalpha. Proc. Spring Conf. Soc. Biochem. Mol. Biol. 2006, 2006, 262. [Google Scholar]
- Civitarese, A.E.; Ukropcova, B.; Carling, S.; Hulver, M.; DeFronzo, R.A.; Mandarino, L.; Ravussin, E.; Smith, S.R. Role of adiponectin in human skeletal muscle bioenergetics. Cell Metab. 2006, 4, 75–87. [Google Scholar] [CrossRef]
- Abdella, N.A.; Mojiminiyi, O.A. Clinical applications of adiponectin measurements in type 2 diabetes mellitus: Screening, diagnosis, and marker of diabetes control. Dis. Markers 2018, 2018, 5187940. [Google Scholar] [CrossRef]
- Fard, A.A.; Hajrezaie, M.; Kadir, F.A.; Sefideh, F.A.; Salama, S.M.; Al-Najar, Z.A.; Qader, S.W.; Alshawsh, M.A.; Abdulla, M.A. The Effects of Combined Adiponectin-Metformin on Glucose and Lipids Levels in Mice and Acute Toxicity and Anti-Ulcerogenic Activity of Adiponectin against Ethanol-Induced Gastric Mucosal Injuries in Rat. Molecules 2011, 16, 9534–9552. [Google Scholar] [CrossRef]
- Min, Q.-Q.; Qin, L.-Q.; Sun, Z.-Z.; Zuo, W.-T.; Zhao, L.; Xu, J.-Y. Effects of Metformin Combined with Lactoferrin on Lipid Accumulation and Metabolism in Mice Fed with High-Fat Diet. Nutrients 2018, 10, 1628. [Google Scholar] [CrossRef] [PubMed]
- Rogers, C.Q.; Ajmo, J.M.; You, M. Adiponectin and alcoholic fatty liver disease. IUBMB Life 2008, 60, 790–797. [Google Scholar] [CrossRef] [PubMed]
- Cantó, C.; Auwerx, J. AMP-activated protein kinase and its downstream transcriptional pathways. Cell. Mol. Life Sci. 2010, 67, 3407–3423. [Google Scholar] [CrossRef] [PubMed]
- Aden, D.P.; Fogel, A.; Plotkin, S.; Damjanov, I.; Knowles, B.B. Controlled synthesis of HBsAg in a differentiated human liver carcinoma-derived cell line. Nature 1979, 282, 615–616. [Google Scholar] [CrossRef]
- López-Terrada, D.; Cheung, S.W.; Finegold, M.J.; Knowles, B.B. Hep G2 is a hepatoblastoma-derived cell line. Hum. Pathol. 2009, 40, 1512. [Google Scholar] [CrossRef]
- Young, L.; Sung, J.; Stacey, G.; Masters, J.R. Detection of Mycoplasma in cell cultures. Nat. Protoc. 2010, 5, 929–934. [Google Scholar] [CrossRef]



Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Boachie, J.; Zammit, V.; Saravanan, P.; Adaikalakoteswari, A. Metformin Inefficiency to Lower Lipids in Vitamin B12 Deficient HepG2 Cells Is Alleviated via Adiponectin-AMPK Axis. Nutrients 2023, 15, 5046. https://doi.org/10.3390/nu15245046
Boachie J, Zammit V, Saravanan P, Adaikalakoteswari A. Metformin Inefficiency to Lower Lipids in Vitamin B12 Deficient HepG2 Cells Is Alleviated via Adiponectin-AMPK Axis. Nutrients. 2023; 15(24):5046. https://doi.org/10.3390/nu15245046
Chicago/Turabian StyleBoachie, Joseph, Victor Zammit, Ponnusamy Saravanan, and Antonysunil Adaikalakoteswari. 2023. "Metformin Inefficiency to Lower Lipids in Vitamin B12 Deficient HepG2 Cells Is Alleviated via Adiponectin-AMPK Axis" Nutrients 15, no. 24: 5046. https://doi.org/10.3390/nu15245046