
S-(−)-Oleocanthal Ex Vivo Modulatory Effects on Gut Microbiota




Abstract
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
- (1)
- We designed and validated a detailed standard operating protocol, thereby achieving a rapid-analysis workflow for studying OC effects on GM via an ex vivo mouse fecal fermentation model, optimizing all associated factors to maximally reflect and closely mimic the living gut system.
- (2)
- We developed protocols for the tailored use of shallow whole metagenome sequencing of optimized ex vivo fecal samples to characterize the GM constitution upon addition of OC, which was seen to comprehensively cause qualitative and quantitative GM changes. Further analysis documented OC effects on various GM species. With information linking these species to corresponding distribution sites (known or predicted) within the human microbiome, it becomes possible to envision prospective human health impacts of such changes.
2. Materials and Methods
2.1. Oleocanthal Extraction and Purification Process
2.2. Housing, Diets, and Animal Model’s Experimental Setup
2.3. Ethics Statement
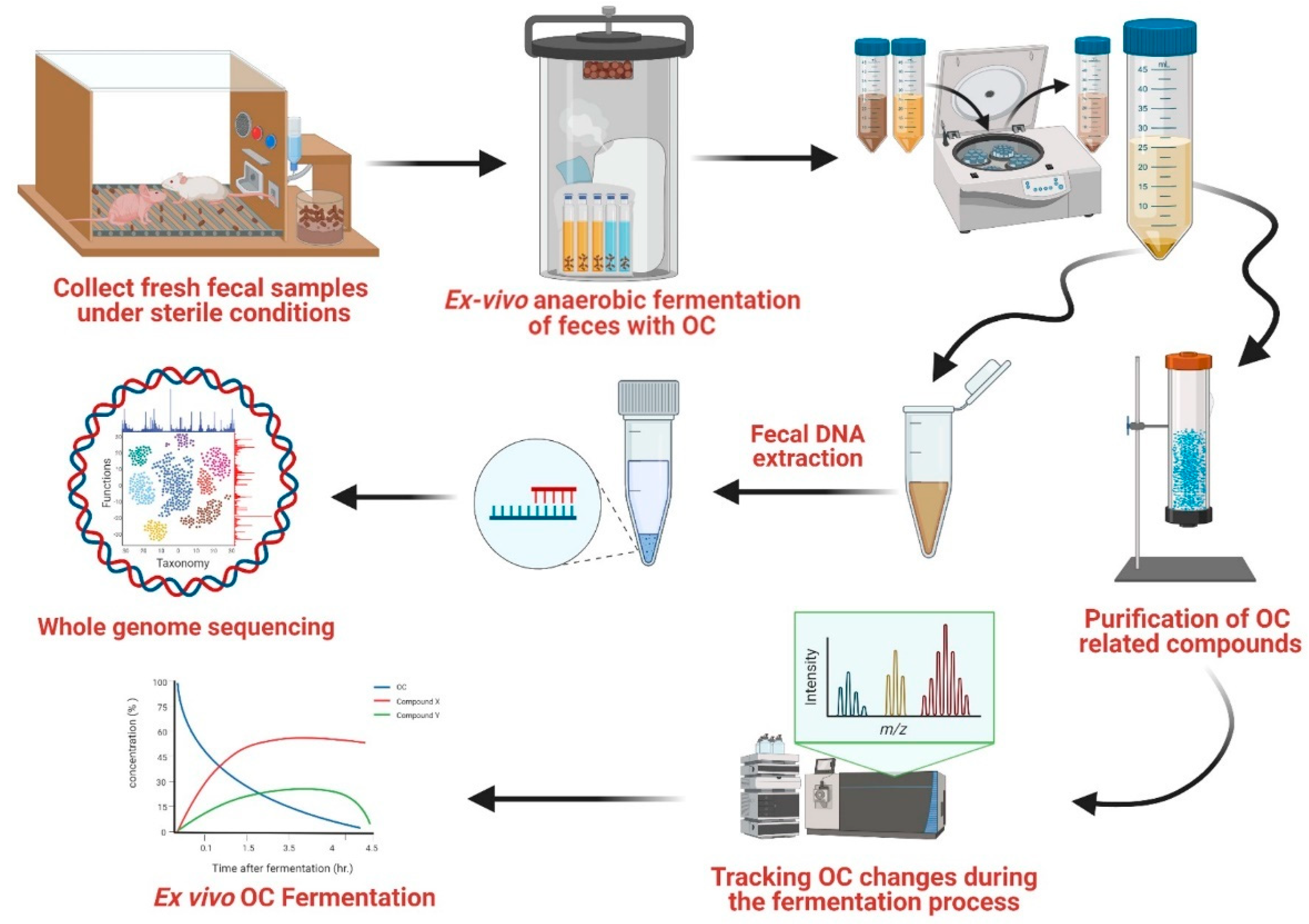
2.4. Ex Vivo Fecal Fermentation
2.5. Fermentation Samples Preparation
2.6. DNA Extraction of Fermented Fecal Samples
2.7. Taxonomy Profiling by Shotgun-Based Next-Generation Sequencing
2.8. Diversity and Differential Abundance Analysis
2.9. Correlation of the OC-Affected GM with the Human Microbiota
3. Results
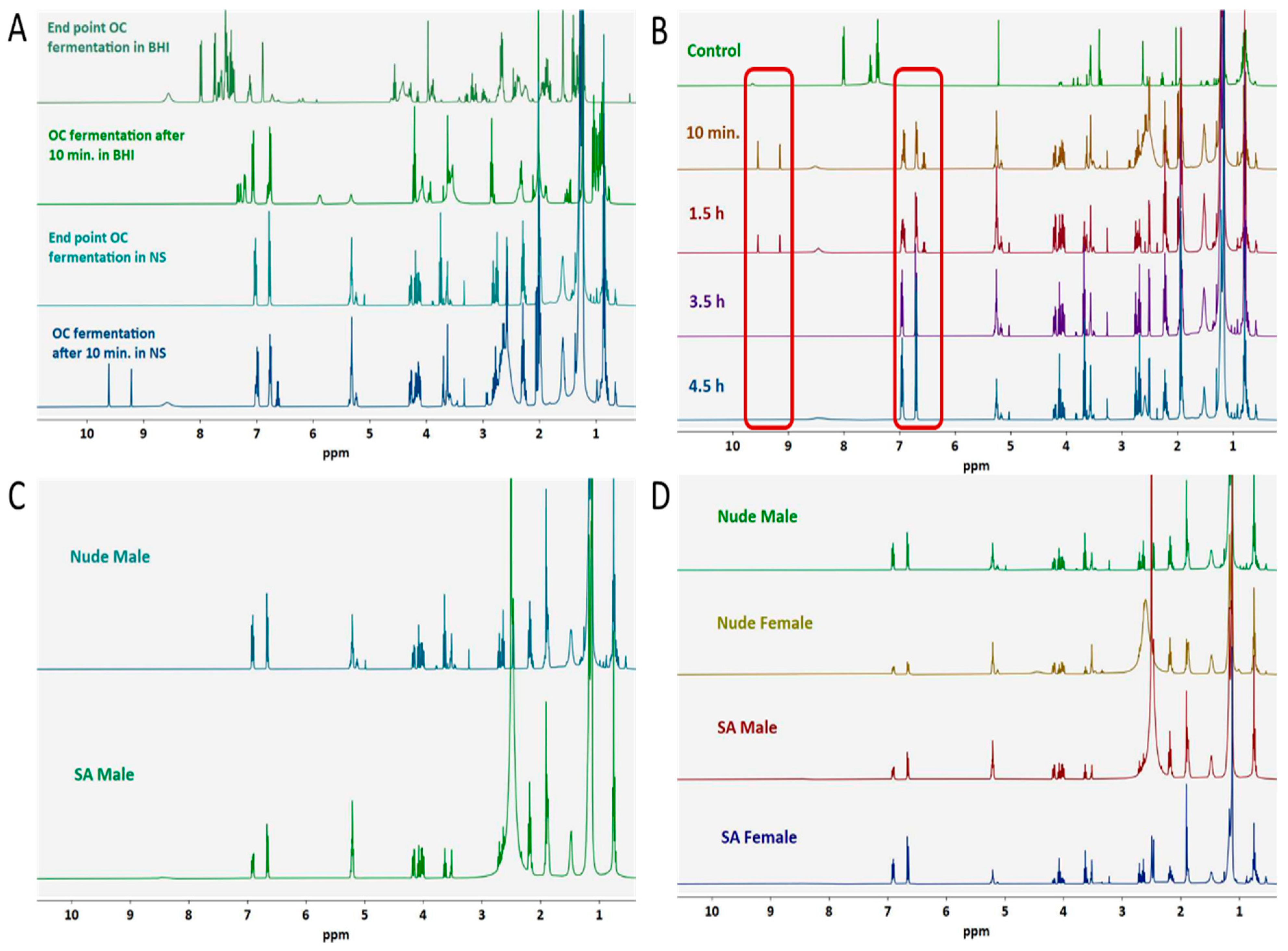
3.1. Ex Vivo Fecal Fermentation Model for Identification of GM Metabolites
3.2. Optimization of OC-GM Interactive Model
3.3. Microbial Taxonomy Profiling Using Shallow Shotgun Metagenomic Sequencing
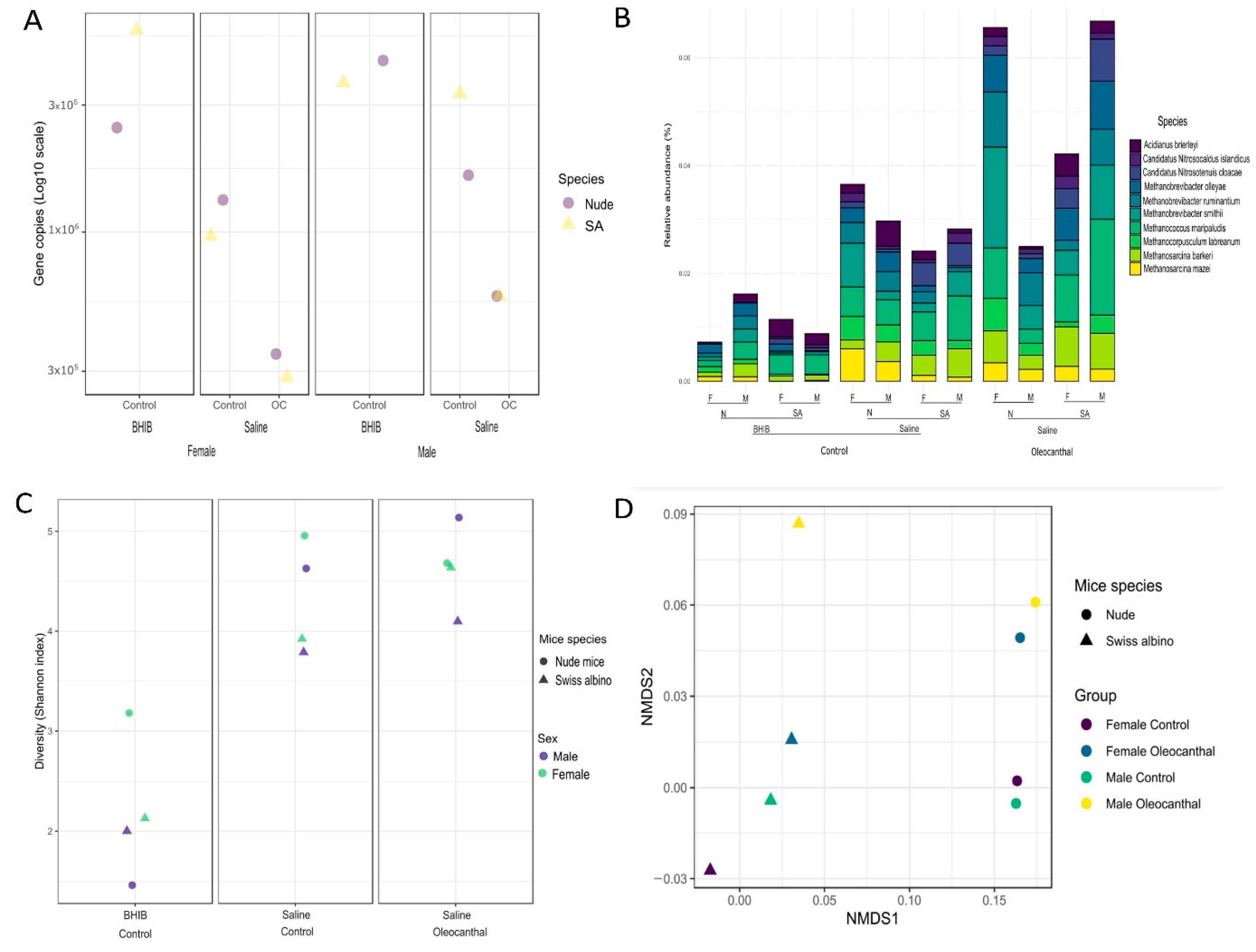
3.4. Absolute Microbial Abundances by qPCR
3.5. Microbial Diversities and Variation Analysis
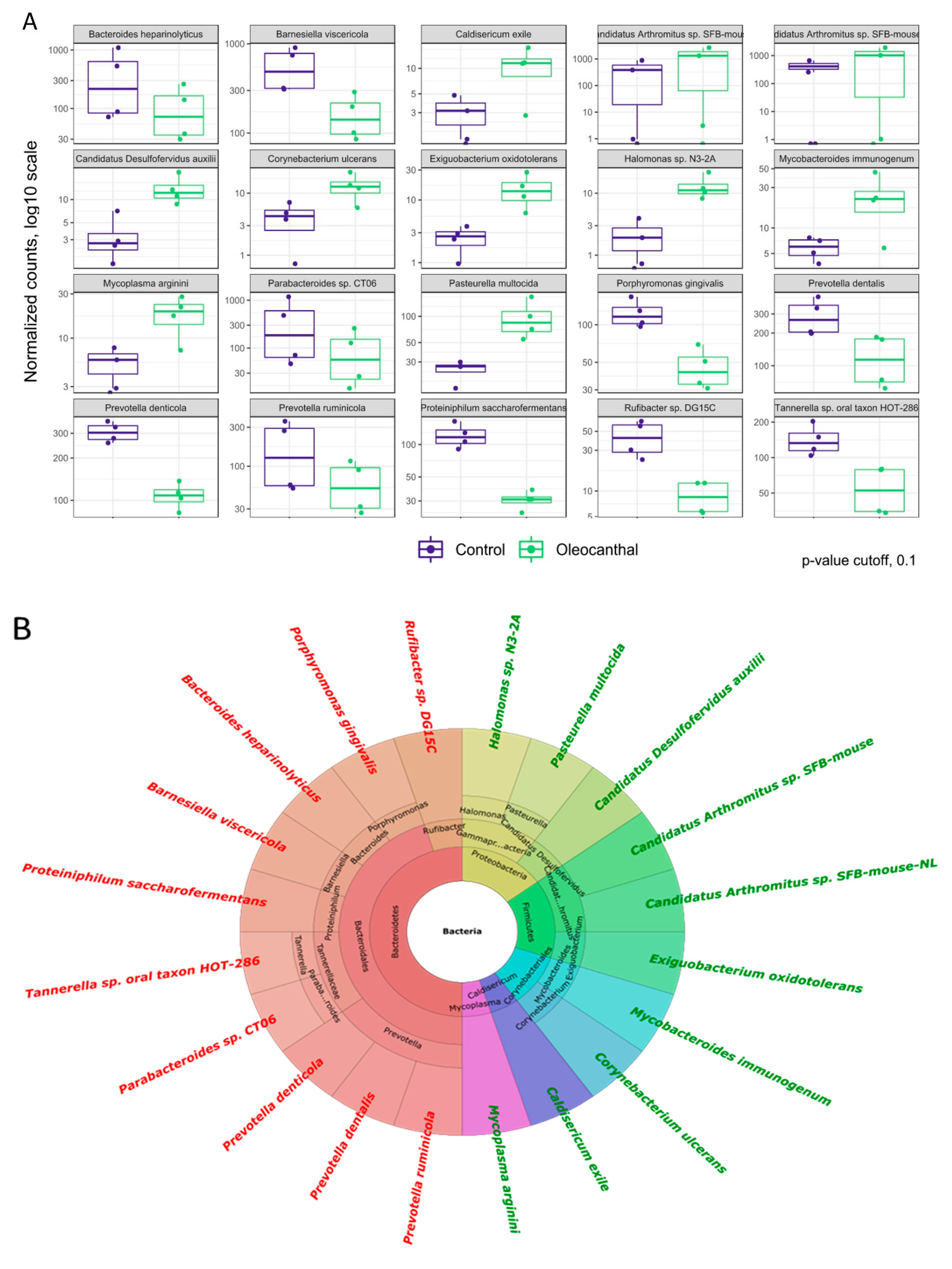
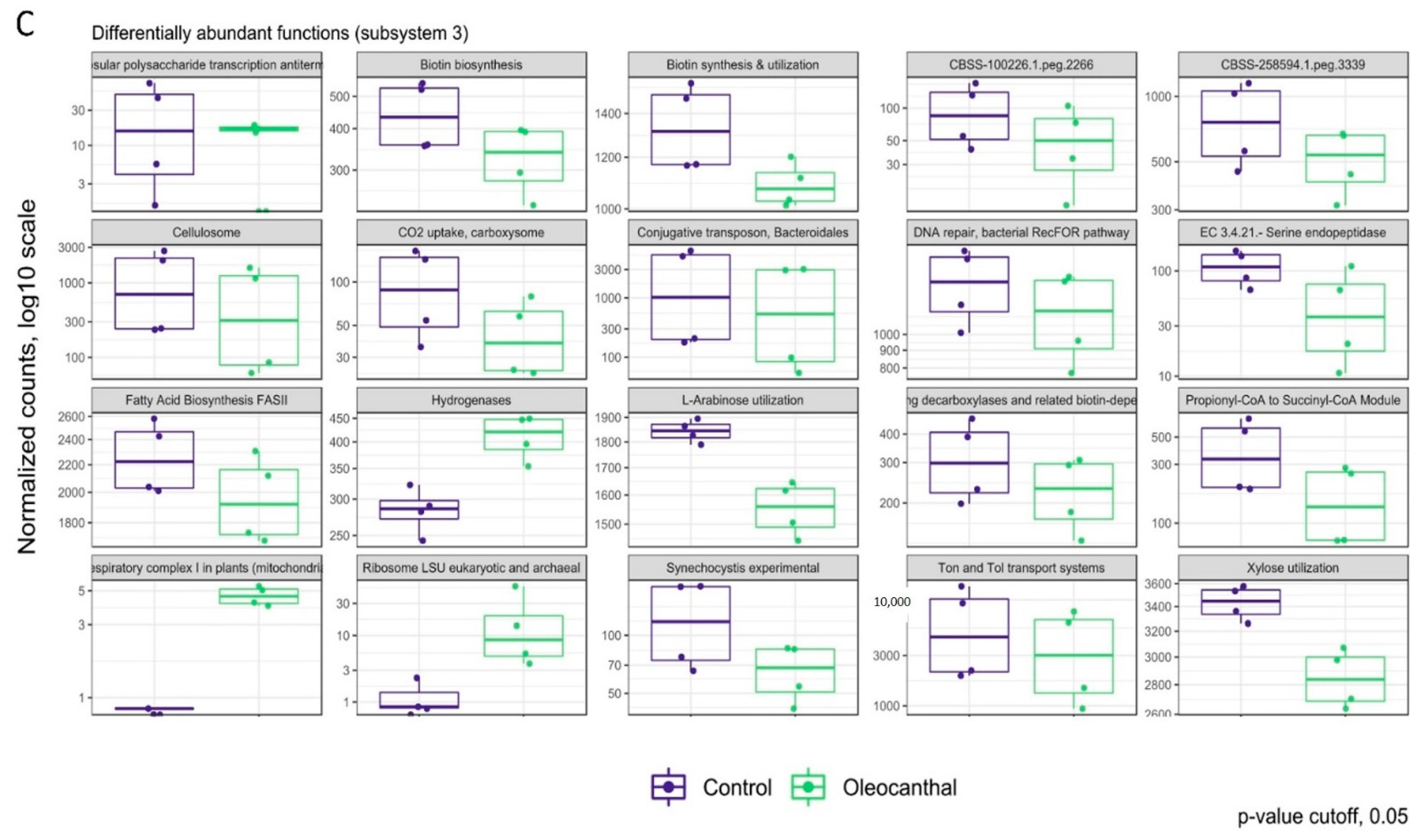
3.6. Analysis of Differential Abundance and Functions of OC-Affected GM
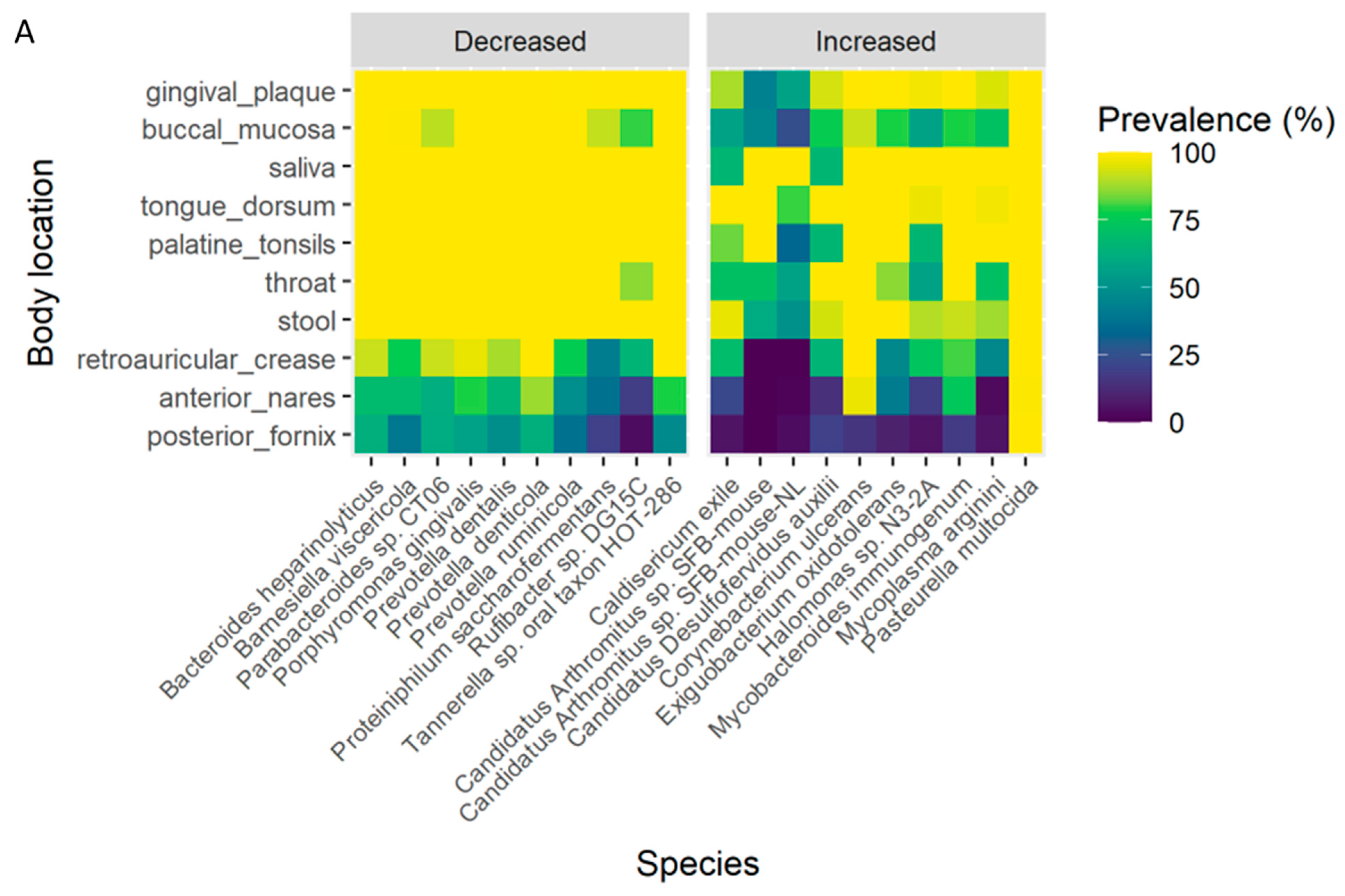
3.7. Association of the OC-Affected Bacteria with the Human Body
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Romani, A.; Ieri, F.; Urciuoli, S.; Noce, A.; Marrone, G.; Nediani, C.; Bernini, R. Health effects of phenolic compounds found in extra-virgin olive oil, by-products, and leaf of Olea europaea L. Nutrients 2019, 11, 1776. [Google Scholar] [CrossRef] [Green Version]
- Segura-Carretero, A.; Curiel, J.A. Current disease-targets for oleocanthal as promising natural therapeutic agent. Int. J. Mol. Sci. 2018, 19, 2899. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Qosa, H.; Batarseh, Y.S.; Mohyeldin, M.M.; El Sayed, K.A.; Keller, J.N.; Kaddoumi, A. Oleocanthal enhances amyloid-β clearance from the brains of TgSwDI mice and in vitro across a human blood-brain barrier model. ACS Chem. Neurosci. 2015, 6, 1849–1859. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mohyeldin, M.M.; Busnena, B.A.; Akl, M.R.; Dragoi, A.M.; Cardelli, J.A.; El Sayed, K.A. Novel c-Met inhibitory olive secoiridoid semisynthetic analogs for the control of invasive breast cancer. Eur. J. Med. Chem. 2016, 118, 299–315. [Google Scholar] [CrossRef] [PubMed]
- Ayoub, N.M.; Siddique, A.B.; Ebrahim, H.Y.; Mohyeldin, M.M.; El Sayed, K.A. The olive oil phenolic (−)-oleocanthal modulates estrogen receptor expression in luminal breast cancer in vitro and in vivo and synergizes with tamoxifen treatment. Eur. J. Pharmacol. 2017, 810, 100–111. [Google Scholar] [CrossRef] [PubMed]
- López-Yerena, A.; Vallverdú-Queralt, A.; Mols, R.; Augustijns, P.; Lamuela-Raventós, R.M.; Escribano-Ferrer, E. Absorption and intestinal metabolic profile of oleocanthal in rats. Pharmaceutics 2020, 12, 134. [Google Scholar] [CrossRef] [Green Version]
- López-Yerena, A.; Vallverdú-Queralt, A.; Jáuregui, O.; Garcia-Sala, X.; Lamuela-Raventós, R.M.; Escribano-Ferrer, E. Tissue distribution of oleocanthal and its metabolites after oral ingestion in rats. Antioxidants 2021, 10, 688. [Google Scholar] [CrossRef]
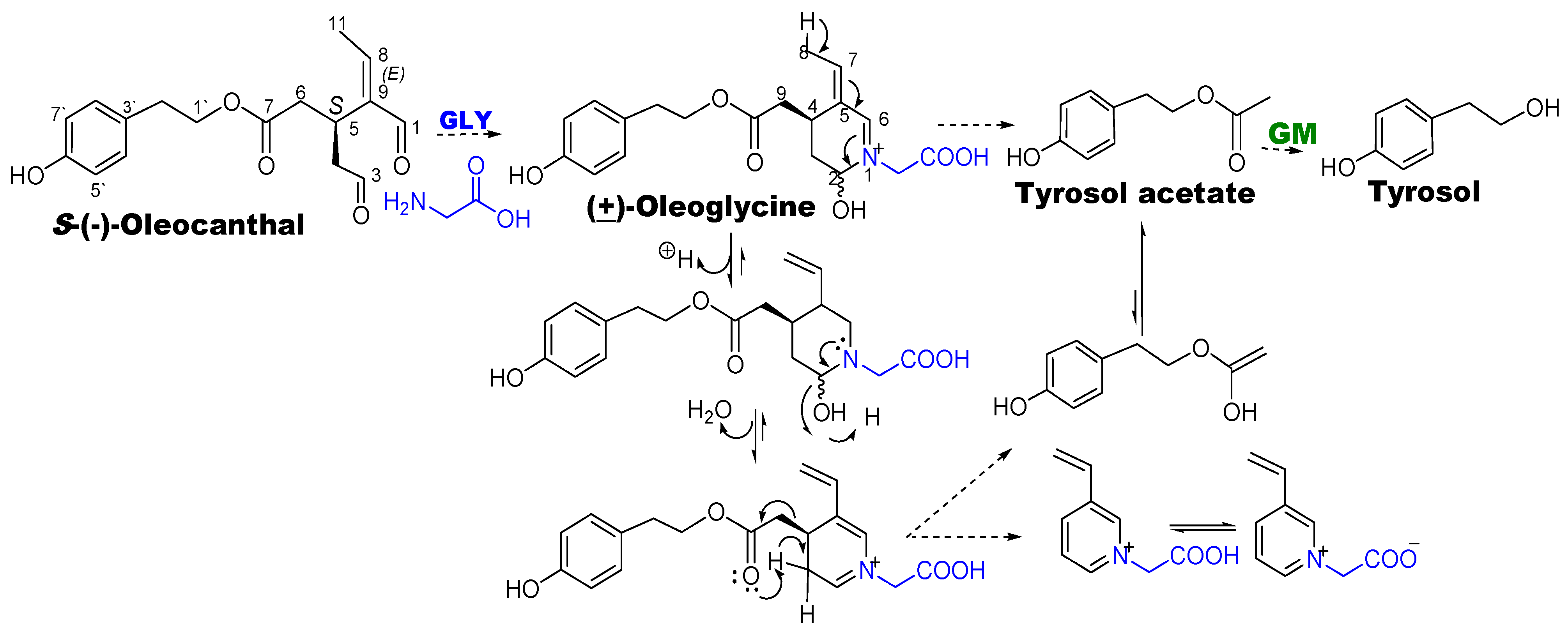
- Darakjian, L.I.; Rigakou, A.; Brannen, A.; Qusa, M.H.; Tasiakou, N.; Diamantakos, P.; Reed, M.N.; Panizzi, P.; Boersma, M.D.; Melliou, E.; et al. Spontaneous in vitro and in vivo interaction of (−)-oleocanthal with glycine in biological fluids: Novel pharmacokinetic markers. ACS Pharmacol. Transl. Sci. 2021, 4, 179–192. [Google Scholar] [CrossRef]
- Farràs, M.; Martinez-Gili, L.; Portune, K.; Arranz, S.; Frost, G.; Tondo, M.; Blanco-Vaca, F. Modulation of the gut microbiota by olive oil phenolic compounds: Implications for lipid metabolism, immune system, and obesity. Nutrients 2020, 12, 2200. [Google Scholar] [CrossRef]
- Millman, J.F.; Okamoto, S.; Teruya, T.; Uema, T.; Ikematsu, S.; Shimabukuro, M.; Masuzaki, H. Extra-virgin olive oil and the gut-brain axis: Influence on gut microbiota, mucosal immunity, and cardiometabolic and cognitive health. Nutr. Rev. 2021, 79, 1362–1374. [Google Scholar] [CrossRef]
- Iglesias-Aguirre, C.E.; Cortés-Martín, A.; Ávila-Gálvez, M.Á.; Giménez-Bastida, J.A.; Selma, M.V.; González-Sarrías, A.; Espín, J.C. Main drivers of (poly)phenol effects on human health: Metabolite production and/or gut microbiota-associated metabotypes? Food Funct. 2021, 12, 10324–10355. [Google Scholar] [CrossRef] [PubMed]
- López-Salazar, V.; Tapia, M.S.; Tobón-Cornejo, S.; Díaz, D.; Alemán-Escondrillas, G.; Granados-Portillo, O.; Noriega, L.; Tovar, A.R.; Torres, N. Consumption of soybean or olive oil at recommended concentrations increased the intestinal microbiota diversity and insulin sensitivity and prevented fatty liver compared to the effects of coconut oil. J. Nutr. Biochem. 2021, 94, 108751. [Google Scholar] [CrossRef] [PubMed]
- Rocchetti, G.; Luisa Callegari, M.; Senizza, A.; Giuberti, G.; Ruzzolini, J.; Romani, A.; Urciuoli, S.; Nediani, C.; Lucini, L. Oleuropein from olive leaf extracts and extra-virgin olive oil provides distinctive phenolic profiles and modulation of microbiota in the large intestine. Food Chem. 2022, 380, 132187. [Google Scholar] [CrossRef] [PubMed]
- Borzì, A.M.; Biondi, A.; Basile, F.; Luca, S.; Vicari, E.; Vacante, M. Olive oil effects on colorectal cancer. Nutrients 2018, 11, 32. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Siddique, A.B.; King, J.A.; Meyer, S.A.; Abdelwahed, K.; Busnena, B.; El Sayed, K. Safety evaluations of single dose of the olive secoiridoid S-(−)-oleocanthal in Swiss albino mice. Nutrients 2020, 12, 314. [Google Scholar] [CrossRef] [Green Version]
- Lu, K.; Mahbub, R.; Fox, J.G. Xenobiotics: Interaction with the intestinal microflora. ILAR J. 2015, 56, 218–227. [Google Scholar] [CrossRef]
- Corrêa, T.; Rogero, M.M.; Hassimotto, N.; Lajolo, F.M. The two-way polyphenols-microbiota interactions and their effects on obesity and related metabolic diseases. Front. Nutr. 2019, 6, 188. [Google Scholar] [CrossRef] [Green Version]
- Van de Steeg, E.; Schuren, F.; Obach, R.S.; van Woudenbergh, C.; Walker, G.S.; Heerikhuisen, M.; Nooijen, I.; Vaes, W. An ex vivo fermentation screening platform to study drug metabolism by human gut microbiota. Drug Metab. Dispos. Biol. Fate Chem. 2018, 46, 1596–1607. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Piwowarski, J.P.; Granica, S.; Stefańska, J.; Kiss, A.K. Differences in metabolism of ellagitannins by human gut microbiota ex vivo cultures. J. Nat. Prod. 2016, 79, 3022–3030. [Google Scholar] [CrossRef]
- Vollmer, M.; Esders, S.; Farquharson, F.M.; Neugart, S.; Duncan, S.H.; Schreiner, M.; Louis, P.; Maul, R.; Rohn, S. Mutual interaction of phenolic compounds and microbiota: Metabolism of complex phenolic apigenin-C- and kaempferol-O-derivatives by human fecal samples. J. Agric. Food Chem. 2018, 66, 485–497. [Google Scholar] [CrossRef] [Green Version]
- Weersma, R.K.; Zhernakova, A.; Fu, J. Interaction between drugs and the gut microbiome. Gut 2020, 69, 1510–1519. [Google Scholar] [CrossRef]
- Zhang, J.; Zhang, J.; Wang, R. Gut microbiota modulates drug pharmacokinetics. Drug Metab. Rev. 2018, 50, 357–368. [Google Scholar] [CrossRef]
- Rowland, I.; Gibson, G.; Heinken, A.; Scott, K.; Swann, J.; Thiele, I.; Tuohy, K. Gut microbiota functions: Metabolism of nutrients and other food components. Eur. J. Nutr. 2018, 57, 1–24. [Google Scholar] [CrossRef] [Green Version]
- Nissen, L.; Casciano, F.; Gianotti, A. Intestinal fermentation in vitro models to study food-induced gut microbiota shift: An updated review. FEMS Microbiol. Lett. 2020, 367, fnaa097. [Google Scholar] [CrossRef]
- Siddique, A.B.; Ebrahim, H.; Mohyeldin, M.; Qusa, M.; Batarseh, Y.; Fayyad, A.; Tajmim, A.; Nazzal, S.; Kaddoumi, A.; El Sayed, K. Novel liquid-liquid extraction and self-emulsion methods for simplified isolation of extra-virgin olive oil phenolics with emphasis on (−)-oleocanthal and its oral anti-breast cancer activity. PLoS ONE 2019, 14, e0214798. [Google Scholar] [CrossRef] [Green Version]
- Pearce, S.C.; Coia, H.G.; Karl, J.P.; Pantoja-Feliciano, I.G.; Zachos, N.C.; Racicot, K. Intestinal in vitro and ex vivo models to study host-microbiome interactions and acute stressors. Front. Physiol. 2018, 9, 1584. [Google Scholar] [CrossRef] [Green Version]
- Tang, L. In vitro intestine model for gut microbiome. Nat. Methods 2019, 16, 578. [Google Scholar] [CrossRef] [PubMed]
- Qusa, M.H.; Siddique, A.B.; Nazzal, S.; El Sayed, K.A. Novel olive oil phenolic (−)-oleocanthal (+)-xylitol-based solid dispersion formulations with potent oral anti-breast cancer activities. Int. J. Pharm. 2019, 569, 118596. [Google Scholar] [CrossRef] [PubMed]
- Knüpfer, M.; Braun, P.; Baumann, K.; Rehn, A.; Antwerpen, M.; Grass, G.; Wölfel, A.R. Evaluation of a highly efficient DNA extraction method for Bacillus anthracis endospores. Microorganisms 2020, 8, 763. [Google Scholar] [CrossRef] [PubMed]
- Clarke, E.L.; Taylor, L.J.; Zhao, C.; Connell, A.; Lee, J.J.; Fett, B.; Bushman, F.D.; Bittinger, K. Sunbeam: An extensible pipeline for analyzing metagenomic sequencing experiments. Microbiome 2019, 7, 46. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Silva, G.G.; Green, K.T.; Dutilh, B.E.; Edwards, R.A. SUPER-FOCUS: A tool for agile functional analysis of shotgun metagenomic data. Bioinformatics 2016, 32, 354–361. [Google Scholar] [CrossRef] [Green Version]
- Kolodziejczyk, A.A.; Zheng, D.; Elinav, E. Diet-microbiota interactions and personalized nutrition. Nat. Rev. Microbiol. 2019, 17, 742–753. [Google Scholar] [CrossRef]
- Leeming, E.R.; Louca, P.; Gibson, R.; Menni, C.; Spector, T.D.; Le Roy, C.I. The complexities of the diet-microbiome relationship: Advances and perspectives. Genome Med. 2021, 13, 10. [Google Scholar] [CrossRef]
- Debédat, J.; Le Roy, T.; Voland, L.; Belda, E.; Alili, R.; Adriouch, S.; Bel Lassen, P.; Kasahara, K.; Hutchison, E.; Genser, L.; et al. The human gut microbiota contributes to type-2 diabetes non-resolution 5-years after Roux-en-Y gastric bypass. Gut Microbes 2022, 14, 2050635. [Google Scholar] [CrossRef]
- Pan, Y.; Zhang, X. Diet and gut microbiome in fatty liver and its associated liver cancer. J. Gastroenterol. Hepatol. 2022, 37, 7–14. [Google Scholar] [CrossRef] [PubMed]
- Kim, C.C.; Kelly, W.J.; Patchett, M.L.; Tannock, G.W.; Jordens, Z.; Stoklosinski, H.M.; Taylor, J.W.; Sims, I.M.; Bell, T.J.; Rosendale, D.I. Monoglobus pectinilyticus gen. nov., sp. nov., a pectinolytic bacterium isolated from human feces. Int. J. Syst. Evol. Microbiol. 2017, 67, 4992–4998. [Google Scholar] [CrossRef] [PubMed]
- Kim, C.C.; Healey, G.R.; Kelly, W.J.; Patchett, M.L.; Jordens, Z.; Tannock, G.W.; Sims, I.M.; Bell, T.J.; Hedderley, D.; Henrissat, B.; et al. Genomic insights from Monoglobus pectinilyticus: A pectin-degrading specialist bacterium in the human colon. ISME J. 2019, 13, 1437–1456. [Google Scholar] [CrossRef] [PubMed]
- Precup, G.; Vodnar, D.C. Gut Prevotella as a possible biomarker of diet and its eubiotic versus dysbiotic roles: A comprehensive literature review. Br. J. Nutr. 2019, 122, 131–140. [Google Scholar] [CrossRef]
- Vanden Eede, H.; Norris, E.; Torfs, M.; Vanderveken, O. Life threatening abscess in the visceral space with penicillin and metronidazole resistant Prevotella Denticola following use of a laryngeal mask airway: Case report. BMC Anesthesiol. 2021, 21, 102. [Google Scholar] [CrossRef]
- Coyne, M.J.; Zitomersky, N.L.; McGuire, A.M.; Earl, A.M.; Comstock, L.E. Evidence of extensive DNA transfer between bacteroidales species within the human gut. mBio 2014, 5, e01305-14. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wesslund, N.A.; Wang, G.R.; Song, B.; Shoemaker, N.B.; Salyers, A.A. Integration and excision of a newly discovered bacteroides conjugative transposon, CTnBST. J. Bacteriol. 2007, 189, 1072–1082. [Google Scholar] [CrossRef] [Green Version]
- García-Bayona, L.; Coyne, M.J.; Comstock, L.E. Mobile Type VI secretion system loci of the gut Bacteroidales display extensive intra-ecosystem transfer, multi-species spread and geographical clustering. PLoS Genet. 2021, 17, e1009541. [Google Scholar] [CrossRef]
- Wolf, P.G.; Biswas, A.; Morales, S.E.; Greening, C.; Gaskins, H.R. H2 metabolism is widespread and diverse among human colonic microbes. Gut Microbes 2016, 7, 235–245. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Smith, N.W.; Shorten, P.R.; Altermann, E.H.; Roy, N.C.; McNabb, W.C. Hydrogen cross-feeders of the human gastrointestinal tract. Gut Microbes 2019, 10, 270–288. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Greening, C.; Biswas, A.; Carere, C.R.; Jackson, C.J.; Taylor, M.C.; Stott, M.B.; Cook, G.M.; Morales, S.E. Genomic and metagenomic surveys of hydrogenase distribution indicate H2 is a widely utilized energy source for microbial growth and survival. ISME J. 2016, 10, 761–777. [Google Scholar] [CrossRef] [Green Version]
- Rajakovich, L.J.; Balskus, E.P. Metabolic functions of the human gut microbiota: The role of metalloenzymes. Nat. Prod. Rep. 2019, 36, 593–625. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nagpal, R.; Wang, S.; Solberg Woods, L.C.; Seshie, O.; Chung, S.T.; Shively, C.A.; Register, T.C.; Craft, S.; McClain, D.A.; Yadav, H. Comparative microbiome signatures and short-chain fatty acids in mouse, rat, non-human primate, and human feces. Front. Microbiol. 2018, 9, 2897. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Daniel, N.; Lécuyer, E.; Chassaing, B. Host/microbiota interactions in health and diseases-Time for mucosal microbiology! Mucosal Immunol. 2021, 14, 1006–1016. [Google Scholar] [CrossRef]
- Zheng, D.; Liwinski, T.; Elinav, E. Interaction between microbiota and immunity in health and disease. Cell Res. 2020, 30, 492–506. [Google Scholar] [CrossRef]
- Ogunrinola, G.A.; Oyewale, J.O.; Oshamika, O.O.; Olasehinde, G.I. The human microbiome and its impacts on health. Int. J. Microbiol. 2020, 2020, 8045646. [Google Scholar] [CrossRef]



Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Qusa, M.H.; Abdelwahed, K.S.; Hill, R.A.; El Sayed, K.A. S-(−)-Oleocanthal Ex Vivo Modulatory Effects on Gut Microbiota. Nutrients 2023, 15, 618. https://doi.org/10.3390/nu15030618
Qusa MH, Abdelwahed KS, Hill RA, El Sayed KA. S-(−)-Oleocanthal Ex Vivo Modulatory Effects on Gut Microbiota. Nutrients. 2023; 15(3):618. https://doi.org/10.3390/nu15030618
Chicago/Turabian StyleQusa, Mohammed H., Khaldoun S. Abdelwahed, Ronald A. Hill, and Khalid A. El Sayed. 2023. "S-(−)-Oleocanthal Ex Vivo Modulatory Effects on Gut Microbiota" Nutrients 15, no. 3: 618. https://doi.org/10.3390/nu15030618
APA StyleQusa, M. H., Abdelwahed, K. S., Hill, R. A., & El Sayed, K. A. (2023). S-(−)-Oleocanthal Ex Vivo Modulatory Effects on Gut Microbiota. Nutrients, 15(3), 618. https://doi.org/10.3390/nu15030618