
Non-Nutritive Sweeteners Acesulfame Potassium and Sucralose Are Competitive Inhibitors of the Human P-glycoprotein/Multidrug Resistance Protein 1 (PGP/MDR1)

Abstract
1. Introduction
2. Materials and Methods
2.1. Cell Culture
2.2. NNS and Other Reagents
2.3. RNA Extraction, cDNA Synthesis, and RT-qPCR
2.4. Protein Extraction, SDS Page, and Western Blot
2.5. Calcein-AM Retention Assay
2.6. PGP ATPase Activation Assay
2.7. Molecular Docking
2.8. Statistical Analysis
3. Results
3.1. Acesulfame Potassium and Sucralose Impact the Expression of Detoxification Actors in Human Liver Cell Line
3.2. Acesulfame Potassium and Sucralose Inhibit Efflux of PGP Substrates
3.3. Sucr and AceK Stimulate PGP Efflux Activity
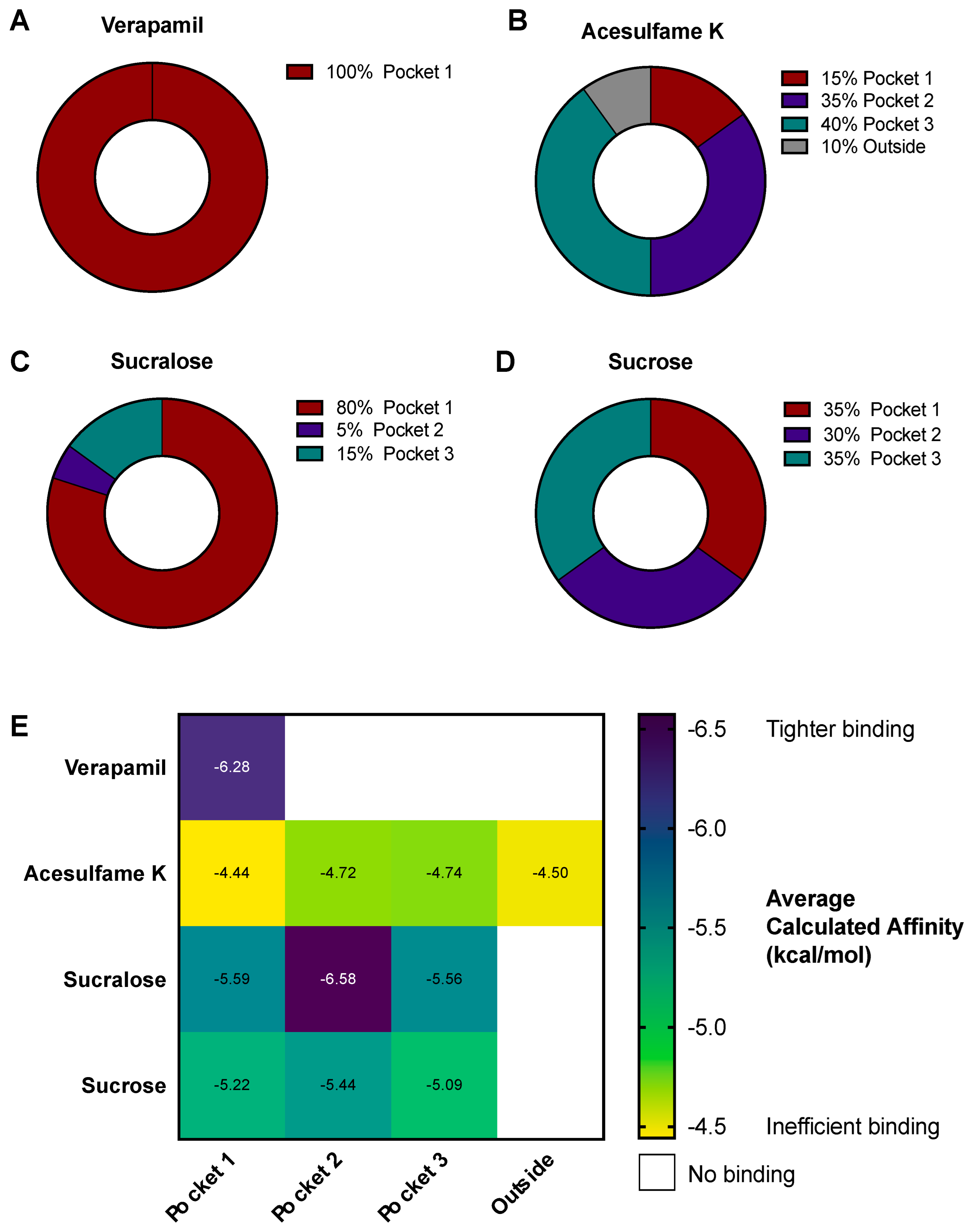
3.4. AceK and Sucr Show Unique PGP Binding Patterns In Silico
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Magnuson, B.A.; Carakostas, M.C.; Moore, N.H.; Poulos, S.P.; Renwick, A.G. Biological Fate of Low-Calorie Sweeteners. Nutr. Rev. 2016, 74, 670–689. [Google Scholar] [CrossRef] [PubMed]
- FDA. Additional Information about High-Intensity Sweeteners Permitted for Use in Food in the United States; FDA: Silver Spring, MD, USA, 2020. [Google Scholar]
- Sylvetsky, A.C.; Jin, Y.; Clark, E.J.; Welsh, J.A.; Rother, K.I.; Talegawkar, S.A. Consumption of Low-Calorie Sweeteners among Children and Adults in the United States. J. Acad. Nutr. Diet. 2017, 117, 441–448.e2. [Google Scholar] [CrossRef] [PubMed]
- Dunford, E.K.; Miles, D.R.; Ng, S.W.; Popkin, B. Types and Amounts of Nonnutritive Sweeteners Purchased by US Households: A Comparison of 2002 and 2018 Nielsen Homescan Purchases. J. Acad. Nutr. Diet. 2020, 120, 1662–1671.e10. [Google Scholar] [CrossRef] [PubMed]
- Harwood, J.J. Molecular Markers for Identifying Municipal, Domestic and Agricultural Sources of Organic Matter in Natural Waters. Chemosphere 2014, 95, 3–8. [Google Scholar] [CrossRef]
- Stichelen, S.O.-V.; Rother, K.I.; Hanover, J.A. Maternal Exposure to Non-Nutritive Sweeteners Impacts Progeny’s Metabolism and Microbiome. Front. Microbiol. 2019, 10, 1360. [Google Scholar] [CrossRef]
- Roberts, A.; Renwick, A.G.; Sims, J.; Snodin, D.J. Sucralose Metabolism and Pharmacokinetics in Man. Food Chem. Toxicol. 2000, 38, 31–41. [Google Scholar] [CrossRef]
- Gardner, C.; Wylie-Rosett, J.; Gidding, S.S.; Steffen, L.M.; Johnson, R.K.; Reader, D.; Lichtenstein, A.H. Nonnutritive Sweeteners: Current Use and Health Perspectives: A Scientific Statement from the American Heart Association and the American Diabetes Association. Diabetes Care 2012, 35, 1798–1808. [Google Scholar] [CrossRef]
- Swithers, S.E.; Laboy, A.F.; Clark, K.; Cooper, S.; Davidson, T.L. Experience with the High-Intensity Sweetener Saccharin Impairs Glucose Homeostasis and GLP-1 Release in Rats. Behav. Brain Res. 2012, 233, 1–14. [Google Scholar] [CrossRef]
- Schiffman, S.S.; Rother, K.I. Sucralose, A Synthetic Organochlorine Sweetener: Overview of Biological Issues. J. Toxicol. Environ. Health Part B 2013, 16, 399–451. [Google Scholar] [CrossRef]
- Rother, K.I.; Conway, E.M.; Sylvetsky, A.C. How Non-Nutritive Sweeteners Influence Hormones and Health. Trends Endocrinol. Metab. 2018, 29, 455–467. [Google Scholar] [CrossRef]
- Green, C.H.; Syn, W.-K. Non-Nutritive Sweeteners and Their Association with the Metabolic Syndrome and Non-Alcoholic Fatty Liver Disease: A Review of the Literature. Eur. J. Nutr. 2019, 58, 1785–1800. [Google Scholar] [CrossRef]
- Brown, R.J.; Rother, K.I. Non-Nutritive Sweeteners and Their Role in the Gastrointestinal Tract. J. Clin. Endocrinol. Metab. 2012, 97, 2597–2605. [Google Scholar] [CrossRef]
- Bian, X.; Chi, L.; Gao, B.; Tu, P.; Ru, H.; Lu, K. Gut Microbiome Response to Sucralose and Its Potential Role in Inducing Liver Inflammation in Mice. Front. Physiol. 2017, 8, 487. [Google Scholar] [CrossRef]
- Kroll, T.; Prescher, M.; Smits, S.H.J.; Schmitt, L. Structure and Function of Hepatobiliary ATP Binding Cassette Transporters. Chem. Rev. 2021, 121, 5240–5288. [Google Scholar] [CrossRef]
- Abou-Donia, M.B.; El-Masry, E.M.; Abdel-Rahman, A.A.; McLendon, R.E.; Schiffman, S.S. Splenda Alters Gut Microflora and Increases Intestinal P-Glycoprotein and Cytochrome P-450 in Male Rats. J. Toxicol. Environ. Health A 2008, 71, 1415–1429. [Google Scholar] [CrossRef]
- Fromm, M.F. Importance of P-Glycoprotein at Blood–Tissue Barriers. Trends Pharmacol. Sci. 2004, 25, 423–429. [Google Scholar] [CrossRef]
- Schinkel, A.H. The Physiological Function of Drug-Transporting P-Glycoproteins. Semin. Cancer Biol. 1997, 8, 161–170. [Google Scholar] [CrossRef]
- Katayama, K.; Kapoor, K.; Ohnuma, S.; Patel, A.; Swaim, W.; Ambudkar, I.S.; Ambudkar, S.V. Revealing the Fate of Cell Surface Human P-Glycoprotein (ABCB1): The Lysosomal Degradation Pathway. Biochim. Biophys. Acta BBA Mol. Cell Res. 2015, 1853, 2361–2370. [Google Scholar] [CrossRef] [PubMed]
- Sylvetsky, A.C.; Bauman, V.; Blau, J.E.; Garraffo, H.M.; Walter, P.J.; Rother, K.I. Plasma Concentrations of Sucralose in Children and Adults. Toxicol. Environ. Chem. 2016, 99, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Rother, K.I.; Sylvetsky, A.C.; Walter, P.J.; Garraffo, H.M.; Fields, D.A. Pharmacokinetics of Sucralose and Acesulfame-Potassium in Breast Milk Following Ingestion of Diet Soda. J. Pediatr. Gastroenterol. Nutr. 2018, 66, 466–470. [Google Scholar] [CrossRef] [PubMed]
- Stampe, S.; Leth-Møller, M.; Greibe, E.; Hoffmann-Lücke, E.; Pedersen, M.; Ovesen, P. Artificial Sweeteners in Breast Milk: A Clinical Investigation with a Kinetic Perspective. Nutrients 2022, 14, 2635. [Google Scholar] [CrossRef] [PubMed]
- Livak, K.J.; Schmittgen, T.D. Analysis of Relative Gene Expression Data Using Real-Time Quantitative PCR and the 2−ΔΔCT Method. Methods 2001, 25, 402–408. [Google Scholar] [CrossRef] [PubMed]
- Nosol, K.; Romane, K.; Irobalieva, R.N.; Alam, A.; Kowal, J.; Fujita, N.; Locher, K.P. Cryo-EM Structures Reveal Distinct Mechanisms of Inhibition of the Human Multidrug Transporter ABCB1. Proc. Natl. Acad. Sci. USA 2020, 117, 26245–26253. [Google Scholar] [CrossRef] [PubMed]
- Ravindranath, P.A.; Forli, S.; Goodsell, D.S.; Olson, A.J.; Sanner, M.F. AutoDockFR: Advances in Protein-Ligand Docking with Explicitly Specified Binding Site Flexibility. PLoS Comput. Biol. 2015, 11, e1004586. [Google Scholar] [CrossRef]
- Eberhardt, J.; Santos-Martins, D.; Tillack, A.F.; Forli, S. AutoDock Vina 1.2.0: New Docking Methods, Expanded Force Field, and Python Bindings. J. Chem. Inf. Model. 2021, 61, 3891–3898. [Google Scholar] [CrossRef]
- Irwin, J.J.; Shoichet, B.K. ZINC—A Free Database of Commercially Available Compounds for Virtual Screening. J. Chem. Inf. Model. 2005, 45, 177–182. [Google Scholar] [CrossRef]
- Trott, O.; Olson, A.J. AutoDock Vina: Improving the Speed and Accuracy of Docking with a New Scoring Function, Efficient Optimization and Multithreading. J. Comput. Chem. 2010, 31, 455–461. [Google Scholar] [CrossRef]
- Lomize, M.A.; Pogozheva, I.D.; Joo, H.; Mosberg, H.I.; Lomize, A.L. OPM Database and PPM Web Server: Resources for Positioning of Proteins in Membranes. Nucleic Acids Res. 2012, 40, D370–D376. [Google Scholar] [CrossRef]
- Mohana, S.; Ganesan, M.; Agilan, B.; Karthikeyan, R.; Srithar, G.; Beaulah Mary, R.; Ananthakrishnan, D.; Velmurugan, D.; Rajendra Prasad, N.; Ambudkar, S.V. Screening Dietary Flavonoids for the Reversal of P-Glycoprotein-Mediated Multidrug Resistance in Cancer. Mol. Biosyst. 2016, 12, 2458–2470. [Google Scholar] [CrossRef]
- Liao, D.; Zhang, W.; Gupta, P.; Lei, Z.-N.; Wang, J.-Q.; Cai, C.-Y.; Vera, A.A.D.; Zhang, L.; Chen, Z.-S.; Yang, D.-H. Tetrandrine Interaction with ABCB1 Reverses Multidrug Resistance in Cancer Cells Through Competition with Anti-Cancer Drugs Followed by Downregulation of ABCB1 Expression. Molecules 2019, 24, 4383. [Google Scholar] [CrossRef]
- Xing, J.; Huang, S.; Heng, Y.; Mei, H.; Pan, X. Computational Insights into Allosteric Conformational Modulation of P-Glycoprotein by Substrate and Inhibitor Binding. Molecules 2020, 25, 6006. [Google Scholar] [CrossRef] [PubMed]
- Schrödinger LLC. The PyMOL Molecular Graphics System, Version 2.0; Schrödinger LLC: New York, NY, USA, 2010. [Google Scholar]
- Wang, X.; Deng, R.; Lu, Y.; Xu, Q.; Yan, M.; Ye, D.; Chen, W. Gambogic Acid as a Non-Competitive Inhibitor of ATP-Binding Cassette Transporter B1 Reverses the Multidrug Resistance of Human Epithelial Cancers by Promoting ATP-Binding Cassette Transporter B1 Protein Degradation. Basic Clin. Pharmacol. Toxicol. 2013, 112, 25–33. [Google Scholar] [CrossRef]
- Dewanjee, S.; Dua, T.; Bhattacharjee, N.; Das, A.; Gangopadhyay, M.; Khanra, R.; Joardar, S.; Riaz, M.; Feo, V.; Zia-Ul-Haq, M. Natural Products as Alternative Choices for P-Glycoprotein (P-Gp) Inhibition. Molecules 2017, 22, 871. [Google Scholar] [CrossRef] [PubMed]
- Yasui-Furukori, N.; Uno, T.; Sugawara, K.; Tateishi, T. Different Effects of Three Transporting Inhibitors, Verapamil, Cimetidine, and Probenecid, on Fexofenadine Pharmacokinetics. Clin. Pharmacol. Ther. 2005, 77, 17–23. [Google Scholar] [CrossRef] [PubMed]
- Calabrese, E.J. P-Glycoprotein Efflux Transporter Activity Often Displays Biphasic Dose-Response Relationships. Crit. Rev. Toxicol. 2008, 38, 473–487. [Google Scholar] [CrossRef] [PubMed]
- Evseenko, D.A.; Paxton, J.W.; Keelan, J.A. ABC Drug Transporter Expression and Functional Activity in Trophoblast-like Cell Lines and Differentiating Primary Trophoblast. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2006, 290, R1357–R1365. [Google Scholar] [CrossRef]
- Roy, U.; Chakravarty, G.; Honer Zu Bentrup, K.; Mondal, D. Montelukast Is a Potent and Durable Inhibitor of Multidrug Resistance Protein 2-Mediated Efflux of Taxol and Saquinavir. Biol. Pharm. Bull. 2009, 32, 2002–2009. [Google Scholar] [CrossRef]
- Masereeuw, R.; Notenboom, S.; Smeets, P.H.E.; Wouterse, A.C.; Russel, F.G.M. Impaired Renal Secretion of Substrates for the Multidrug Resistance Protein 2 in Mutant Transport–Deficient (TR−) Rats. J. Am. Soc. Nephrol. 2003, 14, 2741–2749. [Google Scholar] [CrossRef]
- Uhlén, M.; Fagerberg, L.; Hallström, B.M.; Lindskog, C.; Oksvold, P.; Mardinoglu, A.; Sivertsson, Å.; Kampf, C.; Sjöstedt, E.; Asplund, A.; et al. Tissue-Based Map of the Human Proteome. Science 2015, 347, 1260419. [Google Scholar] [CrossRef]
- Kim, Y.; Chen, J. Molecular Structure of Human P-Glycoprotein in the ATP-Bound, Outward-Facing Conformation. Science 2018, 359, 915–919. [Google Scholar] [CrossRef]
- Urbatsch, I.L.; Al-Shawi, M.K.; Senior, A.E. Characterization of the ATPase Activity of Purified Chinese Hamster P-Glycoprotein. Biochemistry 1994, 33, 7069–7076. [Google Scholar] [CrossRef] [PubMed]
- Shapiro, A.B.; Ling, V. ATPase Activity of Purified and Reconstituted P-Glycoprotein from Chinese Hamster Ovary Cells. J. Biol. Chem. 1994, 269, 3745–3754. [Google Scholar] [CrossRef] [PubMed]
- Aller, S.G.; Yu, J.; Ward, A.; Weng, Y.; Chittaboina, S.; Zhuo, R.; Harrell, P.M.; Trinh, Y.T.; Zhang, Q.; Urbatsch, I.L.; et al. Structure of P-Glycoprotein Reveals a Molecular Basis for Poly-Specific Drug Binding. Science 2009, 323, 1718–1722. [Google Scholar] [CrossRef] [PubMed]
- Juliano, R.L.; Ling, V. A Surface Glycoprotein Modulating Drug Permeability in Chinese Hamster Ovary Cell Mutants. Biochim. Biophys. Acta BBA Biomembr. 1976, 455, 152–162. [Google Scholar] [CrossRef] [PubMed]
- Yang, C.P.H.; DePinho, S.G.; Greenberger, L.M.; Arceci, R.J.; Horwitz, S.B. Progesterone Interacts with P-Glycoprotein in Multidrug-Resistant Cells and in the Endometrium of Gravid Uterus. J. Biol. Chem. 1989, 264, 782–788. [Google Scholar] [CrossRef] [PubMed]
- van Helvoort, A.; Smith, A.J.; Sprong, H.; Fritzsche, I.; Schinkel, A.H.; Borst, P.; van Meer, G. MDR1 P-Glycoprotein Is a Lipid Translocase of Broad Specificity, While MDR3 P-Glycoprotein Specifically Translocates Phosphatidylcholine. Cell 1996, 87, 507–517. [Google Scholar] [CrossRef] [PubMed]
- Watchko, J.F.; Daood, M.J.; Mahmood, B.; Vats, K.; Hart, C.; Ahdab-Barmada, M. P-Glycoprotein and Bilirubin Disposition. J. Perinatol. 2001, 21, S43–S47. [Google Scholar] [CrossRef]
- Iqbal, M.; Ho, H.L.; Petropoulos, S.; Moisiadis, V.G.; Gibb, W.; Matthews, S.G. Pro-Inflammatory Cytokine Regulation of P-Glycoprotein in the Developing Blood-Brain Barrier. PLoS ONE 2012, 7, e43022. [Google Scholar] [CrossRef]
- Schinkel, A.H.; Mayer, U.; Wagenaar, E.; Mol, C.A.A.M.; van Deemter, L.; Smit, J.J.M.; van der Valk, M.A.; Voordouw, A.C.; Spits, H.; van Tellingen, O.; et al. Normal Viability and Altered Pharmacokinetics in Mice Lacking Mdr1-Type (Drug-Transporting) P-Glycoproteins. Proc. Natl. Acad. Sci. USA 1997, 94, 4028–4033. [Google Scholar] [CrossRef]
- Liang, C.; Zhao, J.; Lu, J.; Zhang, Y.; Ma, X.; Shang, X.; Li, Y.; Ma, X.; Liu, M.; Wang, X. Development and Characterization of MDR1 (Mdr1a/b) CRISPR/Cas9 Knockout Rat Model. Drug Metab. Dispos. 2019, 47, 71–79. [Google Scholar] [CrossRef]
- Ambudkar, S.V.; Kimchi-Sarfaty, C.; Sauna, Z.E.; Gottesman, M.M. P-Glycoprotein: From Genomics to Mechanism. Oncogene 2003, 22, 7468–7485. [Google Scholar] [CrossRef] [PubMed]
- Smit, J.W.; Huisman, M.T.; van Tellingen, O.; Wiltshire, H.R.; Schinkel, A.H. Absence or Pharmacological Blocking of Placental P-Glycoprotein Profoundly Increases Fetal Drug Exposure. J. Clin. Investig. 1999, 104, 1441–1447. [Google Scholar] [CrossRef] [PubMed]
- Lankas, G.R.; Wise, L.D.; Cartwright, M.E.; Pippert, T.; Umbenhauer, D.R. Placental P-Glycoprotein Deficiency Enhances Susceptibility to Chemically Induced Birth Defects in Mice. Reprod. Toxicol. 1998, 12, 457–463. [Google Scholar] [CrossRef] [PubMed]
- Engström, K.; Love, T.M.; Watson, G.E.; Zareba, G.; Yeates, A.; Wahlberg, K.; Alhamdow, A.; Thurston, S.W.; Mulhern, M.; McSorley, E.M.; et al. Polymorphisms in ATP-Binding Cassette Transporters Associated with Maternal Methylmercury Disposition and Infant Neurodevelopment in Mother-Infant Pairs in the Seychelles Child Development Study. Environ. Int. 2016, 94, 224–229. [Google Scholar] [CrossRef] [PubMed]
- Olagunju, A.; Owen, A.; Cressey, T.R. Potential Effect of Pharmacogenetics on Maternal, Fetal and Infant Antiretroviral Drug Exposure during Pregnancy and Breastfeeding. Pharmacogenomics 2012, 13, 1501–1522. [Google Scholar] [CrossRef] [PubMed]
- Daud, A.N.; Bergman, J.E.; Bakker, M.K.; Wang, H.; de Walle, H.E.; Plösch, T.; Wilffert, B. Pharmacogenetics of Drug-Induced Birth Defects: The Role of Polymorphisms of Placental Transporter Proteins. Pharmacogenomics 2014, 15, 1029–1041. [Google Scholar] [CrossRef]
- Llop, S.; Engström, K.; Ballester, F.; Franforte, E.; Alhamdow, A.; Pisa, F.; Tratnik, J.S.; Mazej, D.; Murcia, M.; Rebagliato, M.; et al. Polymorphisms in ABC Transporter Genes and Concentrations of Mercury in Newborns—Evidence from Two Mediterranean Birth Cohorts. PLoS ONE 2014, 9, e97172. [Google Scholar] [CrossRef]
- Ellfolk, M.; Tornio, A.; Niemi, M.; Leinonen, M.K.; Lahesmaa-Korpinen, A.; Malm, H. Placental Transporter-mediated Drug Interactions and Offspring Congenital Anomalies. Br. J. Clin. Pharmacol. 2020, 86, 868–879. [Google Scholar] [CrossRef]
- Glaeser, H. Importance of P-Glycoprotein for Drug–Drug Interactions. In Drug Transporters; Fromm, M.F., Kim, R.B., Eds.; Handbook of Experimental Pharmacology; Springer: Berlin/Heidelberg, Germany, 2011; Volume 201, pp. 285–297. ISBN 978-3-642-14540-7. [Google Scholar]
- Kapoor, A.; Iqbal, M.; Petropoulos, S.; Ho, H.L.; Gibb, W.; Matthews, S.G. Effects of Sertraline and Fluoxetine on P-Glycoprotein at Barrier Sites: In Vivo and In Vitro Approaches. PLoS ONE 2013, 8, e56525. [Google Scholar] [CrossRef]
- O, B.Y.S.; Coyle, D.H.; Dunford, E.K.; Wu, J.H.Y.; Louie, J.C.Y. The Use of Non-Nutritive and Low-Calorie Sweeteners in 19,915 Local and Imported Pre-Packaged Foods in Hong Kong. Nutrients 2021, 13, 1861. [Google Scholar] [CrossRef]
- Nunn, R.; Young, L.; Ni Mhurchu, C. Prevalence and Types of Non-Nutritive Sweeteners in the New Zealand Food Supply, 2013 and 2019. Nutrients 2021, 13, 3228. [Google Scholar] [CrossRef] [PubMed]
- Sylvetsky, A.C.; Greenberg, M.; Zhao, X.; Rother, K.I. What Parents Think about Giving Nonnutritive Sweeteners to Their Children: A Pilot Study. Int. J. Pediatr. 2014, 2014, 819872. [Google Scholar] [CrossRef] [PubMed]
- Products—Data Briefs—Number 377—September 2020. Available online: https://www.cdc.gov/nchs/products/databriefs/db377.htm (accessed on 16 January 2023).
- Sharma, M.; Nazareth, I.; Petersen, I. Trends in Incidence, Prevalence and Prescribing in Type 2 Diabetes Mellitus between 2000 and 2013 in Primary Care: A Retrospective Cohort Study. BMJ Open 2016, 6, e010210. [Google Scholar] [CrossRef] [PubMed]
- Soric, M.M.; Moorman, J.M.; Boyle, J.A.; Dengler-Crish, C.M. Prevalence and Predictors of Metformin Prescribing in Adults with Type 2 Diabetes Mellitus: A National Cross-Sectional Study. Pharmacother. J. Hum. Pharmacol. Drug Ther. 2016, 36, 715–722. [Google Scholar] [CrossRef]
- US Department of Health and Human Services. Antibiotic Use in the United States, 2018: Progress and Opportunities; US Department of Health and Human Services: Atlanta, GA, USA, 2019.
- Debras, C.; Chazelas, E.; Srour, B.; Druesne-Pecollo, N.; Esseddik, Y.; de Edelenyi, F.S.; Agaësse, C.; Sa, A.D.; Lutchia, R.; Gigandet, S.; et al. Artificial Sweeteners and Cancer Risk: Results from the NutriNet-Santé Population-Based Cohort Study. PLoS Med. 2022, 19, e1003950. [Google Scholar] [CrossRef]
- Honda, Y.; Ushigome, F.; Koyabu, N.; Morimoto, S.; Shoyama, Y.; Uchiumi, T.; Kuwano, M.; Ohtani, H.; Sawada, Y. Effects of Grapefruit Juice and Orange Juice Components on P-Glycoprotein- and MRP2-Mediated Drug Efflux. Br. J. Pharmacol. 2004, 143, 856–864. [Google Scholar] [CrossRef]
- Konishi, T.; Satsu, H.; Hatsugai, Y.; Aizawa, K.; Inakuma, T.; Nagata, S.; Sakuda, S.; Nagasawa, H.; Shimizu, M. Inhibitory Effect of a Bitter Melon Extract on the P-Glycoprotein Activity in Intestinal Caco-2 Cells. Br. J. Pharmacol. 2004, 143, 379–387. [Google Scholar] [CrossRef]
- Yu, C.P.; Hsieh, Y.W.; Lin, S.P.; Chi, Y.C.; Hariharan, P.; Chao, P.D.L.; Hou, Y.C. Potential Modulation on P-Glycoprotein and CYP3A by Soymilk and Miso: In Vivo and Ex-Vivo Studies. Food Chem. 2014, 149, 25–30. [Google Scholar] [CrossRef]
- Pedersen, K.E.; Dorph-Pedersen, A.; Hvidt, S.; Klitgaard, N.A.; Pedersen, K.K. The Long-Term Effect of Verapamil on Plasma Digoxin Concentration and Renal Digoxin Clearance in Healthy Subjects. Eur. J. Clin. Pharmacol. 1982, 22, 123–127. [Google Scholar] [CrossRef]
- Belz, G.G.; Doering, W.; Munkes, R.; Matthews, J. Interaction between Digoxin and Calcium Antagonists and Antiarrhythmic Drugs. Clin. Pharmacol. Ther. 1983, 33, 410–417. [Google Scholar] [CrossRef]
- Briguglio, M.; Hrelia, S.; Malaguti, M.; Serpe, L.; Canaparo, R.; Dell’Osso, B.; Galentino, R.; De Michele, S.; Zanaboni Dina, C.; Porta, M.; et al. Food Bioactive Compounds and Their Interference in Drug Pharmacokinetic/Pharmacodynamic Profiles. Pharmaceutics 2018, 10, 277. [Google Scholar] [CrossRef] [PubMed]
- Yi, Z.-M.; Zhi, X.-J.; Yang, L.; Sun, S.-S.; Zhang, Z.; Sun, Z.-M.; Zhai, S.-D. Identify Practice Gaps in Medication Education through Surveys to Patients and Physicians. Patient Prefer. Adherence 2015, 9, 1423–1430. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Hussain, S. Patient Counseling about Herbal-Drug Interactions. Afr. J. Tradit. Complement. Altern. Med. 2011, 8, 152–163. [Google Scholar] [CrossRef] [PubMed]
- Ilias, I.; Rizzo, M.; Zabuliene, L. Metformin: Sex/Gender Differences in Its Uses and Effects—Narrative Review. Medicina 2022, 58, 430. [Google Scholar] [CrossRef] [PubMed]
- Ceckova-Novotna, M.; Pavek, P.; Staud, F. P-Glycoprotein in the Placenta: Expression, Localization, Regulation and Function. Reprod. Toxicol. 2006, 22, 400–410. [Google Scholar] [CrossRef] [PubMed]
- Rubinchik-Stern, M.; Eyal, S. Drug Interactions at the Human Placenta: What Is the Evidence? Front. Pharmacol. 2012, 3, 126. [Google Scholar] [CrossRef] [PubMed]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Acesulfame Potassium | Max Concentration ng/mL (nM) |
|---|---|
| Human plasma [22] | 1500 (7500) |
| Human breastmilk [21] | 1000 (5000) |
| Mouse plasma [6] | 250 (1250) |
| Sucralose | Max Concentration ng/mL (nM) |
| Human plasma [20] | 300 (750) |
| Human breastmilk [21] | 1200 (3000) |
| Mouse plasma [6] | 80 (200) |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Danner, L.; Malard, F.; Valdes, R.; Olivier-Van Stichelen, S. Non-Nutritive Sweeteners Acesulfame Potassium and Sucralose Are Competitive Inhibitors of the Human P-glycoprotein/Multidrug Resistance Protein 1 (PGP/MDR1). Nutrients 2023, 15, 1118. https://doi.org/10.3390/nu15051118
Danner L, Malard F, Valdes R, Olivier-Van Stichelen S. Non-Nutritive Sweeteners Acesulfame Potassium and Sucralose Are Competitive Inhibitors of the Human P-glycoprotein/Multidrug Resistance Protein 1 (PGP/MDR1). Nutrients. 2023; 15(5):1118. https://doi.org/10.3390/nu15051118
Chicago/Turabian StyleDanner, Laura, Florian Malard, Raquel Valdes, and Stephanie Olivier-Van Stichelen. 2023. "Non-Nutritive Sweeteners Acesulfame Potassium and Sucralose Are Competitive Inhibitors of the Human P-glycoprotein/Multidrug Resistance Protein 1 (PGP/MDR1)" Nutrients 15, no. 5: 1118. https://doi.org/10.3390/nu15051118
APA StyleDanner, L., Malard, F., Valdes, R., & Olivier-Van Stichelen, S. (2023). Non-Nutritive Sweeteners Acesulfame Potassium and Sucralose Are Competitive Inhibitors of the Human P-glycoprotein/Multidrug Resistance Protein 1 (PGP/MDR1). Nutrients, 15(5), 1118. https://doi.org/10.3390/nu15051118