
Identification of Metabolism-Related Proteins as Biomarkers of Insulin Resistance and Potential Mechanisms of m6A Modification

Abstract
:1. Introduction
2. Materials and Methods
2.1. Design of the Study
2.2. Data Acquisition and Processing
2.3. Selection of MP-DEGs
2.4. Functional and Pathway Enrichment Analyses of the MP-DEGs
2.5. Construction of the Protein–Protein Interaction Network of the MP-DEGs and Gene Expression Analysis
2.6. LASSO Regression Analysis
2.7. Clinical Sample Collection and Processing
2.8. Grouping of Study Subjects
2.9. Gene-Specific MeRIP-qPCR
2.10. Statistical Analysis
3. Results
3.1. Identification of Differentially Expressed Genes
3.2. Selection of the MP-DEGs
3.3. Functional and Pathway Enrichment of the MP-DEGs
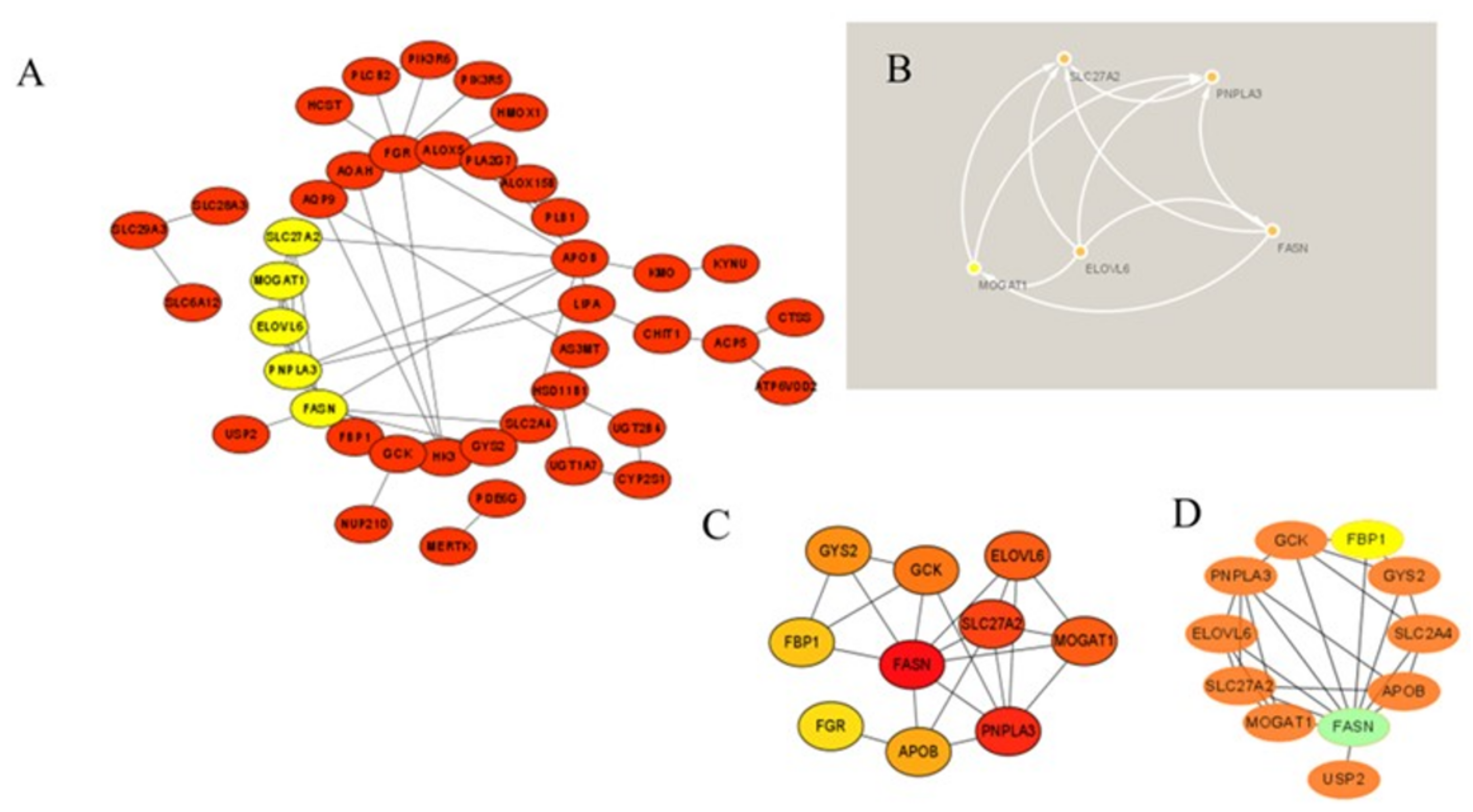
3.4. Protein–Protein Interaction Network Construction and Module Analysis
3.5. Selection and Validation of the Primary Hub Genes
3.6. FASN Is Lower in IR and Associated with FPG and HbA1c
3.7. Correlations of Candidate Gene Expression with m6A Modification in Insulin Resistance
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Tabák, A.G.; Herder, C.; Rathmann, W.; Brunner, E.J.; Kivimäki, M. Prediabetes: A high-risk state for diabetes development. Lancet 2012, 379, 2279–2290. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Knowler, W.C.; Fowler, S.E.; Hamman, R.F.; Christophi, C.A.; Hoffman, H.J.; Brenneman, A.T.; Brown-Friday, J.O.; Goldberg, R.; Venditti, E.; Nathan, D.M. 10-year follow-up of diabetes incidence and weight loss in the Diabetes Prevention Program Outcomes Study. Lancet 2009, 374, 1677–1686. [Google Scholar] [PubMed] [Green Version]
- Petersen, M.C.; Shulman, G.I. Mechanisms of Insulin Action and Insulin Resistance. Physiol. Rev. 2018, 98, 2133–2223. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Staten, M.A.; Stern, M.P.; Miller, W.G.; Steffes, M.W.; Campbell, S.E. Insulin assay standardization: Leading to measures of insulin sensitivity and secretion for practical clinical care. Diabetes Care 2010, 33, 205–206. [Google Scholar] [CrossRef] [Green Version]
- Dranse, H.J.; Waise, T.M.Z.; Hamr, S.C.; Bauer, P.V.; Abraham, M.A.; Rasmussen, B.A.; Lam, T.K.T. Physiological and therapeutic regulation of glucose homeostasis by upper small intestinal PepT1-mediated protein sensing. Nat. Commun. 2018, 9, 1118. [Google Scholar] [CrossRef] [Green Version]
- Gustafson, B.; Hedjazifar, S.; Gogg, S.; Hammarstedt, A.; Smith, U. Insulin resistance and impaired adipogenesis. Trends Endocrinol. Metab. 2015, 26, 193–200. [Google Scholar] [CrossRef]
- Burhans, M.S.; Hagman, D.K.; Kuzma, J.N.; Schmidt, K.A.; Kratz, M. Contribution of Adipose Tissue Inflammation to the Development of Type 2 Diabetes Mellitus. Compr. Physiol. 2018, 9, 1–58. [Google Scholar]
- Roden, M.; Shulman, G.I. The integrative biology of type 2 diabetes. Nature 2019, 576, 51–60. [Google Scholar] [CrossRef] [Green Version]
- Hu, E.; Liang, P.; Spiegelman, B.M. AdipoQ is a novel adipose-specific gene dysregulated in obesity. J. Biol. Chem. 1996, 271, 10697–10703. [Google Scholar] [CrossRef] [Green Version]
- Dua, A.; Hennes, M.I.; Hoffmann, R.G.; Maas, D.L.; Krakower, G.R.; Sonnenberg, G.E.; Kissebah, A.H. Leptin: A significant indicator of total body fat but not of visceral fat and insulin insensitivity in African-American women. Diabetes 1996, 45, 1635–1637. [Google Scholar] [CrossRef] [Green Version]
- Randle, P.J.; Garland, P.B.; Hales, C.N.; Newsholme, E.A. The glucose fatty-acid cycle. Its role in insulin sensitivity and the metabolic disturbances of diabetes mellitus. Lancet 1963, 1, 785–789. [Google Scholar] [CrossRef] [PubMed]
- Hrdlickova, R.; Toloue, M.; Tian, B. RNA-Seq methods for transcriptome analysis. Wiley Interdiscip. Rev. RNA 2017, 8, e1364. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kleinstein, S.E.; McCorrison, J.; Ahmed, A.; Hasturk, H.; Van Dyke, T.E.; Freire, M. Transcriptomics of type 2 diabetic and healthy human neutrophils. BMC Immunol. 2021, 22, 37. [Google Scholar] [CrossRef] [PubMed]
- Herder, C.; Karakas, M.; Koenig, W. Biomarkers for the prediction of type 2 diabetes and cardiovascular disease. Clin. Pharmacol. Ther. 2011, 90, 52–66. [Google Scholar] [CrossRef]
- Padilla-Martinez, F.; Wojciechowska, G.; Szczerbinski, L.; Kretowski, A. Circulating Nucleic Acid-Based Biomarkers of Type 2 Diabetes. Int. J. Mol. Sci. 2021, 23, 295. [Google Scholar] [CrossRef]
- Yang, Y.; Shen, F.; Huang, W.; Qin, S.; Huang, J.T.; Sergi, C.; Yuan, B.F.; Liu, S.M. Glucose Is Involved in the Dynamic Regulation of m6A in Patients with Type 2 Diabetes. J. Clin. Endocrinol. Metab. 2019, 104, 665–673. [Google Scholar] [CrossRef] [Green Version]
- De Jesus, D.F.; Zhang, Z.; Kahraman, S.; Brown, N.K.; Chen, M.; Hu, J.; Gupta, M.K.; He, C.; Kulkarni, R.N. m(6)A mRNA Methylation Regulates Human β-Cell Biology in Physiological States and in Type 2 Diabetes. Nat. Metab. 2019, 1, 765–774. [Google Scholar] [CrossRef]
- Xie, W.; Ma, L.L.; Xu, Y.Q.; Wang, B.H.; Li, S.M. METTL3 inhibits hepatic insulin sensitivity via N6-methyladenosine modification of Fasn mRNA and promoting fatty acid metabolism. Biochem. Biophys. Res. Commun. 2019, 518, 120–126. [Google Scholar] [CrossRef]
- Emont, M.P.; Jacobs, C.; Essene, A.L.; Pant, D.; Tenen, D.; Colleluori, G.; Di Vincenzo, A.; Jørgensen, A.M.; Dashti, H.; Stefek, A.; et al. A single-cell atlas of human and mouse white adipose tissue. Nature 2022, 603, 926–933. [Google Scholar] [CrossRef]
- Ritchie, M.E.; Phipson, B.; Wu, D.; Hu, Y.; Law, C.W.; Shi, W.; Smyth, G.K. limma powers differential expression analyses for RNA-sequencing and microarray studies. Nucleic Acids Res. 2015, 43, e47. [Google Scholar] [CrossRef]
- Uhlén, M.; Fagerberg, L.; Hallström, B.M.; Lindskog, C.; Oksvold, P.; Mardinoglu, A.; Sivertsson, Å.; Kampf, C.; Sjöstedt, E.; Asplund, A.; et al. Proteomics. Tissue-based map of the human proteome. Science 2015, 347, 1260419. [Google Scholar] [CrossRef] [PubMed]
- Szklarczyk, D.; Gable, A.L.; Lyon, D.; Junge, A.; Wyder, S.; Huerta-Cepas, J.; Simonovic, M.; Doncheva, N.T.; Morris, J.H.; Bork, P.; et al. STRING v11: Protein-protein association networks with increased coverage, supporting functional discovery in genome-wide experimental datasets. Nucleic Acids Res. 2019, 47, D607–D613. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bader, G.D.; Hogue, C.W. An automated method for finding molecular complexes in large protein interaction networks. BMC Bioinform. 2003, 4, 2. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chin, C.H.; Chen, S.H.; Wu, H.H.; Ho, C.W.; Ko, M.T.; Lin, C.Y. cytoHubba: Identifying hub objects and sub-networks from complex interactome. BMC Syst. Biol. 2014, 8 (Suppl. S4), S11. [Google Scholar] [CrossRef] [Green Version]
- McEligot, A.J.; Poynor, V.; Sharma, R.; Panangadan, A. Logistic LASSO Regression for Dietary Intakes and Breast Cancer. Nutrients 2020, 12, 2652. [Google Scholar] [CrossRef]
- Polymeris, A.; Papapetrou, P.D. Anthropometric indicators of insulin resistance. Hormones 2022, 21, 51–52. [Google Scholar] [CrossRef]
- Li, Y.; Xiao, J.; Bai, J.; Tian, Y.; Qu, Y.; Chen, X.; Wang, Q.; Li, X.; Zhang, Y.; Xu, J. Molecular characterization and clinical relevance of m(6)A regulators across 33 cancer types. Mol. Cancer 2019, 18, 137. [Google Scholar] [CrossRef]
- Carling, D. AMPK signalling in health and disease. Curr. Opin. Cell Biol. 2017, 45, 31–37. [Google Scholar] [CrossRef]
- Wang, N.; Zhang, S.; Zhu, F.; Yang, Y.; Chen, L.; Lü, P.; Yu, L.; Chen, K. Proteomic Study on the New Potential Mechanism and Biomarkers of Diabetes. Proteom. Clin. Appl. 2019, 13, e1800043. [Google Scholar] [CrossRef]
- Osbak, K.K.; Colclough, K.; Saint-Martin, C.; Beer, N.L.; Bellanné-Chantelot, C.; Ellard, S.; Gloyn, A.L. Update on mutations in glucokinase (GCK), which cause maturity-onset diabetes of the young, permanent neonatal diabetes, and hyperinsulinemic hypoglycemia. Hum. Mutat. 2009, 30, 1512–1526. [Google Scholar] [CrossRef]
- Li, C.; Yang, Y.; Liu, X.; Li, Z.; Liu, H.; Tan, Q. Glucose metabolism-related gene polymorphisms as the risk predictors of type 2 diabetes. Diabetol. Metab. Syndr. 2020, 12, 97. [Google Scholar] [CrossRef] [PubMed]
- Karaglani, M.; Panagopoulou, M.; Cheimonidi, C.; Tsamardinos, I.; Maltezos, E.; Papanas, N.; Papazoglou, D.; Mastorakos, G.; Chatzaki, E. Liquid Biopsy in Type 2 Diabetes Mellitus Management: Building Specific Biosignatures via Machine Learning. J. Clin. Med. 2022, 11, 1045. [Google Scholar] [CrossRef] [PubMed]
- Cruz, M.D.; Wali, R.K.; Bianchi, L.K.; Radosevich, A.J.; Crawford, S.E.; Jepeal, L.; Goldberg, M.J.; Weinstein, J.; Momi, N.; Roy, P.; et al. Colonic mucosal fatty acid synthase as an early biomarker for colorectal neoplasia: Modulation by obesity and gender. Cancer Epidemiol. Biomark. Prev. 2014, 23, 2413–2421. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fazli, H.R.; Moradzadeh, M.; Mehrbakhsh, Z.; Sharafkhah, M.; Masoudi, S.; Pourshams, A.; Mohamadkhani, A. Diagnostic Significance of Serum Fatty Acid Synthase in Patients with Pancreatic Cancer. Middle East J. Dig. Dis. 2021, 13, 115–120. [Google Scholar] [CrossRef] [PubMed]
- Jiang, W.; Xing, X.L.; Zhang, C.; Yi, L.; Xu, W.; Ou, J.; Zhu, N. MET and FASN as Prognostic Biomarkers of Triple Negative Breast Cancer: A Systematic Evidence Landscape of Clinical Study. Front. Oncol. 2021, 11, 604801. [Google Scholar] [CrossRef]
- Rabionet, M.; Polonio-Alcalá, E.; Relat, J.; Yeste, M.; Sims-Mourtada, J.; Kloxin, A.M.; Planas, M.; Feliu, L.; Ciurana, J.; Puig, T. Fatty acid synthase as a feasible biomarker for triple negative breast cancer stem cell subpopulation cultured on electrospun scaffolds. Mater. Today Biol. 2021, 12, 100155. [Google Scholar] [CrossRef]
- Rhode, P.; Mehdorn, M.; Lyros, O.; Kahlert, C.; Kurth, T.; Venus, T.; Schierle, K.; Estrela-Lopis, I.; Jansen-Winkeln, B.; Lordick, F.; et al. Characterization of Total RNA, CD44, FASN, and PTEN mRNAs from Extracellular Vesicles as Biomarkers in Gastric Cancer Patients. Cancers 2021, 13, 5975. [Google Scholar] [CrossRef]
- Ricklefs, F.L.; Maire, C.L.; Matschke, J.; Dührsen, L.; Sauvigny, T.; Holz, M.; Kolbe, K.; Peine, S.; Herold-Mende, C.; Carter, B.; et al. FASN Is a Biomarker Enriched in Malignant Glioma-Derived Extracellular Vesicles. Int. J. Mol. Sci. 2020, 21, 2084. [Google Scholar] [CrossRef] [Green Version]
- Nadler, S.T.; Stoehr, J.P.; Schueler, K.L.; Tanimoto, G.; Yandell, B.S.; Attie, A.D. The expression of adipogenic genes is decreased in obesity and diabetes mellitus. Proc. Natl. Acad. Sci. USA 2000, 97, 11371–11376. [Google Scholar] [CrossRef] [Green Version]
- Menendez, J.A.; Vazquez-Martin, A.; Ortega, F.J.; Fernandez-Real, J.M. Fatty Acid Synthase: Association with Insulin Resistance, Type 2 Diabetes, and Cancer. Clin. Chem. 2009, 55, 425–438. [Google Scholar] [CrossRef] [Green Version]
- Sievert, H.; Krause, C.; Geißler, C.; Grohs, M.; El-Gammal, A.T.; Wolter, S.; Mann, O.; Lehnert, H.; Kirchner, H. Epigenetic Downregulation of FASN in Visceral Adipose Tissue of Insulin Resistant Subjects. Exp. Clin. Endocrinol. Diabetes 2020, 129, 674–682. [Google Scholar] [CrossRef] [PubMed]
- Fernandez-Real, J.M.; Menendez, J.A.; Moreno-Navarrete, J.M.; Blüher, M.; Vazquez-Martin, A.; Vázquez, M.J.; Ortega, F.; Diéguez, C.; Frühbeck, G.; Ricart, W.; et al. Extracellular fatty acid synthase: A possible surrogate biomarker of insulin resistance. Diabetes 2010, 59, 1506–1511. [Google Scholar] [CrossRef] [Green Version]
- Hillgartner, F.B.; Salati, L.M.; Goodridge, A.G. Physiological and molecular mechanisms involved in nutritional regulation of fatty acid synthesis. Physiol. Rev. 1995, 75, 47–76. [Google Scholar] [CrossRef] [PubMed]
- Moreno-Indias, I.; Tinahones, F.J. Impaired adipose tissue expandability and lipogenic capacities as ones of the main causes of metabolic disorders. J. Diabetes Res. 2015, 2015, 970375. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, J.; Song, Y.; Shi, Q.; Fu, L. Research progress on FASN and MGLL in the regulation of abnormal lipid metabolism and the relationship between tumor invasion and metastasis. Front. Med. 2021, 15, 649–656. [Google Scholar] [CrossRef] [PubMed]
- Sun, D.; Zhao, T.; Zhang, Q.; Wu, M.; Zhang, Z. Fat mass and obesity-associated protein regulates lipogenesis via m(6) A modification in fatty acid synthase mRNA. Cell Biol. Int. 2021, 45, 334–344. [Google Scholar] [CrossRef] [PubMed]
- Zhou, J.; Wan, J.; Gao, X.; Zhang, X.; Jaffrey, S.R.; Qian, S.B. Dynamic m(6)A mRNA methylation directs translational control of heat shock response. Nature 2015, 526, 591–594. [Google Scholar] [CrossRef] [Green Version]
- Wang, X.; Lu, Z.; Gomez, A.; Hon, G.C.; Yue, Y.; Han, D.; Fu, Y.; Parisien, M.; Dai, Q.; Jia, G.; et al. N6-methyladenosine-dependent regulation of messenger RNA stability. Nature 2014, 505, 117–120. [Google Scholar] [CrossRef] [Green Version]
- Zhao, T.; Sun, D.; Xiong, W.; Man, J.; Zhang, Q.; Zhao, M.; Zhang, Z. N(6)-methyladenosine plays a dual role in arsenic carcinogenesis by temporal-specific control of core target AKT1. J. Hazard. Mater. 2023, 445, 130468. [Google Scholar] [CrossRef]










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| FASN | GCK | |
|---|---|---|
| FPG, mmol/L | −0.309 ** | −0.230 * |
| HbA1c, % | −0.296 ** | −0.096 |
| TC, mmol/L | 0.059 | −0.011 |
| TG, mmol/L | 0.153 | 0.033 |
| HDL, mmol/L | −0.121 | 0.060 |
| LDL, mmol/L | 0.081 | −0.080 |
| Homa-IR | −0.279 ** | −0.220 * |
| BMI | 0.097 | 0.144 |
| FIns, mIU/L | −0.325 ** | −0.191 |
| AUC (95% CI) | Sensitivity (%) | Specificity (%) | p | |
|---|---|---|---|---|
| FASN | 0.78 (0.69–0.80) | 85.7 | 65.4 | 0.000 |
| GCK | 0.63 (0.52–0.74) | 88.1 | 40.4 | 0.028 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Li, Y.-L.; Li, L.; Liu, Y.-H.; Hu, L.-K.; Yan, Y.-X. Identification of Metabolism-Related Proteins as Biomarkers of Insulin Resistance and Potential Mechanisms of m6A Modification. Nutrients 2023, 15, 1839. https://doi.org/10.3390/nu15081839
Li Y-L, Li L, Liu Y-H, Hu L-K, Yan Y-X. Identification of Metabolism-Related Proteins as Biomarkers of Insulin Resistance and Potential Mechanisms of m6A Modification. Nutrients. 2023; 15(8):1839. https://doi.org/10.3390/nu15081839
Chicago/Turabian StyleLi, Yan-Ling, Long Li, Yu-Hong Liu, Li-Kun Hu, and Yu-Xiang Yan. 2023. "Identification of Metabolism-Related Proteins as Biomarkers of Insulin Resistance and Potential Mechanisms of m6A Modification" Nutrients 15, no. 8: 1839. https://doi.org/10.3390/nu15081839