
Dietary Trace Elements and the Pathogenesis of Neurodegenerative Diseases

Abstract
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Characteristics of Trace Elements and Their Roles in Biological Functions
2.1. Iron
2.2. Zinc
2.3. Copper
2.4. Manganese
2.5. Aluminum
3. Roles of Trace Elements in the Pathogenesis of Neurodegenerative Diseases
3.1. Alzheimer’s Disease
3.2. Prion Diseases
3.3. Lewy Body Diseases
3.4. Vascular-Type Senile Dementia
4. Hypothesis: Cross-Talk Occurs between Trace Elements and Amyloidogenic Proteins at Synapses
5. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Becker, J.S.S.; Matusch, A.; Palm, C.; Salber, D.; Morton, K.; Becker, S. Bioimaging of metals in brain tissue by laser ablation inductively coupled plasma mass spectrometry (LA-ICP-MS) and metallomics. Metallomics 2010, 2, 104–111. [Google Scholar] [CrossRef]
- Cobley, J.N.; Fiorello, M.L.; Bailey, D.M. 13 reasons why the brain is susceptible to oxidative stress. Redox Biol. 2018, 15, 490–503. [Google Scholar] [CrossRef]
- Dirandeh, E.; Palizgir, A.; Kassiri, N. An overview of the relationship between occupational manganese exposure and Parkinsonism. Cureus 2022, 14, e32161. [Google Scholar] [CrossRef]
- Kawahara, M.; Tanaka, K.-I.; Kato-Negishi, M. Copper as a collaborative partner of zinc-induced neurotoxicity in the pathogenesis of vascular dementia. Int. J. Mol. Sci. 2021, 22, 7242. [Google Scholar] [CrossRef] [PubMed]
- Adlard, P.A.; Bush, A.I. Metals and Alzheimer’s disease: How far have we come in the clinic? J. Alzheimers Dis. 2018, 62, 1369–1379. [Google Scholar] [CrossRef]
- Brown, D.R. Metalloproteins and neuronal death. Metallomics 2010, 2, 186–194. [Google Scholar] [CrossRef]
- Jiang, H.; Song, N.; Jiao, Q.; Shi, L.; Du, X. Iron pathophysiology in Parkinson diseases. Adv. Exp. Med. Biol. 2019, 1173, 45–66. [Google Scholar] [PubMed]
- Weiss, J.H.; Sensi, S.L.; Koh, J.Y. Zn(2+): A novel ionic mediator of neural injury in brain disease. Trends Pharmacol. Sci. 2000, 21, 395–401. [Google Scholar] [CrossRef] [PubMed]
- Carrell, R.W.; Lomas, D.A. Conformational disease. Lancet 1997, 350, 134–138. [Google Scholar] [CrossRef]
- Kawahara, M.; Kato-Negishi, M.; Tanaka, K.I. Amyloids: Regulators of metal homeostasis in the synapse. Molecules 2020, 25, 1441. [Google Scholar] [CrossRef]
- Moshtaghie, A.A.; Ani, M.; Bazrafshan, M.R. Comparative binding study of aluminum and chromium to human transferrin. Biol. Trace Elem. Res. 1992, 32, 39–46. [Google Scholar] [CrossRef]
- Nyarko-Danquah, I.; Pajarillo, E.; Digman, A.; Soliman, K.F.A.; Aschner, M.; Lee, E. Manganese accumulation in the brain via various transporters and its neurotoxicity mechanisms. Molecules 2020, 25, 5880. [Google Scholar] [CrossRef]
- Fujishiro, H.; Kambe, T. Manganese transport in mammals by zinc transporter family proteins, ZNT and ZIP. J. Pharmacol. Sci. 2022, 148, 125–133. [Google Scholar] [CrossRef]
- Thirupathi, A.; Chang, Y.Z. Brain iron metabolism and CNS diseases. Adv. Exp. Med. Biol. 2019, 1173, 1–19. [Google Scholar] [PubMed]
- Wang, Y.; Wu, Y.; Li, T.; Wang, X.; Zhu, C. Iron metabolism and brain development in premature infants. Front. Physiol. 2019, 10, 463. [Google Scholar] [CrossRef] [PubMed]
- Silitonga, H.T.S.; Salim, L.A.; Nurmala, I.; Wartiningsih, M. Compliance of iron supplementation and determinants among adolescent girls: A systematic review. Iran J. Public Health 2023, 52, 37–48. [Google Scholar] [CrossRef]
- Beck, K.L.; von Hurst, P.R.; O’Brien, W.J.; Badenhorst, C.E. Micronutrients and athletic performance: A review. Food Chem. Toxicol. 2021, 158, 112618. [Google Scholar] [CrossRef] [PubMed]
- Wang, C.; Liu, H.; Xu, S.; Deng, Y.; Xu, B.; Yang, T.; Liu, W. Ferroptosis and neurodegenerative diseases: Insights into the regulatory roles of SLC7A11. Cell Mol. Neurobiol. 2023; ahead of print. [Google Scholar] [CrossRef]
- Pasini, A.M.F.; Stranieri, C.; Busti, F.; Di Leo, E.G.; Girelli, D.; Cominacini, L. New insights into the role of ferroptosis in cardiovascular diseases. Cells 2023, 12, 867. [Google Scholar] [CrossRef] [PubMed]
- Collings, R.; Harvey, L.; Hooper, L.; Hurst, R.; Brown, T.; Ansett, J.; King, M.; Fairweather-Tait, S. The absorption of iron from whole diets: A systematic review. Am. J. Clin. Nutr. 2013, 98, 65–81. [Google Scholar] [CrossRef]
- Du, F.; Qian, Z.M.; Luo, Q.; Yung, W.H.; Ke, Y. Hepcidin suppresses brain iron accumulation by downregulating iron transport proteins in iron-overloaded rats. Mol. Neurobiol. 2015, 52, 101–114. [Google Scholar] [CrossRef]
- Skjørringe, T.; Burkhart, A.; Johnsen, K.B.; Moos, T. Divalent metal transporter 1 (DMT1) in the brain: Implications for a role in iron transport at the blood-brain barrier, and neuronal and glial pathology. Front. Mol. Neurosci. 2015, 8, 19. [Google Scholar]
- Zhou, Z.D.; Tan, E.K. Iron regulatory protein (IRP)-iron responsive element (IRE) signaling pathway in human neurodegenerative diseases. Mol. Neurodegener. 2017, 12, 75. [Google Scholar] [CrossRef]
- World Health Organization. Assessing the Iron Status of Populations: Including Literature Reviews; World Health Organization: Geneva, Switzerland, 2007; ISBN 9789241596107. [Google Scholar]
- Ministry of Health, Labor and Welfare. National Health and Nutrition Survey Japan 2019. Available online: https://www.mhlw.go.jp/stf/seisakunitsuite/bunya/kenkou_iryou/kenkou/eiyou/r1-houkoku_00002.html (accessed on 1 April 2023).
- Olynyk, J.K.; Ramm, G.A. Hemochromatosis. N. Engl. J. Med. 2022, 387, 2159–2170. [Google Scholar] [CrossRef]
- Frederickson, C.J.; Suh, S.W.; Silva, D.; Frederickson, C.J.; Thompson, R.B. Importance of zinc in the central nervous system: The zinc-containing neuron. J. Nutr. 2000, 130, 1471S–1483S. [Google Scholar] [CrossRef] [PubMed]
- Prasad, A.S. Impact of the discovery of human zinc deficiency on health. J. Am. Coll. Nutr. 2009, 28, 257–265. [Google Scholar] [CrossRef] [PubMed]
- Hambidge, M. Human zinc deficiency. J. Nutr. 2000, 130, 1344S–1349S. [Google Scholar] [CrossRef]
- Takeda, A.; Tamano, H. Subclinical zinc deficiency impairs human brain function. J. Trace Elem. Med. Biol. 2012, 26, 70–73. [Google Scholar]
- Takeda, A.; Tamano, H. The impact of synaptic Zn2+ dynamics on cognition and its decline. Int. J. Mol. Sci. 2017, 18, E2411. [Google Scholar] [CrossRef]
- Ueno, S.; Tsukamoto, M.; Hirano, T.; Kikuchi, K.; Yamada, K.-M.; Nishiyama, N.; Nagano, N.; Matsuki, N.; Ikegaya, Y. Mossy fiber Zn2+ spillover modulates heterosynaptic N-methyl-D-aspartate receptor activity in hippocampal CA3 circuits. J. Cell Biol. 2002, 158, 215–220. [Google Scholar] [CrossRef] [PubMed]
- Vogler, N.W.; Betti, V.M.; Goldberg, J.M.; Tzounopoulos, T. Mechanisms underlying long-term synaptic zinc plasticity at mouse dorsal cochlear nucleus glutamatergic synapses. J. Neurosci. 2020, 40, 4981–4996. [Google Scholar] [CrossRef]
- Hamer, D.H. Metallothionein. Annu. Rev. Biochem. 1986, 55, 913–951. [Google Scholar] [CrossRef] [PubMed]
- Hara, T.; Yoshigai, E.; Ohashi, T.; Fukada, T. Zinc transporters as potential therapeutic targets: An updated review. J. Pharmacol. Sci. 2022, 148, 221–228. [Google Scholar] [CrossRef] [PubMed]
- Joint statement by the World Health Organization, the World Food Programme and the United Nations Children’s Fund. Preventing and Controlling Micronutrient Deficiencies in Populations Affected by an Emergency: Multiple Vitamin and Mineral Supplements for Pregnant and Lactating Women, and for Children Aged 6 to 59 Months. 7 April 2006. Available online: https://www.who.int/publications/m/item/WHO-WFP-UNICEF-statement-micronutrients-deficiencies-emergency (accessed on 1 April 2023).
- Kogirima, M.; Kurasawa, R.; Kubori, S.; Sarukura, N.; Nakamori, M.; Okada, S.; Kamioka, S.; Yamamoto, S. Ratio of low serum zinc levels in elderly Japanese people living in the central part of Japan. Eur. J. Clin. Nutr. 2007, 61, 375–381. [Google Scholar] [CrossRef]
- An, Y.; Li, S.; Huang, X.; Chen, X.; Shan, H.; Zhang, M. The role of copper homeostasis in brain disease. Int. J. Mol. Sci. 2022, 23, 13850. [Google Scholar] [CrossRef] [PubMed]
- Helman, S.L.; Zhou, J.; Fuqua, B.K.; Lu, Y.; Collins, J.F.; Chen, H.; Vulpe, C.D.; Anderson, G.J.; Frazer, D.M. The biology of mammalian multi-copper ferroxidases. Biometals 2023, 36, 263–281. [Google Scholar] [CrossRef]
- D’Ambrosi, N.; Rossi, L. Copper at synapse: Release, binding and modulation of neurotransmission. Neurochem. Inter. 2015, 90, 36–45. [Google Scholar] [CrossRef]
- Kardos, J.; Héja, L.; Simon, A.; Jablonkai, I.; Kovács, R.; Jemnitz, K. Copper signalling: Causes and consequences. Cell Commun. Signal 2018, 16, 71. [Google Scholar] [CrossRef]
- Kodama, H.; Fujisawa, C.; Bhadhprasit, W. Inherited copper transport disorders: Biochemical mechanisms, diagnosis, and treatment. Curr. Drug Metab. 2012, 13, 237–250. [Google Scholar] [CrossRef]
- Hartwig, C.; Méndez, G.M.; Bhattacharjee, S.; Vrailas-Mortimer, A.D.; Zlatic, S.A.; Freeman, A.A.H.; Gokhale, A.; Concilli, M.; Werner, E.; Savas, C.S. Golgi-dependent copper homeostasis sustains synaptic development and mitochondrial content. J. Neurosci. 2021, 41, 215–233. [Google Scholar] [CrossRef]
- Zietz, B.P.; Dieter, H.H.; Lakomek, M.; Schneider, H.; Kessler-Gaedtke, B.; Dunkelberg, H. Epidemiological investigation on chronic copper toxicity to children exposed via the public drinking water supply. Sci. Total Environ. 2003, 302, 127–144. [Google Scholar] [CrossRef]
- Chen, P.; Chakraborty, S.; Mukhopadhyay, S.; Lee, E.; Paoliello, M.M.B.; Bowman, A.B.; Aschner, M. Manganese homeostasis in the nervous system. J. Neurochem. 2015, 134, 601–610. [Google Scholar] [CrossRef]
- Martins, A.C.; Krum, B.N.; Queirós, L.; Tinkoval, A.A.; Skalny, A.V.; Bowman, A.B.; Aschner, M. Manganese in the diet: Bioaccessibility, adequate intake, and neurotoxicological effects. J. Agric. Food Chem. 2020, 68, 12893–12903. [Google Scholar] [CrossRef]
- Roels, H.A.; Bowler, R.M.; Kim, Y.; Henn, B.C.; Mergler, D.; Hoet, P.; Gocheva, V.V.; Bellinger, D.C.; Wright, R.O.; Harris, M.G.; et al. Manganese exposure and cognitive deficits: A growing concern for manganese neurotoxicity. Neurotoxicology 2012, 33, 872–880. [Google Scholar] [CrossRef] [PubMed]
- Peres, T.V.; Schettinger, M.R.C.; Chen, P.; Carvalho, F.; Avila, D.S.; Bowman, A.B.; Aschner, M. Manganese-induced neurotoxicity: A review of its behavioral consequences and neuroprotective strategies. BMC Pharmacol. Toxicol. 2016, 17, 57. [Google Scholar] [CrossRef] [PubMed]
- Venkataramani, V.; Doeppner, T.R.; Willkommen, D.; Cahill, C.M.; Xin, Y.; Ye, G.; Liu, Y.; Southon, A.; Aron, A.; Au-Yeung, H.Y.; et al. Manganese causes neurotoxic iron accumulation via translational repression of amyloid precursor protein and H-Ferritin. J. Neurochem. 2018, 147, 831–848. [Google Scholar] [CrossRef] [PubMed]
- Kawahara, M.; Kato-Negishi, M. Link between aluminum and the pathogenesis of Alzheimer’s disease: The integration of the aluminum and amyloid cascade hypotheses. Int. J. Alzheimers Dis. 2011, 2011, 276393. [Google Scholar] [CrossRef] [PubMed]
- Kawahara, M.; Tanaka, K.-I.; Kato-Negishi, M. Neurotoxicity of Aluminum and its link with neurodegenerative diseases. Metallomics Res. 2021, 1, MR202104. [Google Scholar]
- Niu, Q. Overview of the relationship between aluminum exposure and health of human being. Adv. Exp. Med. Biol. 2018, 1091, 1–31. [Google Scholar] [PubMed]
- Jouhanneau, P.; Raisbeck, G.M.; Yiou, F.; Lacour, B.; Banide, H.; Drueke, T.B. Gastrointestinal absorption, tissue retention, and urinary excretion of dietary aluminum in rats determined by using 26Al. Clin. Chem. 1997, 43, 1023–1028. [Google Scholar] [CrossRef]
- Kobayashi, K.; Yumoto, S.; Nagai, H.; Hosoyama, Y.; Imamura, M.; Masuzawa, S.; Koizumi, Y.; Yamashita, H. 26Al tracer experiment by accelerator mass spectrometry and its application to the studies for amyotrophic lateral sclerosis and Alzheimer’s disease. I. Proc. Japan Acad. Ser. B 1990, 66, 189–192. [Google Scholar] [CrossRef]
- Nakahori, N.; Sekine, M.; Yamada, M.; Tatsuse, T.; Kido, H.; Suzuki, M. Future projections of the prevalence of dementia in Japan: Results from the Toyama Dementia Survey. BMC Geriatr. 2021, 21, 602. [Google Scholar] [CrossRef] [PubMed]
- Selkoe, D.J. The molecular pathology of Alzheimer’s disease. Neuron 1991, 6, 487–498. [Google Scholar] [CrossRef]
- Kawahara, M.; Negishi-Kato, M.; Sadakane, Y. Calcium dyshomeostasis and neurotoxicity of Alzheimer’s beta-amyloid protein. Expert Rev. Neurother. 2009, 9, 681–693. [Google Scholar] [CrossRef] [PubMed]
- Wirths, O.; Multhaup, G.; Bayer, T.A. A modified ß-amyloid hypothesis: Intraneuronal accumulation of the beta-amyloid peptide--the first step of a fatal cascade. J. Neurochem. 2004, 91, 513–520. [Google Scholar] [CrossRef] [PubMed]
- Fukuyama, R.; Mizuno, T.; Mori, S.; Nakajima, K.; Fushiki, S.; Yanagisawa, K. Age-dependent change in the levels of Aß40 and Aß42 in cerebrospinal fluid from control subjects, and a decrease in the ratio of Aß42 to Aß40 level in cerebrospinal fluid from Alzheimer’s disease patients. Eur. Neurol. 2000, 43, 155–160. [Google Scholar] [CrossRef]
- Dyrks, T.; Dyrks, E.; Masters, C.L.; Beyreuther, K. Amyloidogenicity of rodent and human beta A4 sequences. FEBS Lett. 1993, 324, 231–236. [Google Scholar] [CrossRef]
- Exley, C.; Price, N.C.; Kelly, S.M.; Birchal, J.D. An interaction of beta-amyloid with aluminium in vitro. FEBS Lett. 1993, 324, 293–295. [Google Scholar] [CrossRef]
- Kawahara, M.; Muramoto, K.; Kobayashi, K.; Mori, H.; Kuroda, Y. Aluminum promotes the aggregation of Alzheimer’s amyroid ß-protein in vitro. Biochem. Biophys. Res. Commun. 1994, 198, 531–535. [Google Scholar] [CrossRef]
- Mantyh, P.W.; Ghilardi, J.R.; Rogers, S.; DeMaster, E.; Allen, C.J.; Stimson, E.R.; Maggio, J.E. Aluminum, iron, and zinc ions promote aggregation of physiological concentrations of beta-amyloid peptide. J. Neurochem. 1993, 61, 1171–1174. [Google Scholar] [CrossRef]
- Bush, A.I.; Pettingell, W.H.; Multhaup, G.; d Paradis, M.; Vonsattel, J.P.; Gusella, J.F.; Beyreuther, K.; Masters, C.L.; Tanzi, R.E. Rapid induction of Alzheimer Aβ amyloid formation by zinc. Science 1994, 265, 1464–1467. [Google Scholar] [CrossRef]
- Atwood, C.S.; Moir, R.D.; Huang, X.; Scarpa, R.C.; Bacarra, N.M.; Romano, D.M.; Hartshorn, M.A.; Tanzi, R.E.; Bush, A.I. Dramatic aggregation of Alzheimer aß by Cu(II) is induced by conditions representing physiological acidosis. J. Biol. Chem. 1998, 273, 12817–12826. [Google Scholar] [CrossRef] [PubMed]
- Wallin, C.; Kulkarni, Y.S.; Abelein, A.; Jarvet, J.; Liao, Q.; Strodel, B.; Olsson, L.; Luo, J.; Abrahams, J.P.; Sholts, S.B.; et al. Characterization of Mn(II) ion binding to the amyloid-β peptide in Alzheimer’s disease. J. Trace Elem. Med. Biol. 2016, 38, 183–193. [Google Scholar] [CrossRef] [PubMed]
- Mold, M.; Linhart, C.; Gómez-Ramírez, J.; Villegas-Lanau, A.; Exley, C. Aluminum and Amyloid-β in Familial Alzheimer’s Disease. J Alzheimers Dis. 2020, 73, 1627–1635. [Google Scholar] [CrossRef]
- James, S.A.; Churches, Q.I.; de Jonge, M.D.; Birchall, I.E.; Streltsov, V.; McColl, G.; Adlard, P.A.; Hare, D.J. Iron, copper, and zinc concentration in Aβ Plaques in the APP/PS1 mouse model of Alzheimer’s Disease correlates with metal levels in the surrounding neuropil. ACS Chem. Neurosci. 2017, 8, 629–637. [Google Scholar] [CrossRef] [PubMed]
- White, A.R.; Multhaup, G.; Maher, F.; Bellingham, S.; Camakaris, J.; Zheng, H.; Bush, A.I.; Beyreuther, K.; Masters, C.L.; Cappai, R. The Alzheimer’s disease amyloid precursor protein modulates copper-induced toxicity and oxidative stress in primary neuronal cultures. J. Neurosci. 1999, 19, 9170–9179. [Google Scholar] [CrossRef]
- Baumkötter, F.; Schmidt, N.; Vargas, C.; Schilling, S.; Weber, R.; Wagner, K.; Fiedler, S.; Klug, W.; Radzimanowski, J.; Nickolaus, S.; et al. Amyloid precursor protein dimerization and synaptogenic function depend on copper binding to the growth factor-like domain. J. Neurosci. 2014, 34, 11159–11172. [Google Scholar] [CrossRef]
- Spoerri, L.; Vella, L.J.; Pham, C.L.L.; Barnham, K.J.; Cappai, R. The amyloid precursor protein copper binding domain histidine residues 149 and 151 mediate APP stability and metabolism. J. Biol. Chem. 2012, 287, 26840–26853. [Google Scholar] [CrossRef]
- Multhaup, G.; Schlicksupp, A.; Hesse, L.; Beher, D.; Ruppert, T.; Masters, C.K.; Beyreuther, K. The amyloid precursor protein of Alzheimer’s disease in the reduction of copper(II) to copper(I). Science 1996, 271, 1406–1409. [Google Scholar] [CrossRef]
- Wong, B.X.; Tsatsanis, A.; Lim, L.Q.; Adlard, P.A.; Bush, A.I.; Duce, J.A. β-Amyloid precursor protein does not possess ferroxidase activity but does stabilize the cell surface ferrous iron exporter ferroportin. PLoS ONE 2014, 9, e114174. [Google Scholar] [CrossRef]
- Pizzo, P.; Basso, E.; Filadi, R.; Greotti, E.; Leparulo, A.; Pendin, D.; Redolfi, N.; Rossini, M.; Vajente, N.; Pozzan, T.; et al. Presenilin-2 and calcium handling: Molecules, organelles, cells and brain networks. Cells 2020, 9, 2166. [Google Scholar] [CrossRef]
- Greenough, M.A.; Volitakis, I.; Li, Q.X.; Laughton, K.; Evin, G.; Ho, M.; Dalziel, A.H.; Camakaris, J.; Bush, A.I. Presenilins promote the cellular uptake of copper and zinc and maintain copper chaperone of SOD1-dependent copper/zinc superoxide dismutase activity. J. Biol. Chem. 2011, 286, 9776–9786. [Google Scholar] [CrossRef] [PubMed]
- Gerber, H.; Wu, F.; Dimitrov, M.; Garcia Osuna, G.M.; Fraering, P.C. Zinc and copper differentially modulate amyloid precursor protein processing by γ-secretase and amyloid-β peptide production. J. Biol. Chem. 2017, 292, 3751–3767. [Google Scholar] [CrossRef] [PubMed]
- Rogers, J.T.; Randall, J.D.; Cahill, C.M.; Eder, P.S.; Huang, X.; Gunshin, H.; Leiter, L.; McPhee, J.; Sarang, S.S.; Utsuki, T.; et al. An iron-responsive element type II in the 5′-untranslated region of the Alzheimer’s amyloid precursor protein transcript. J. Biol. Chem. 2002, 277, 45518–45528. [Google Scholar] [CrossRef] [PubMed]
- Chen, M.; Zheng, J.; Liu, G.; Zeng, C.; Xu, E.; Zhu, W.; Anderson, G.; Chen, H. High dietary iron disrupts iron homeostasis and induces amyloid-β and phospho-tau expression in the hippocampus of adult wild-type and APP/PS1 transgenic mice. J. Nutr. 2019, 149, 2247–2254. [Google Scholar] [CrossRef] [PubMed]
- Namekata, K.; Imagawa, M.; Terashi, A.; Ohta, S.; Oyama, F.; Ihara, Y. Association of transferrin C2 allele with late-onset Alzheimer’s disease. Hum. Genet. 1997, 101, 126–129. [Google Scholar] [CrossRef]
- Imagawa, M.; Naruse, S.; Tsuji, S.; Fujioka, A.; Yamaguchi, H. Coenzyme Q10, iron, and vitamin B6 in genetically-confirmed Alzheimer’s disease. Lancet 1992, 340, 671. [Google Scholar] [CrossRef]
- Lu, L.N.; Qian, Z.M.; Wu, K.C.; Yung, W.H.; Ke, Y. Expression of iron transporters and pathological hallmarks of Parkinson’s and Alzheimer’s diseases in the brain of young, adult, and aged rats. Mol. Neurobiol. 2017, 54, 5213–5224. [Google Scholar] [CrossRef]
- Bao, W.D.; Pang, P.; Zhou, X.T.; Hu, F.; Xiong, W.; Chen, K.; Wang, J.; Wang, F.; Xie, D.; Hu, Y.Z.; et al. Loss of ferroportin induces memory impairment by promoting ferroptosis in Alzheimer’s disease. Cell Death Differ. 2021, 28, 1548–1562. [Google Scholar] [CrossRef]
- Prusiner, S.B. Biology and genetics of prions causing neurodegeneration. Annu. Rev. Genet. 2013, 47, 601–623. [Google Scholar] [CrossRef]
- Brown, D.R.; Qin, K.; Herms, J.W.; Madlung, A.; Manson, J.; Strome, R.; Fraser, P.E.; Kruck, T.; von Bohlen, A.; Schulz-Schaeffer, W.; et al. The cellular prion protein binds copper in vivo. Nature 1997, 390, 684–687. [Google Scholar] [CrossRef]
- Jackson, G.S.; Murray, I.; Hosszu, L.L.; Gibbs, N.; Waltho, J.P.; Clarke, A.R.; Collinge, J. Location and properties of metal-binding sites on the human prion protein. Proc. Natl. Acad. Sci. USA 2001, 98, 8531–8535. [Google Scholar] [CrossRef]
- Huang, S.; Chen, L.; Bladen, C.; Stys, P.K.; Zamponi, G.W. Differential modulation of NMDA and AMPA receptors by cellular prion protein and copper ions. Mol. Brain 2018, 11, 62. [Google Scholar] [CrossRef]
- Forloni, G.; Chiesa, R.; Bugiani, O.; Salmona, M.; Tagliavini, F. Review: PrP 106-126-25 years after. Neuropathol Appl. Neurobiol. 2019, 45, 430–440. [Google Scholar] [CrossRef] [PubMed]
- Kawahara, M.; Koyama, H.; Nagata, T.; Sadakane, Y. Zinc, copper, and carnosine attenuate neurotoxicity of prion fragment PrP106-126. Metallomics 2011, 3, 726–734. [Google Scholar] [CrossRef]
- Nguyen, X.T.A.; Tran, T.H.; Cojoc, D.; Legname, G. Copper binding regulates cellular prion protein function. Mol. Neurobiol. 2019, 56, 6121–6133. [Google Scholar] [CrossRef] [PubMed]
- Salzano, G.; Giachin, G.; Legname, G. Structural Consequences of Copper Binding to the Prion Protein. Cells 2019, 8, 770. [Google Scholar] [CrossRef] [PubMed]
- Siggs, O.M.; Cruite, J.T.; Du, X.; Rutschmann, S.; Masliah, E.; Beutler, B.; Oldstone, M.B.A. Disruption of copper homeostasis due to a mutation of Atp7a delays the onset of prion disease. Proc. Natl. Acad. Sci. USA 2012, 109, 13733–13738. [Google Scholar] [CrossRef]
- Spevacek, A.R.; Evans, E.G.B.; Miller, J.L.; Meyer, H.C.; Pelton, J.G.; Millhauser, G.L. Zinc drives a tertiary fold in the prion protein with familial disease mutation sites at the interface. Structure 2013, 21, 236–246. [Google Scholar] [CrossRef]
- Schmitt-Ulms, G.; Ehsani, S.; Watts, J.C.; Westaway, D.; Wille, H. Evolutionary descent of prion genes from the ZIP family of metal ion transporters. PLoS ONE 2009, 4, e7208. [Google Scholar] [CrossRef]
- Watt, N.T.; Griffiths, H.H.; Hooper, N.M. Neuronal zinc regulation and the prion protein. Prion 2013, 7, 203–208. [Google Scholar] [CrossRef]
- Singh, A.; Haldar, S.; Horback, K.; Tom, C.; Zhou, L.; Meyerson, H.; Singh, N. Prion protein regulates iron transport by functioning as a ferrireductase. J. Alzheimers Dis. 2013, 35, 541–552. [Google Scholar] [CrossRef]
- Tripathi, A.K.; Haldar, S.; Qian, J.; Beserra, A.; Suda, S.; Singh, A.; Hopfer, U.; Chen, S.G.; Garrick, M.D.; Turner, J.R.; et al. Prion protein functions as a ferrireductase partner for ZIP14 and DMT1. Free Radic. Biol. Med. 2015, 84, 322–330. [Google Scholar] [CrossRef]
- Singh, A.; Kong, Q.; Luo, X.; Petersen, R.B.; Meyerson, H.; Singh, N. Prion protein (PrP) knock-out mice show altered iron metabolism: A functional role for PrP in iron uptake and transport. PLoS ONE 2009, 4, e6115. [Google Scholar] [CrossRef]
- Rogers, J.T.; Cahill, C.M. Iron-responsive-like elements and neurodegenerative ferroptosis. Learn Mem. 2020, 27, 395–413. [Google Scholar] [CrossRef]
- Brown, D.R. Prions and manganese: A maddening beast. Metallomics 2011, 3, 229–238. [Google Scholar] [CrossRef]
- Davies, P.; Brown, D.R. Manganese enhances prion protein survival in model soils and increases prion infectivity to cells. PLoS ONE 2009, 4, e7518. [Google Scholar] [CrossRef]
- White, S.N.; O’Rourke, K.I.; Gidlewski, T.; VerCauteren, K.C.; Mousel, M.R.; Phillips, G.E.; Spraker, T.R. Increased risk of chronic wasting disease in Rocky Mountain elk associated with decreased magnesium and increased manganese in brain tissue. Can. J. Vet. Res. 2010, 74, 50–53. [Google Scholar]
- Hesketh, S.; Sassoon, J.; Knight, R.; Hopkins, J.; Brown, D.R. Elevated manganese levels in blood and central nervous system occur before onset of clinical signs in scrapie and bovine spongiform encephalopathy. J. Anim. Sci. 2007, 85, 1596–1609. [Google Scholar] [CrossRef]
- Hesketh, S.; Sassoon, J.; Knight, R.; Brown, D.R. Elevated manganese levels in blood and CNS in human prion disease. Mol. Cell Neurosci. 2008, 37, 590–598. [Google Scholar] [CrossRef]
- Slivarichová, D.; Mitrová, E.; Ursínyová, M.; Uhnáková, I.; Koscová, S.; Wsólová, L. Geographic accumulation of Creutzfeldt-Jakob disease in Slovakia--environmental metal imbalance as a possible cofactor. Cent. Eur. J. Public Health 2011, 19, 158–164. [Google Scholar] [CrossRef]
- Masánová, V.; Mitrova, E.; Ursinyova, M.; Uhnakova, I.; Slivarichova, D. Manganese and copper imbalance in the food chain constituents in relation to Creutzfeldt-Jakob disease. Int. J. Environ. Health Res. 2007, 17, 419–428. [Google Scholar] [CrossRef]
- Bernal-Conde, L.D.; Ramos-Acevedo, R.; Reyes-Hernández, M.A.; Balbuena-Olvera, A.J.; Morales-Moreno, I.D.; Argüero-Sánchez, R.; Schüle, B.; Guerra-Crespo, M. Alpha-synuclein physiology and pathology: A perspective on cellular structures and organelles. Front. Neurosci. 2020, 13, 1399. [Google Scholar] [CrossRef]
- Brookes, A.J.; St Clair, D. Synuclein proteins and Alzheimer’s disease. Trends Neurosci. 1994, 14, 404–405. [Google Scholar] [CrossRef]
- Uversky, V.N.; Li, J.; Fink, A.L. Metal-triggered structural transformations, aggregation, and fibrillation of human alpha-synuclein. A possible molecular NK between Parkinson’s disease and heavy metal exposure. J. Biol. Chem. 2001, 276, 44284–44296. [Google Scholar] [CrossRef]
- Carboni, E.; Lingor, P. Insights on the interaction of alpha-synuclein and metals in the pathophysiology of Parkinson’s disease. Metallomics 2015, 7, 395–404. [Google Scholar] [CrossRef]
- Wang, H.; Mörman, C.; Sternke-Hoffmann, R.; Huang, C.Y.; Prota, A.; Ma, P.; Luo, J. Cu2+ ions modulate the interaction between α-synuclein and lipid membranes. J. Inorg. Biochem. 2022, 236, 111945. [Google Scholar] [CrossRef]
- Miotto, M.C.; Binolfi, A.; Zweckstetter, M.; Griesinger, C.; Fernández, C.O. Bioinorganic chemistry of synucleinopathies: Deciphering the binding features of Met motifs and His-50 in AS-Cu(I) interactions. J. Inorg. Biochem. 2014, 141, 208–211. [Google Scholar] [CrossRef]
- Davies, P.; Moualla, D.; Brown, D.R. Alpha-synuclein is a cellular ferrireductase. PLoS ONE 2011, 6, e15814. [Google Scholar] [CrossRef]
- Kienzl, E.; Jellinger, K.; Stachelberger, H.; Linert, W. Iron as catalyst for oxidative stress in the pathogenesis of Parkinson’s disease? Life Sci. 1999, 65, 1973–1976. [Google Scholar] [CrossRef]
- Sofic, E.; Riederer, P.; Heinsen, H.; Beckmann, H.; Reynolds, G.P.; Hebenstreit, G.; Youdim, M.B. Increased iron (III) and total iron content in post mortem substantia nigra of parkinsonian brain. J. Neural Transm. 1988, 74, 199–205. [Google Scholar] [CrossRef]
- McDowall, J.S.; Ntai, I.; Honeychurch, K.C.; Hart, J.P.; Colin, P.; Schneider, B.L.; Brown, D.R. Alpha-synuclein ferrireductase activity is detectible in vivo, is altered in Parkinson’s disease and increases the neurotoxicity of DOPAL. Mol. Cell. Neurosci. 2017, 85, 1–11. [Google Scholar] [CrossRef]
- Cahill, C.M.; Lahiri, D.K.; Huang, X.; Rogers, J.T. Amyloid precursor protein and alpha synuclein translation, implications for iron and inflammation in neurodegenerative diseases. Biochim. Biophys. Acta 2009, 1790, 615–628. [Google Scholar] [CrossRef]
- Wang, T.Y.; Ma, Z.; Wang, C.; Liu, C.; Yan, D.Y.; Deng, Y.; Liu, W.; Xu, Z.F.; Xu, B. Manganese-induced alpha-synuclein overexpression impairs synaptic vesicle fusion by disrupting the Rab3 cycle in primary cultured neurons. Toxicol. Lett. 2018, 285, 34–42. [Google Scholar] [CrossRef]
- Dugan, L.L.; Choi, D.W. Excitotoxicity, free radicals, and cell membrane changes. Ann. Neurol. 1994, 35 (Suppl. 1), S17–S21. [Google Scholar] [CrossRef]
- Koh, J.Y.; Suh, S.W.; Gwag, B.J.; He, Y.Y.; Hsu, C.Y.; Choi, D.W. The role of zinc in selective neuronal death after transient global cerebral ischemia. Science 1996, 272, 1013–1016. [Google Scholar] [CrossRef]
- Calderone, A.; Jover, T.; Mashiko, T.; Noh, K.M.; Tanaka, H.; Bennett, M.V.; Zukin, R.S. Late calcium EDTA rescues hippocampal CA1 neurons from global ischemia-induced death. J. Neurosci. 2004, 24, 9903–9913. [Google Scholar] [CrossRef]
- Kawahara, M.; Mizuno, D.; Koyama, H.; Konoha, K.; Ohkawara, S.; Sadakane, Y. Disruption of zinc homeostasis and the pathogenesis of senile dementia. Metallomics 2014, 6, 209–219. [Google Scholar] [CrossRef]
- Tanaka, K.I.; Kawahara, M. Copper enhances zinc-induced neurotoxicity and the endoplasmic reticulum stress response in a neuronal model of vascular dementia. Front. Neurosci. 2017, 11, 58. [Google Scholar] [CrossRef]
- Kawahara, M.; Tanaka, K.I.; Kato-Negishi, M. Crosstalk of copper and zinc in the pathogenesis of vascular dementia. J. Clin. Biochem. Nutr. 2022, 71, 7–15. [Google Scholar] [CrossRef]
- Del Prete, D.; Lombino, F.; Liu, X.; D’Adamio, L. APP is cleaved by Bace1 in pre-synaptic vesicles and establishes a pre-synaptic interactome, via its intracellular domain, with molecular complexes that regulate pre-synaptic vesicles functions. PLoS ONE 2014, 9, e108576. [Google Scholar] [CrossRef]
- Adle-Biassette, H.; Verney, C.; Peoc’h, K.; Dauge, M.C.; Razavi, F.; Choudat, L.; Gressens, P.; Budka, H.; Henin, D. Immunohistochemical expression of prion protein (PrPC) in the human forebrain during development. J. Neuropathol. Exp. Neurol. 2006, 65, 698–706. [Google Scholar] [CrossRef]
- Shen, J. Function and dysfunction of presenilin. Neurodegener. Dis. 2014, 13, 61–63. [Google Scholar] [CrossRef] [PubMed]
- Tzioras, M.; McGeachan, R.I.; Durrant, C.S.; Spires-Jones, T.L. Synaptic degeneration in Alzheimer disease. Nat. Rev. Neurol. 2023, 19, 19–38. [Google Scholar] [CrossRef] [PubMed]
- Roos, P.M.; Vesterberg, O.; Syversen, T.; Flaten, T.P.; Nordberg, M. Metal concentrations in cerebrospinal fluid and blood plasma from patients with amyotrophic lateral sclerosis. Biol. Trace Elem. Res. 2013, 151, 159–170. [Google Scholar] [CrossRef]
- Schikorski, T.; Stevens, C.F. Quantitative ultrastructural analysis of hippocampal excitatory synapses. J. Neurosci. 1997, 17, 5858–5867. [Google Scholar] [CrossRef]
- Vogt, K.; Mellor, J.; Tong, G.; Nicoll, R. The actions of synaptically released zinc at hippocampal mossy fiber synapses. Neuron 2000, 26, 187–196. [Google Scholar] [CrossRef] [PubMed]
- Kardos, J.; Kovács, I.; Hajós, F.; Kálmán, N.; Simonyi, M. Nerve endings from rat brain tissue release copper upon depolarization. A possible role in regulating neuronal excitability. Neurosci. Lett. 1989, 103, 139–144. [Google Scholar] [CrossRef]
- Hopt, A.; Korte, S.; Fink, H.; Panneal, U.; Niessner, R.; Jahn, R.; Kretzschmar, H.; Herms, J. Methods for studying synaptosomal copper release. J. Neurosci. Methods 2003, 128, 159–172. [Google Scholar] [CrossRef]
- Faria-Pereira, A.; Morais, V.A. Synapses: The brain’s energy-demanding sites. Int. J. Mol. Sci. 2022, 23, 3627. [Google Scholar] [CrossRef]
- Wu, L.J.; Leenders, A.G.M.; Cooperman, S.; Meyron-Holtz, E.; Smith, S.; Land, W.; Tsai, R.Y.L.; Berger, U.V.; Sheng, Z.H.; Rouault, T.A. Expression of the iron transporter ferroportin in synaptic vesicles and the blood-brain barrier. Brain Res. 2004, 1001, 108–117. [Google Scholar] [CrossRef]
- Zheng, W.; Xin, N.; Chi, Z.H.; Zhao, B.L.; Zhang, J.; Li, J.X.; Wang, Z.Y. Divalent metal transporter 1 is involved in amyloid precursor protein processing and Abeta generation. FASEB J. 2009, 23, 4207–4217. [Google Scholar] [CrossRef]
- Mellone, M.; Pelucchi, S.; Alberti, L.; Genazzani, A.A.; Di Luca, M.; Gardoni, F. Zinc transporter-1: A novel NMDA receptor-binding protein at the postsynaptic density. J. Neurochem. 2015, 132, 159–168. [Google Scholar] [CrossRef] [PubMed]
- De Benedictis, C.A.; Haffke, C.; Hagmeyer, S.; Sauer, A.K.; Grabrucker, A.M. Expression analysis of zinc transporters in nervous tissue cells reveals neuronal and synaptic localization of ZIP4. Int. J. Mol. Sci. 2021, 22, 4511. [Google Scholar] [CrossRef]
- Purro, S.A.; Nicoll, A.J.; Collinge, J. Prion protein as a toxic acceptor of amyloid ß oligomers. Biol. Psychiatry 2018, 83, 358–368. [Google Scholar] [CrossRef] [PubMed]
- Roberts, H.L.; Schneider, B.L.; Brown, D.R. α-Synuclein increases ß-amyloid secretion by promoting β-/γ-secretase processing of APP. PLoS ONE 2017, 12, e0171925. [Google Scholar] [CrossRef] [PubMed]
- Segal, D.; Ohana, E.; Besser, L.; Hershfinkel, M.; Moran, A.; Sekler, I. A role for ZnT-1 in regulating cellular cation influx. Biochem. Biophy. Res. Commun. 2004, 323, 1145–1150. [Google Scholar] [CrossRef] [PubMed]
- Koh, J.Y.; Lee, S.J. Metallothionein-3 as a multifunctional player in the control of cellular processes and diseases. Mol. Brain 2020, 13, 116. [Google Scholar] [CrossRef] [PubMed]
- Boldyrev, A.A.; Aldini, G.; Derave, W. Physiology and pathophysiology of carnosine. Physiol. Rev. 2013, 93, 1803–1845. [Google Scholar] [CrossRef] [PubMed]
- Bakardjiev, A. Carnosine and beta-alanine release is stimulated by glutamatergic receptors in cultured rat oligodendrocytes. Glia 1998, 24, 346–351. [Google Scholar]
- Wahby, M.M.; Mohammed, D.S.; Newairy, A.A.; Abdou, H.M.; Zaky, A. Aluminum-induced molecular neurodegeneration: The protective role of genistein and chickpea extract. Food Chem. Toxicol. 2017, 107, 57–67. [Google Scholar] [CrossRef]
- Praticò, D.; Uryu, K.; Sung, S.; Tang, S.; Trojanowski, J.Q.; Lee, V.M.Y. Aluminum modulates brain amyloidosis through oxidative stress in APP transgenic mice. FASEB J. 2002, 16, 1138–1140. [Google Scholar] [CrossRef]
- Brewer, G.J. Copper-2 hypothesis for causation of the current Alzheimer’s disease. Epidemic Together with dietary changes that enhance the epidemic. Chem. Res. Toxicol. 2017, 30, 763–768. [Google Scholar] [CrossRef]
- Xu, J.; Xu, G.; Fang, J. Association between serum copper and stroke risk factors in adults: Evidence from the National Health and Nutrition Examination Survey, 2011–2016. Biol. Trace Elem. Res. 2022, 200, 1089–1094. [Google Scholar] [CrossRef]
- Zhang, M.; Li, W.; Wang, Y.; Wang, T.; Ma, M.; Tian, C. Association between the change of serum copper and ischemic stroke: A sys tematic review and meta-analysis. J. Mol. Neurosci. 2020, 70, 475–480. [Google Scholar] [CrossRef] [PubMed]
- Arispe, N.; Rojas, E.; Pollard, H.B. Alzheimer disease amyloid ß protein forms calcium channels in bilayer membranes: Blockade by tromethamine and aluminum. Proc. Natl. Acad. Sci. USA 1993, 90, 567–571. [Google Scholar] [CrossRef]
- Lee, J.; Kim, Y.H.; Arce, F.T.; Gillman, A.G.; Jang, H.; Kagan, B.L.; Nussinov, B.; Yang, J.; Lal, R. Amyloid β ion channels in a membrane comprising brain total lipid extracts. ACS Chem. Neurosci. 2017, 8, 1348–1357. [Google Scholar] [CrossRef] [PubMed]
- Kawahara, M.; Ohtsuka, I.; Yokoyama, S.; Kato-Negishi, M.; Sadakane, Y. Membrane incorporation, channel formation, and disruption of calcium homeostasis by Alzheimer’s ß-amyloid protein. Int. J. Alzheimer Dis. 2011, 2011, 304583. [Google Scholar]
- Parodi, J.; Ormeño, D.; Ochoa-de la Paz, L.D. Amyloid pore-channel hypothesis: Effect of ethanol on aggregation state using frog oocytes for an Alzheimer’s disease study. BMB Rep. 2015, 48, 13–18. [Google Scholar] [CrossRef] [PubMed]
- Kourie, J.I.; Culverson, A. Prion peptide fragment PrP[106-126] forms distinct cation channel types. J. Neurosci. Res. 2000, 62, 120–133. [Google Scholar] [CrossRef]
- Lashuel, H.A.; Hartley, D.; Petre, B.M.; Walz, T.; Lansbury, P.T., Jr. Neurodegenerative disease: Amyloid pores from pathogenic mutations. Nature 2002, 418, 291. [Google Scholar] [CrossRef]
- Chen, W.T.; Liao, Y.H.; Yu, H.M.; Cheng, I.H.; Chen, Y.R. Distinct effects of Zn2+, Cu2+, Fe3+, and Al3+ on amyloid-beta stability, oligomerization, and aggregation: Amyloid-beta destabilization promotes annular protofibril formation. J. Biol. Chem. 2011, 286, 9646–9656. [Google Scholar] [CrossRef]
- Sharma, A.K.; Pavlova, S.T.; Kim, J.; Kim, J.; Mirica, L.M. The effect of Cu(2+) and Zn(2+) on the Aβ42 peptide aggregation and cellular toxicity. Metallomics 2013, 5, 1529–1536. [Google Scholar] [CrossRef]
- Bolognin, S.; Zatta, P.; Lorenzetto, E.; Valenti, M.T.; Buffelli, M. β-Amyloid-aluminum complex alters cytoskeletal stability and increases ROS production in cortical neurons. Neurochem. Int. 2013, 62, 566–574. [Google Scholar] [CrossRef] [PubMed]
- Faux, N.G.; Ritchie, C.W.; Gunn, A.; Rembach, A.; Tsatsanis, A.; Bedo, J.; Harrison, J.; Lannfelt, L.; Blennow, K.; Zetterberg, H.; et al. PBT2 rapidly improves cognition in Alzheimer’s Disease: Additional phase II analyses. J. Alzheimers Dis. 2010, 20, 509–516. [Google Scholar] [CrossRef] [PubMed]
- Sampson, E.L.; Jenagaratnam, L.; McShane, R. Metal protein attenuating compounds for the treatment of Alzheimer’s dementia. Cochrane Database Syst. Rev. 2014, 2, CD005380. [Google Scholar] [CrossRef] [PubMed]
- Davenward, S.; Bentham, P.; Wright, J.; Crome, P.; Job, D.; Polwart, A.; Exley, C. Silicon-rich mineral water as a non-invasive test of the ’aluminum hypothesis’ in Alzheimer’s disease. J. Alzheimers Dis. 2013, 33, 423–430. [Google Scholar] [CrossRef]
- McLachlan, D.R.C.; Dalton, A.J.; Kruck, T.P.; Bell, M.Y.; Smith, W.L.; Kalow, W.; Andrews, D.F. Intramuscular desferrioxamine in patients with Alzheimer’s disease. Lancet 1991, 337, 1304–1308. [Google Scholar] [CrossRef]
- Percy, M.E.; Kruck, T.P.A.; Pogue, A.I.; Lukiw, W.J. Towards the prevention of potential aluminum toxic effects and an effective treatment for Alzheimer’s disease. J. Inorg. Biochem. 2011, 105, 1505–1512. [Google Scholar] [CrossRef] [PubMed]
- Farr, A.C.; Xiong, M.P. Challenges and opportunities of deferoxamine delivery for treatment of Alzheimer’s disease, Parkinson’s disease, and intracerebral hemorrhage. Mol. Pharm. 2021, 18, 593–609. [Google Scholar] [CrossRef]
- Levi, S.; Volonté, M.A. Iron chelation in early Parkinson’s disease. Lancet Neurol. 2023, 22, 290–291. [Google Scholar] [CrossRef] [PubMed]
- Thapa, K.; Khan, H.; Kanojia, N.; Singh, T.G.; Kaur, A.; Kaur, G. Therapeutic insights on ferroptosis in Parkinson’s disease. Eur. J. Pharmacol. 2022, 930, 175133. [Google Scholar] [CrossRef] [PubMed]
- Bareggi, S.R.; Braida, D.; Pollera, C.; Bondiolotti, G.; Formentin, E.; Puricelli, M.; Poli, G.; Ponti, W.; Sala, M. Effects of clioquinol on memory impairment and the neurochemical modifications induced by scrapie infection in golden hamsters. Brain Res. 2009, 1280, 195–200. [Google Scholar] [CrossRef] [PubMed]
- Sigurdsson, E.M.; Brown, D.R.; Alim, M.A.; Scholtzova, H.; Carp, R.; Meeker, H.C.; Prelli, F.; Frangione, B.; Wisniewski, T. Copper chelation delays the onset of prion disease. J. Biol. Chem. 2003, 278, 46199–46202. [Google Scholar] [CrossRef]
- Kawahara, M.; Tanaka, K.-I.; Kato-Negishi, M. Zinc, Carnosine, and Neurodegenerative Diseases. Nutrients 2018, 10, 147. [Google Scholar] [CrossRef]
- Corona, C.; Frazzini, V.; Silvestri, E.; Lattanzio, R.; La Sorda, R.; Piantelli, M.; Canzoniero, L.M.; Ciavardelli, D.; Rizzarelli, E.; Sensi, S.L. Effects of dietary supplementation of carnosine on mitochondrial dysfunction, amyloid pathology, and cognitive deficits in 3xTg-AD mice. PLoS ONE 2011, 6, e17971. [Google Scholar] [CrossRef]
- Davis, C.K.; Laud, P.J.; Bahor, Z.; Rajanikant, G.K.; Majid, A. Systematic review and stratified meta-analysis of the efficacy of carnosine in animal models of ischemic stroke. J. Cereb. Blood Flow Metab. 2016, 36, 1686–1694. [Google Scholar] [CrossRef] [PubMed]
- Kawahara, M.; Konoha, K.; Nagata, T.; Sadakane, Y. Drugs for prevention or treatment of vascular dementia. Patent No. JP5382633, 11 October 2013. [Google Scholar]
- Kawahara, M.; Kato-Negishi, M.; Tanaka, K.-I. Cross talk between neurometals and amyloidogenic proteins at the synapse and the pathogenesis of neurodegenerative diseases. Metallomics 2017, 9, 619–633. [Google Scholar] [CrossRef] [PubMed]

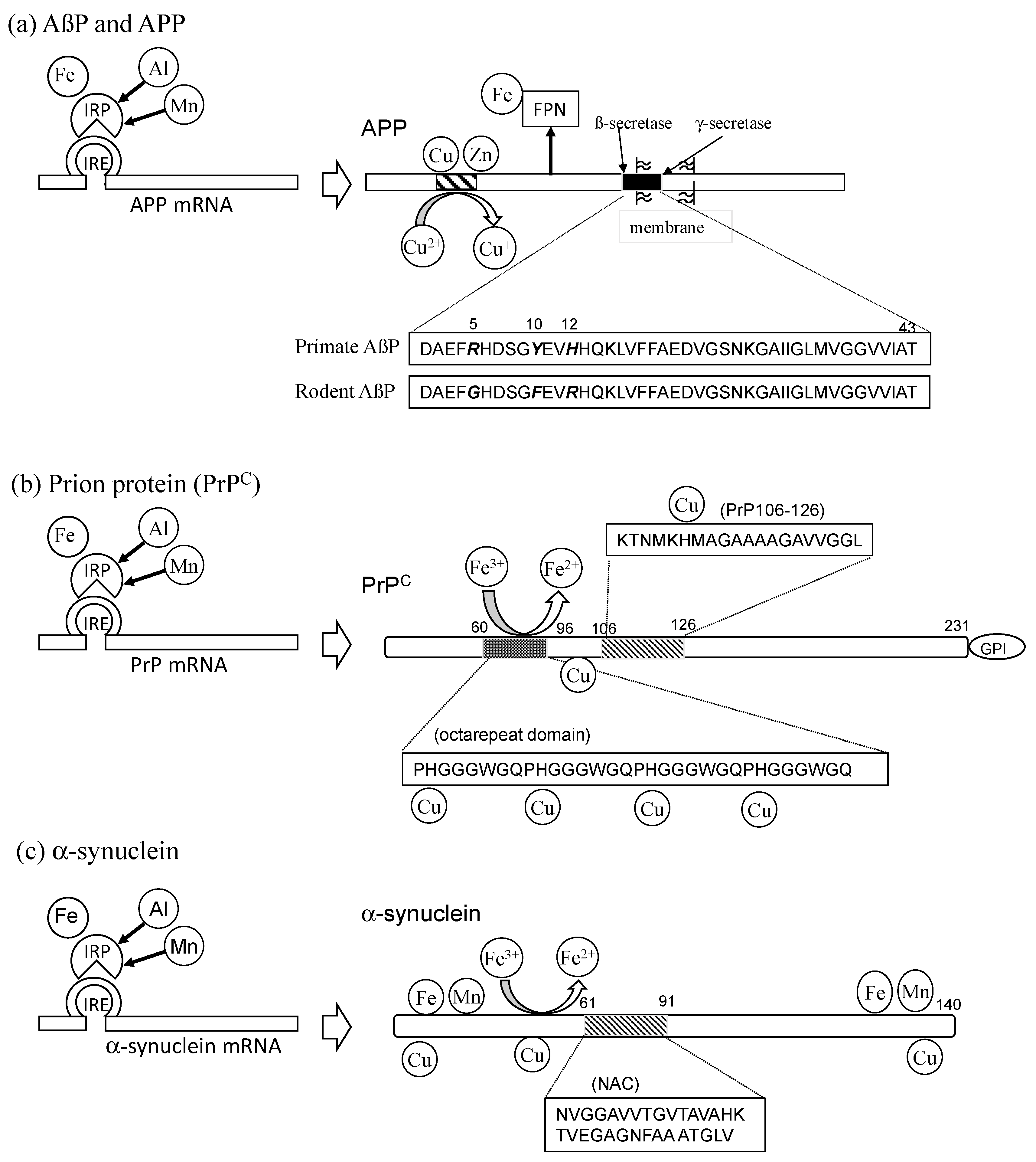
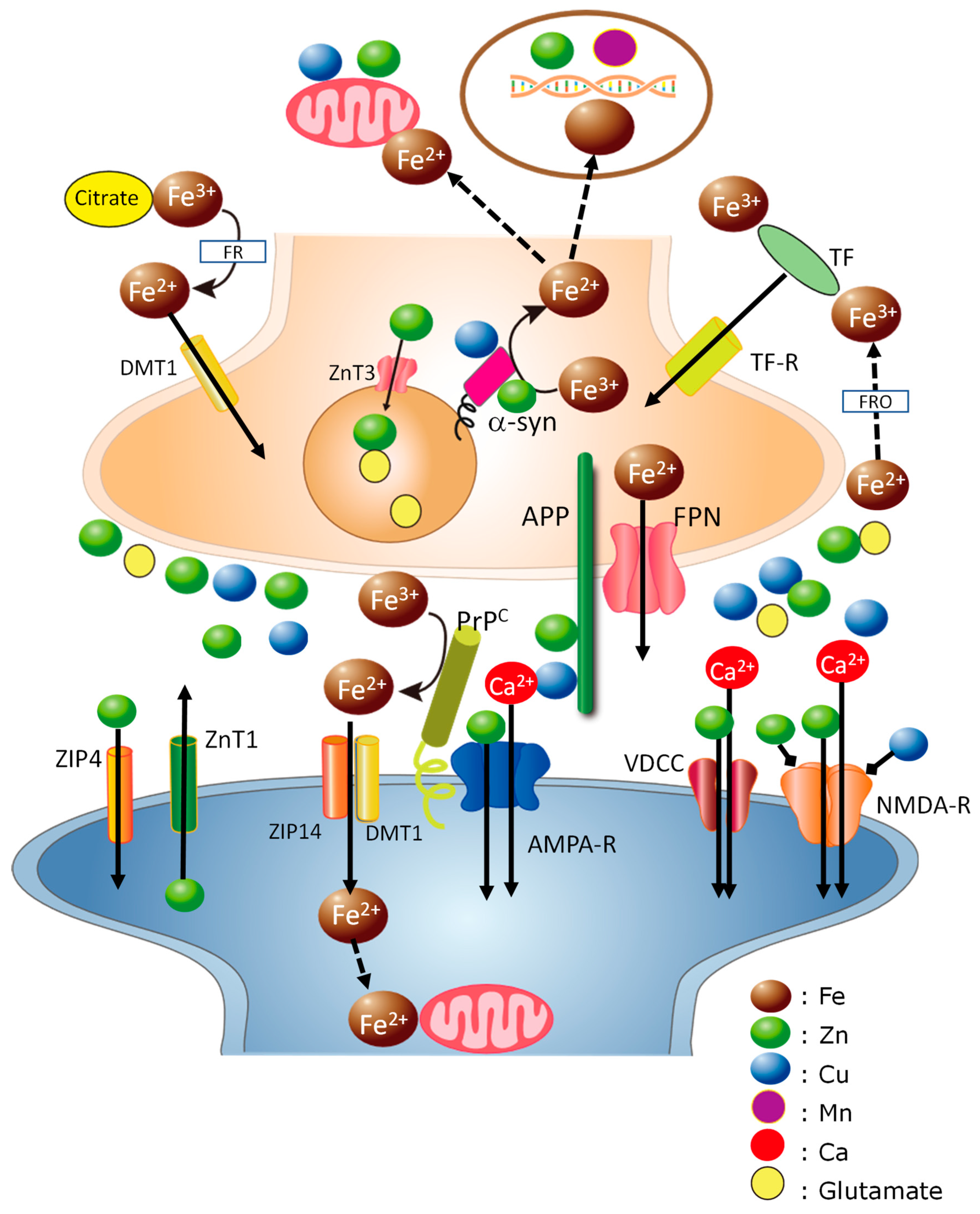
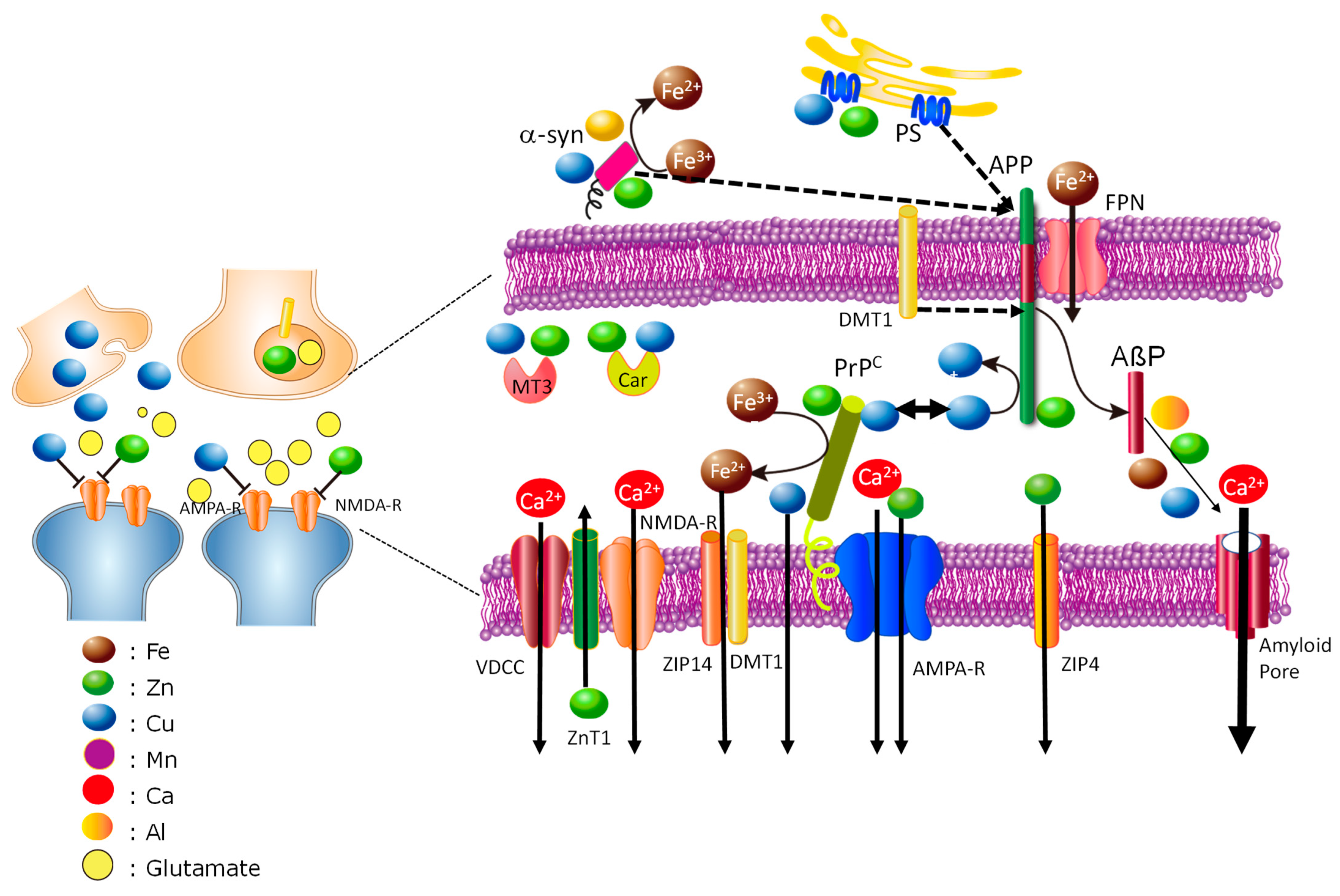
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kawahara, M.; Kato-Negishi, M.; Tanaka, K.-i. Dietary Trace Elements and the Pathogenesis of Neurodegenerative Diseases. Nutrients 2023, 15, 2067. https://doi.org/10.3390/nu15092067
Kawahara M, Kato-Negishi M, Tanaka K-i. Dietary Trace Elements and the Pathogenesis of Neurodegenerative Diseases. Nutrients. 2023; 15(9):2067. https://doi.org/10.3390/nu15092067
Chicago/Turabian StyleKawahara, Masahiro, Midori Kato-Negishi, and Ken-ichiro Tanaka. 2023. "Dietary Trace Elements and the Pathogenesis of Neurodegenerative Diseases" Nutrients 15, no. 9: 2067. https://doi.org/10.3390/nu15092067
APA StyleKawahara, M., Kato-Negishi, M., & Tanaka, K.-i. (2023). Dietary Trace Elements and the Pathogenesis of Neurodegenerative Diseases. Nutrients, 15(9), 2067. https://doi.org/10.3390/nu15092067