Long-Term Management of Patients with Mild Urea Cycle Disorders Identified through the Newborn Screening: An Expert Opinion for Clinical Practice

 ,
, 

 ,
,  and
and
Abstract
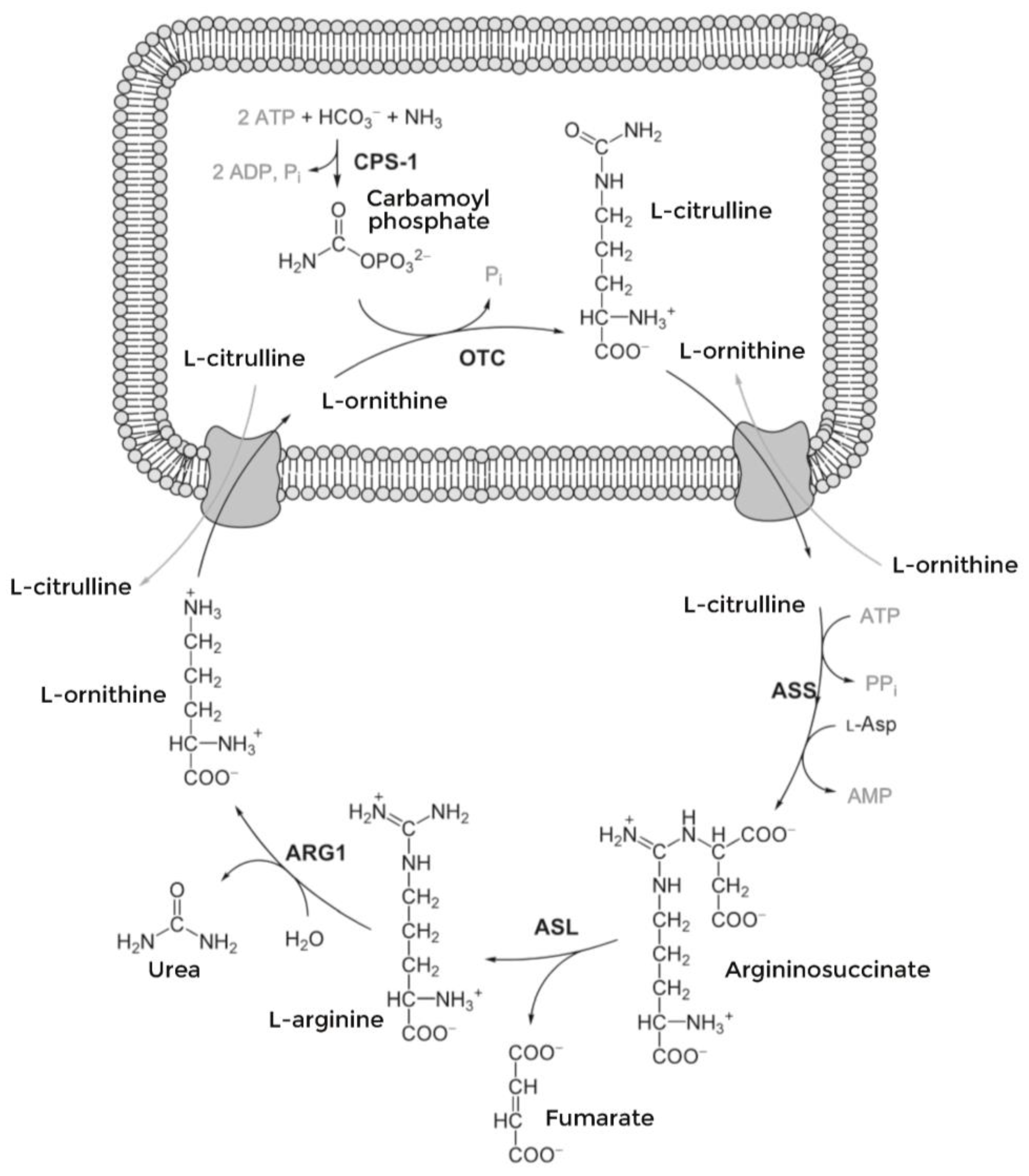
:1. Introduction
2. Methods
2.1. Panel Selection
2.2. Design
3. Results
3.1. The Role of Expanded Newborn Screening in Distal UCDs
3.2. Biochemical Assessment
3.3. Role of Genetic Testing
3.4. Treatment and Follow-Up

{kind=link}
{kind=link}
{kind=link}
| Glycerol Phenylbutyrate | Sodium Phenylbutyrate | Sodium Benzoate/Sodium Phenylbutyrate | |
|---|---|---|---|
| Registered | Yes | Yes | No |
| Indication | Deficiencies of CPS1, OTC, ASS, ASL, ARG and HHH | Deficiencies of CPS1, OTC or ASS | Not available |
| Age | Children and adults, any age | Children and adults, any age | Not available |
| Pharmaceutical form | Oral liquid 1.1 g/mL | Tablets, granules | Many presentations |
| Daily dose | 4.5 mL/m2/day to 11.2 mL/m2/day | 450–600 mg/kg/day in children weighing < 20 kg 9.9–13.0 g/m2/day in children weighing > 20 kg, adolescents and adults | Not available |
| Method of administration | With meals directly into the mouth Possible use of naso-gastric tube | With meals, mix with liquids Possible use of naso-gastric tube | |
| Sodium content | None | 62 mg/tablet 124 mg/g | Yes, variable with different presentations |
| Clinical efficacy | Study 1 (44 adults, controlled, non-inferiority, cross-over, 4 weeks): vs. sodium phenylbutyrate. Results: non-inferior for 24 h AUC for ammonia Study 2 (51 adults, uncontrolled, open, 12 months). Results: Mean fasting venous ammonia values were normal Study 3 and 4 (pediatric patient, open, switch from sodium phenylbutyrate, 14 and 10 days). Results: non-inferior for ammonia control Long-term pediatric studies (49 pediatric patients, part of study 1 and extension of studies 3 and 4, 12 months). Results: mean fasting venous ammonia within limits Study 5 (45 pediatric patients, open, up to 5.9 years). Results: mean fasting venous ammonia within limits at 24 months Study under 2 months age (16 patients, uncontrolled, open, 24 months, naïve patients or switch from sodium phenylbutyrate). Results: successful transition in 3 days, mean normalized venous ammonia values were normal in the long term Study 2 months to 2 years (10 patients, uncontrolled, open, 24 months). Results: nine successful transitions in 4 days | See studies vs. glycerol phenylbutyrate | Not available |
| Elimination | Metabolized by pancreatic enzymes; metabolites are eliminated in the urine; 80–100% of the medicinal product is excreted by the kidneys within 24 h as phenylacetylglutamine | Metabolized to phenylacetate that forms phenylacetylglutamine in liver and kidney; 80–100% of the medicinal product is excreted by the kidneys within 24 h as phenylacetylglutamine | Converted to benzoic acid. The individual maximum rate of metabolism can be estimated from the urinary excretion rate of hippuric acid 1.5 to 3 h after the single oral dose |
4. Discussion and Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Acknowledgments
Conflicts of Interest
References
- Ah Mew, N.; Simpson, K.L.; Gropman, A.L.; Lanpher, B.C.; Chapman, K.A.; Summar, M.L. Urea Cycle Disorders Overview. In GeneReviews; Adam, M.P., Mirzaa, G.M., Pagon, R.A., Wallace, S.E., Bean, L.J.H., Gripp, K.W., Amemiya, A., Eds.; University of Washington: Seattle, WA, USA, 2003; pp. 1993–2023. [Google Scholar]
- Ruoppolo, M.; Malvagia, S.; Boenzi, S.; Carducci, C.; Dionisi-Vici, C.; Teofoli, F.; Burlina, A.; Angeloni, A.; Aronica, T.; Bordugo, A.; et al. Expanded Newborn Screening in Italy Using Tandem Mass Spectrometry: Two Years of National Experience. Int. J. Neonatal Screen. 2022, 8, 47. [Google Scholar] [CrossRef] [PubMed]
- Bin Sawad, A.; Jackimiec, J.; Bechter, M.; Trucillo, A.; Lindsley, K.; Bhagat, A.; Uyei, J.; Diaz, G.A. Epidemiology, methods of diagnosis, and clinical management of patients with arginase 1 deficiency (ARG1-D): A systematic review. Mol. Genet. Metab. 2022, 137, 153–163. [Google Scholar] [CrossRef] [PubMed]
- Dionisi-Vici, C.; Rizzo, C.; Burlina, A.B.; Caruso, U.; Sabetta, G.; Uziel, G.; Abeni, D. Inborn errors of metabolism in the Italian pediatric population: A national retrospective survey. J. Pediatr. 2002, 140, 321–327. [Google Scholar] [CrossRef] [PubMed]
- Taruscio, D.; Taruscio, D.; Piccioli, A.; Piccioli, A. Newborn screening in Italy: A unique program of public health in Europe. Ann Ist Super Sanita 2023, 59, 1–3. [Google Scholar] [CrossRef] [PubMed]
- Häberle, J.; Burlina, A.; Chakrapani, A.; Dixon, M.; Karall, D.; Lindner, M.; Mandel, H.; Martinelli, D.; Pintos-Morell, G.; Santer, R.; et al. Suggested guidelines for the diagnosis and management of urea cycle disorders: First revision. J. Inherit. Metab. Dis. 2019, 42, 1192–1230. [Google Scholar] [CrossRef] [PubMed]
- Gardeitchik, T.; Humphrey, M.; Nation, J.; Boneh, A. Early Clinical Manifestations and Eating Patterns in Patients with Urea Cycle Disorders. J. Pediatr. 2012, 161, 328–332. [Google Scholar] [CrossRef]
- Gallego, D.; Bueno, S. Exploring the application of the Delphi method as a forecasting tool in Information Systems and Tech-nologies research. Technol. Anal. Strateg. Manag. 2014, 26, 987–999. [Google Scholar] [CrossRef]
- Gustafson, D.H.; Shukla, R.K.; Delbecq, A.; Walster, G. A comparative study of differences in subjective likelihood estimates made by individuals, interacting groups, Delphi groups, and nominal groups. Organ. Behav. Hum. Perform. 1973, 9, 280–291. [Google Scholar] [CrossRef]
- Candiani, G.; Colombo, C.; Daghini, R.; Magrini, N. Sistema Nazionale per le Linee Guida. National System for Guidelines; Zadig Editore: Milano, Italy, 2019. [Google Scholar]
- Wilson, J.M.G.; Jungner, G.; World Health Organization. Principles and Practice of Screening for Disease; WHO: Geneva, Switzerland, 1968. Available online: https://apps.who.int/iris/handle/10665/37650 (accessed on 12 December 2023).
- Batshaw, M.L.; Berry, G.T. Use of citrulline as a diagnostic marker in the prospective treatment of urea cycle disorders. J. Pediatr. 1991, 118, 914–917. [Google Scholar] [CrossRef]
- Trinh, M.-U.; Blake, J.; Harrison, J.R.; Gerace, R.; Ranieri, E.; Fletcher, J.M.; Johnson, D.W. Quantification of Glutamine in Dried Blood Spots and Plasma by Tandem Mass Spectrometry for the Biochemical Diagnosis and Monitoring of Ornithine Transcarbamylase Deficiency. Clin. Chem. 2003, 49, 681–684. [Google Scholar] [CrossRef]
- Cavicchi, C.; Malvagia, S.; la Marca, G.; Gasperini, S.; Donati, M.; Zammarchi, E.; Guerrini, R.; Morrone, A.; Pasquini, E. Hypocitrullinemia in expanded newborn screening by LC–MS/MS is not a reliable marker for ornithine transcarbamylase deficiency. J. Pharm. Biomed. Anal. 2009, 49, 1292–1295. [Google Scholar] [CrossRef] [PubMed]
- Hall, P.L.; Wittenauer, A.L.; Wilcox, W.R. Proximal urea cycle defects are challenging to detect with newborn screening: Results of a prospective pilot study using post-analytical tools. Am. J. Med. Genet. Part C Semin. Med. Genet. 2022, 190, 178–186. [Google Scholar] [CrossRef] [PubMed]
- Siri, B.; Olivieri, G.; Angeloni, A.; Cairoli, S.; Carducci, C.; Cotugno, G.; Di Michele, S.; Giovanniello, T.; La Marca, G.; Lepri, F.R.; et al. The diagnostic challenge of mild citrulline elevation at newborn screening. Mol. Genet. Metab. 2022, 135, 327–332. [Google Scholar] [CrossRef]
- Mercimek-Mahmutoglu, S.; Moeslinger, D.; Häberle, J.; Engel, K.; Herle, M.; Strobl, M.; Scheibenreiter, S.; Muehl, A.; Stöckler-Ipsiroglu, S. Long-term outcome of patients with argininosuccinate lyase deficiency diagnosed by newborn screening in Austria. Mol. Genet. Metab. 2010, 100, 24–28. [Google Scholar] [CrossRef] [PubMed]
- Engel, K.; Höhne, W.; Häberle, J. Mutations and polymorphisms in the human argininosuccinate synthetase (ASS1) gene. Hum. Mutat. 2009, 30, 300–307. [Google Scholar] [CrossRef] [PubMed]
- Diez-Fernandez, C.; Rüfenacht, V.; Häberle, J. Mutations in the Human Argininosuccinate Synthetase (ASS1) Gene, Impact on Patients, Common Changes, and Structural Considerations. Hum. Mutat. 2017, 38, 471–484. [Google Scholar] [CrossRef] [PubMed]
- Diez-Fernandez, C.; Rüfenacht, V.; Gemperle, C.; Fingerhut, R.; Häberle, J. Mutations and common variants in the human arginase 1 (ARG1) gene: Impact on patients, diagnostics, and protein structure considerations. Hum. Mutat. 2018, 39, 1029–1050. [Google Scholar] [CrossRef]
- Posset, R.; Zielonka, M.; Gleich, F.; Garbade, S.F.; Hoffmann, G.F.; Kölker, S.; Urea Cycle Disorders Consortium (UCDC); European Registry and Network for Intoxication Type Metabolic Diseases (E-IMD) Consortia Study Group. The challenge of understanding and predicting phenotypic diversity in urea cycle disorders. J. Inherit. Metab. Dis. 2023, 46, 1007–1016. [Google Scholar] [CrossRef]
- WHO Technical Report Series. Protein and Amino Acid Requirement in Human Nutrition; World Health Organization: Geneva, Switzerland, 2007.
- Siddiq, S.; Wilson, B.J.; Graham, I.D.; Lamoureux, M.; Khangura, S.D.; Tingley, K.; Tessier, L.; Chakraborty, P.; Coyle, D.; Canadian Inherited Metabolic Diseases Research Network (CIMDRN); et al. Experiences of caregivers of children with inherited metabolic diseases: A qualitative study. Orphanet. J. Rare Dis. 2016, 11, 168. [Google Scholar] [CrossRef]
- Häberle, J.; Pauli, S.; Schmidt, E.; Schulze-Eilfing, B.; Berning, C.; Koch, H.G. Mild citrullinemia in Caucasians is an allelic variant of argininosuccinate synthetase deficiency (citrullinemia type 1). Mol. Genet. Metab. 2003, 80, 302–306. [Google Scholar] [CrossRef]
- Fabre, A.; Baumstarck, K.; Cano, A.; Loundou, A.; Berbis, J.; Chabrol, B.; Auquier, P. Assessment of quality of life of the children and parents affected by inborn errors of metabolism with restricted diet: Preliminary results of a cross-sectional study. Healt Qual. Life Outcomes 2013, 11, 158. [Google Scholar] [CrossRef] [PubMed]
- Batshaw, M.L.; Brusilow, S.; Waber, L.; Blom, W.; Brubakk, A.M.; Burton, B.K.; Cann, H.M.; Kerr, D.; Mamunes, P.; Matalon, R.; et al. Treatment of inborn errors of urea synthesis: Activation of alternative pathways of waste nitrogen synthesis and excretion. N. Engl. J. Med. 1982, 306, 1387–1392. [Google Scholar] [CrossRef] [PubMed]
- Yeo, M.; Rehsi, P.; Dorman, M.; Grunewald, S.; Baruteau, J.; Chakrapani, A.; Footitt, E.; Prunty, H.; McSweeney, M. Direct replacement of oral sodium benzoate with glycerol phenylbutyrate in children with urea cycle disorders. JIMD Rep. 2022, 63, 137–145. [Google Scholar] [CrossRef] [PubMed]
- Ammonaps SmPC. Available online: https://www.ema.europa.eu/en/medicines/human/EPAR/ammonaps (accessed on 10 December 2023).
- Pherurane SmPC. Available online: https://www.accessdata.fda.gov/drugsatfda_docs/label/2022/216513s000lbl.pdf (accessed on 10 December 2023).
- Martín-Hernández, E.; Quijada-Fraile, P.; Correcher, P.; Meavilla, S.; Sánchez-Pintos, P.; Montero, J.d.L.H.; Blasco-Alonso, J.; Dougherty, L.; Marquez, A.; Peña-Quintana, L.; et al. Switching to Glycerol Phenylbutyrate in 48 Patients with Urea Cycle Disorders: Clinical Experience in Spain. J. Clin. Med. 2022, 11, 5045. [Google Scholar] [CrossRef] [PubMed]
- Ravicti. EMA Summary of Product Characteristics. Available online: www.ema.europa.eu/en/documents/product-information/ravicti-epar-product-information_it.pdf (accessed on 13 December 2023).
- Diaz, G.A.; Krivitzky, L.S.; Mokhtarani, M.; Rhead, W.; Bartley, J.; Feigenbaum, A.; Longo, N.; Berquist, W.; Berry, S.A.; Gallagher, R.; et al. Ammonia control and neurocognitive outcome among urea cycle disorder patients treated with glycerol phenylbutyrate. Hepatology 2013, 57, 2171–2179. [Google Scholar] [CrossRef]
- Yeowell, G.; Burns, D.S.; Fatoye, F. The burden of pharmacological treatment on health-related quality of life in people with a urea cycle disorder: A qualitative study. J. Patient-Rep. Outcomes 2021, 5, 110. [Google Scholar] [CrossRef]
- Yeo, M.; Rehsi, P.; Dorman, M.; Grunewald, S.; Baruteau, J.; Chakrapani, A.; Footitt, E.; Prunty, H.; McSweeney, M. Clinical experience with glycerol phenylbutyrate in 20 patients with urea cycle disorders at a UK paediatric centre. J. Inherit. Metab. Dis. Rep. 2023, 64, 317–326. [Google Scholar] [CrossRef]
- ACMG ACT Sheets and Algorithms. Bethesda (MD): American College of Medical Genetics and Genomics. 2001. Available online: www.ncbi.nlm.nih.gov/books/NBK55832/ (accessed on 13 December 2023).
- Barends, M.; Pitt, J.; Morrissy, S.; Tzanakos, N.; Boneh, A.; Newborn Screening Laboratory Staff. Biochemical and molecular characteristics of patients with organic acidaemias and urea cycle disorders identified through newborn screening. Mol. Genet. Metab. 2014, 113, 46–52. [Google Scholar] [CrossRef]
- Zielonka, M.; Kölker, S.; Gleich, F.; Stützenberger, N.; Nagamani, S.C.S.; Gropman, A.L.; Hoffmann, G.F.; Garbade, S.F.; Posset, R.; Urea Cycle Disorders Consortium (UCDC); et al. Early prediction of phenotypic severity in Citrullinemia Type 1. Ann. Clin. Transl. Neurol. 2019, 6, 1858–1871. [Google Scholar] [CrossRef]
- Posset, R.; Garbade, S.F.; Gleich, F.; Gropman, A.L.; de Lonlay, P.; Hoffmann, G.F.; Garcia-Cazorla, A.; Nagamani, S.C.S.; Baumgartner, M.R.; Schulze, A.; et al. Long-term effects of medical management on growth and weight in individuals with urea cycle disorders. Sci. Rep. 2020, 10, 11948. [Google Scholar] [CrossRef]
- Berning, C.; Bieger, I.; Pauli, S.; Vermeulen, T.; Vogl, T.; Rummel, T.; Höhne, W.; Koch, H.G.; Rolinski, B.; Gempel, K.; et al. Investigation of citrullinemia type I variants by in vitro expression studies. Hum. Mutat. 2008, 29, 1222–1227. [Google Scholar] [CrossRef] [PubMed]
- Urea Cycle Disorders Consortium. Available online: https://www1.rarediseasesnetwork.org/cms/UCDC (accessed on 10 December 2023).
- European Registry and Network for Intoxication Type Metabolic Diseases. Available online: https://www.e-imd.org/ (accessed on 12 December 2023).
- Posset, R.; Gropman, A.L.; Nagamani, S.C.S.; Burrage, L.C.; Bedoyan, J.K.; Wong, D.; Berry, G.T.; Baumgartner, M.R.; Yudkoff, M.; Zielonka, M.; et al. Impact of Diagnosis and Therapy on Cognitive Function in Urea Cycle Disorders. Ann. Neurol. 2019, 86, 116–128. [Google Scholar] [CrossRef] [PubMed]
- Ficicioglu, C.; Mandell, R.; Shih, V. Argininosuccinate lyase deficiency: Longterm outcome of 13 patients detected by newborn screening. Mol. Genet. Metab. 2009, 98, 273–277. [Google Scholar] [CrossRef] [PubMed]


| Final Items | Final Statements | Score (%) | Degree of Consensus (%) | ||||
|---|---|---|---|---|---|---|---|
| Score 1 | Score 2 | Score 3 | Score 4 | Score 5 | |||
| The role of expanded newborn screening in UCDs |
| 0 | 0 | 0 | 12.5 | 87.5 | 100 |
| 0 | 0 | 0 | 50 | 50 | 100 | |
| 0 | 0 | 0 | 0 | 100 | 100 | |
| 0 | 0 | 12.5 | 25 | 62.5 | 87.5 | |
| Clinical genotype/phenotype correlation |
| 0 | 0 | 0 | 25 | 75 | 100 |
| 0 | 0 | 0 | 0 | 100 | 100 | |
| 0 | 12.5 | 0 | 12.5 | 75 | 87.5 | |
| 0 | 0 | 12.5 | 12.5 | 75 | 87.5 | |
| 0 | 0 | 0 | 37.5 | 62.5 | 100 | |
| Diet therapy |
| 0 | 0 | 0 | 0 | 100 | 100 |
| 0 | 0 | 0 | 25 | 75 | 100 | |
| 0 | 0 | 0 | 25 | 75 | 100 | |
| 0 | 0 | 0 | 12.5 | 87.5 | 100 | |
| 0 | 0 | 0 | 0 | 100 | 100 | |
| 0 | 0 | 0 | 0 | 100 | 100 | |
| Pharmacological treatment |
| 0 | 0 | 0 | 12.5 | 87.5 | 100 |
| 0 | 0 | 12.5 | 0 | 87.5 | 87.5 | |
| 0 | 0 | 0 | 0 | 100 | 100 | |
| Follow-up |
| 0 | 0 | 0 | 25 | 75 | 100 |
| 0 | 0 | 0 | 12.5 | 87.5 | 100 | |
| 0 | 0 | 0 | 12.5 | 87.5 | 100 | |
| 0 | 0 | 12.5 | 12.5 | 75 | 87.5 | |
| 0 | 0 | 0 | 50 | 50 | 100 | |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Burlina, A.; Gasperini, S.; la Marca, G.; Pession, A.; Siri, B.; Spada, M.; Ruoppolo, M.; Tummolo, A. Long-Term Management of Patients with Mild Urea Cycle Disorders Identified through the Newborn Screening: An Expert Opinion for Clinical Practice. Nutrients 2024, 16, 13. https://doi.org/10.3390/nu16010013
Burlina A, Gasperini S, la Marca G, Pession A, Siri B, Spada M, Ruoppolo M, Tummolo A. Long-Term Management of Patients with Mild Urea Cycle Disorders Identified through the Newborn Screening: An Expert Opinion for Clinical Practice. Nutrients. 2024; 16(1):13. https://doi.org/10.3390/nu16010013
Chicago/Turabian StyleBurlina, Alberto, Serena Gasperini, Giancarlo la Marca, Andrea Pession, Barbara Siri, Marco Spada, Margherita Ruoppolo, and Albina Tummolo. 2024. "Long-Term Management of Patients with Mild Urea Cycle Disorders Identified through the Newborn Screening: An Expert Opinion for Clinical Practice" Nutrients 16, no. 1: 13. https://doi.org/10.3390/nu16010013
APA StyleBurlina, A., Gasperini, S., la Marca, G., Pession, A., Siri, B., Spada, M., Ruoppolo, M., & Tummolo, A. (2024). Long-Term Management of Patients with Mild Urea Cycle Disorders Identified through the Newborn Screening: An Expert Opinion for Clinical Practice. Nutrients, 16(1), 13. https://doi.org/10.3390/nu16010013