
GPER/PKA-Dependent Enhancement of Hormone-Sensitive Lipase Phosphorylation in 3T3-L1 Adipocytes by Piceatannol

 ,
, {kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Materials
2.2. Cell Cultures
2.3. Western Blotting
2.4. Oil-Red O Staining
2.5. Measurement of Intracellular Triglyceride Concentration
2.6. Statistics
3. Results
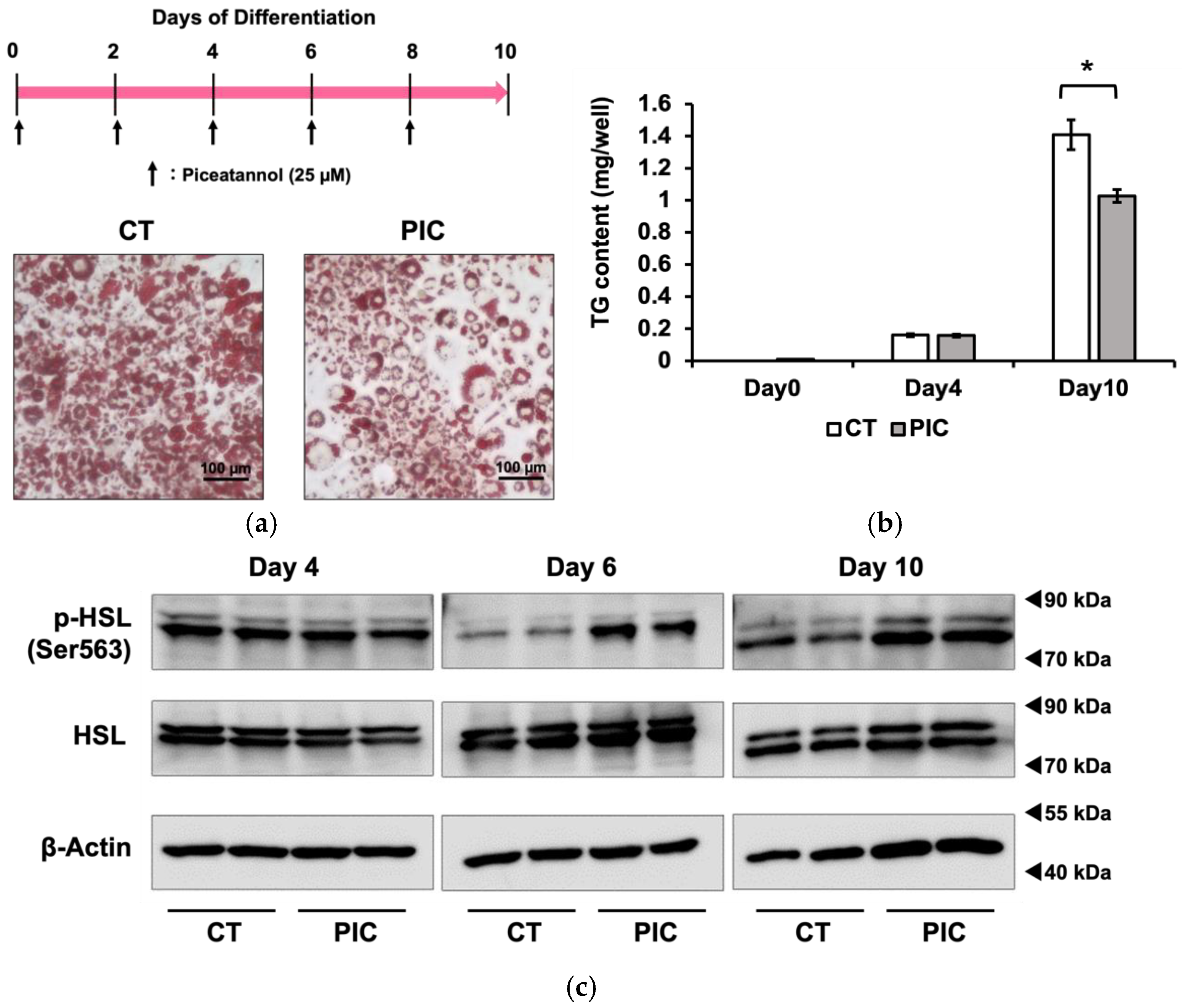
3.1. Inhibition Effects of PIC on Fat Accumulation in 3T3-L1 Adipocytes
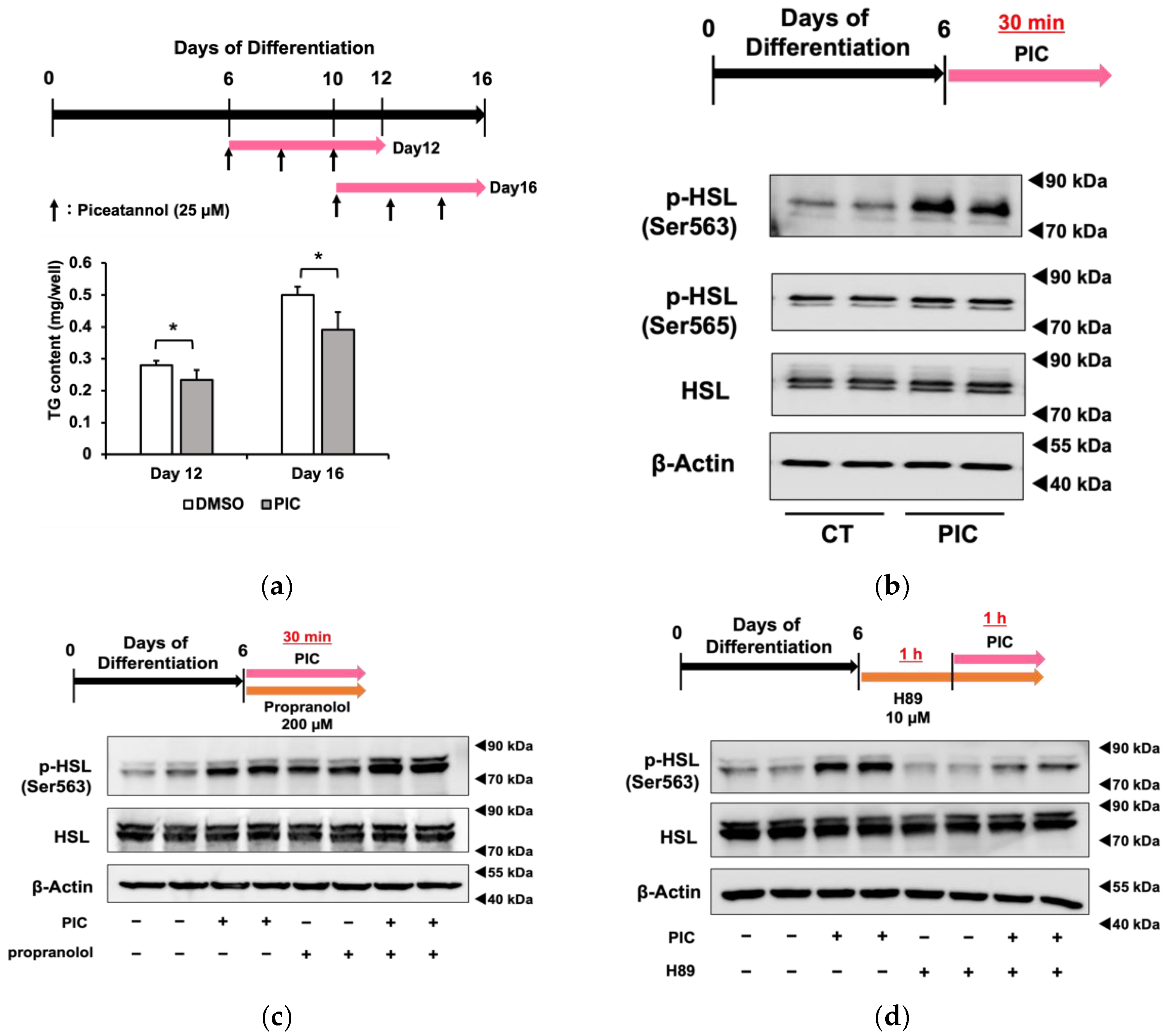
3.2. The Phosphorylation of HSL by PIC Is Regulated Independently of β-Adrenergic Receptors
3.3. Piceatannol Phosphorylates HSL via the GPER Pathway
3.4. PIC Inhibits Akt Phosphorylation
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Tella, S.H.; Gallagher, J.C. Prevention and Treatment of Postmenopausal Osteoporosis. J. Steroid Biochem. Mol. Biol. 2014, 142, 155–170. [Google Scholar] [CrossRef] [PubMed]
- Mauvais-Jarvis, F.; Clegg, D.J.; Hevener, A.L. The Role of Estrogens in Control of Energy Balance and Glucose Homeostasis. Endocr. Rev. 2013, 34, 309–338. [Google Scholar] [CrossRef] [PubMed]
- Maric-Bilkan, C.; Gilbert, E.L.; Ryan, M.J. Impact of Ovarian Function on Cardiovascular Health in Women: Focus on Hypertension. Int. J. Womens Health 2014, 6, 131–139. [Google Scholar] [CrossRef] [PubMed]
- Clegg, D.J. Minireview: The Year in Review of Estrogen Regulation of Metabolism. Mol. Endocrinol. 2012, 26, 1957–1960. [Google Scholar] [CrossRef] [PubMed]
- Leeners, B.; Geary, N.; Tobler, P.N.; Asarian, L. Ovarian Hormones and Obesity. Hum. Reprod. Update 2017, 23, 300–321. [Google Scholar] [CrossRef]
- Heine, P.A.; Taylor, J.A.; Iwamoto, G.A.; Lubahn, D.B.; Cooke, P.S. Increased Adipose Tissue in Male and Female Estrogen Receptor-α Knockout Mice. Proc. Natl. Acad. Sci. USA 2000, 97, 12729–12734. [Google Scholar] [CrossRef]
- Cooke, P.S.; Heine, P.A.; Taylor, J.A.; Lubahn, D.B. The Role of Estrogen and Estrogen Receptor-α in Male Adipose Tissue. Mol. Cell. Endocrinol. 2001, 178, 147–154. [Google Scholar] [CrossRef]
- Chen, J.-Q.; Brown, T.R.; Russo, J. Regulation of Energy Metabolism Pathways by Estrogens and Estrogenic Chemicals and Potential Implications in Obesity Associated with Increased Exposure to Endocrine Disruptors. Biochim. Et Biophys. Acta (BBA) Mol. Cell Res. 2009, 1793, 1128–1143. [Google Scholar] [CrossRef]
- Babaei, P.; Mehdizadeh, R.; Ansar, M.M.; Damirchi, A. Effects of Ovariectomy and Estrogen Replacement Therapy on Visceral Adipose Tissue and Serum Adiponectin Levels in Rats. Menopause Int. 2010, 16, 100–104. [Google Scholar] [CrossRef]
- Papadakis, G.E.; Hans, D.; Gonzalez Rodriguez, E.; Vollenweider, P.; Waeber, G.; Marques-Vidal, P.; Lamy, O. Menopausal Hormone Therapy Is Associated with Reduced Total and Visceral Adiposity: The OsteoLaus Cohort. J. Clin. Endocrinol. Metab. 2018, 103, 1948–1957. [Google Scholar] [CrossRef]
- Wunderle, M.; Pretscher, J.; Brucker, S.Y.; Volz, B.; Hartmann, A.; Fiessler, C.; Hein, A.; Häberle, L.; Jud, S.M.; Lux, M.P.; et al. Association between Breast Cancer Risk Factors and Molecular Type in Postmenopausal Patients with Hormone Receptor-Positive Early Breast Cancer. Breast Cancer Res. Treat. 2019, 174, 453–461. [Google Scholar] [CrossRef] [PubMed]
- Matsui, Y.; Sugiyama, K.; Kamei, M.; Takahashi, T.; Suzuki, T.; Katagata, Y.; Ito, T. Extract of Passion Fruit (Passiflora edulis) Seed Containing High Amounts of Piceatannol Inhibits Melanogenesis and Promotes Collagen Synthesis. J. Agric. Food Chem. 2010, 58, 11112–11118. [Google Scholar] [CrossRef] [PubMed]
- Piotrowska, H.; Kucinska, M.; Murias, M. Biological Activity of Piceatannol: Leaving the Shadow of Resveratrol. Mutat. Res. 2012, 750, 60–82. [Google Scholar] [CrossRef] [PubMed]
- Park, I.S.; Han, Y.; Jo, H.; Lee, K.W.; Song, Y.S. Piceatannol Is Superior to Resveratrol at Suppressing Adipogenesis in Human Visceral Adipose-Derived Stem Cells. Plants 2021, 10, 366. [Google Scholar] [CrossRef] [PubMed]
- Cordova-Gomez, M.; Galano, A.; Raúl Alvarez-Idaboy, J. Piceatannol, a Better Peroxyl Radical Scavenger than Resveratrol. RSC Adv. 2013, 3, 20209–20218. [Google Scholar] [CrossRef]
- Kawakami, S.; Kinoshita, Y.; Maruki-Uchida, H.; Yanae, K.; Sai, M.; Ito, T. Piceatannol and Its Metabolite, Isorhapontigenin, Induce SIRT1 Expression in THP-1 Human Monocytic Cell Line. Nutrients 2014, 6, 4794–4804. [Google Scholar] [CrossRef] [PubMed]
- Setoguchi, Y.; Oritani, Y.; Ito, R.; Inagaki, H.; Maruki-Uchida, H.; Ichiyanagi, T.; Ito, T. Absorption and Metabolism of Piceatannol in Rats. J. Agric. Food Chem. 2014, 62, 2541–2548. [Google Scholar] [CrossRef]
- Arisawa, K.; Kaneko, M.; Matsuoka, A.; Ozawa, N.; Kawawa, R.; Ishikawa, T.; Ichi, I.; Fujiwara, Y. Piceatannol Prevents Obesity and Fat Accumulation Caused by Estrogen Deficiency in Female Mice by Promoting Lipolysis. Nutrients 2023, 15, 1374. [Google Scholar] [CrossRef]
- Mottillo, E.P.; Granneman, J.G. Intracellular Fatty Acids Suppress β-Adrenergic Induction of PKA-Targeted Gene Expression in White Adipocytes. Am. J. Physiol. Endocrinol. Metab. 2011, 301, E122–E131. [Google Scholar] [CrossRef]
- Anthonsen, M.W.; Rönnstrand, L.; Wernstedt, C.; Degerman, E.; Holm, C. Identification of Novel Phosphorylation Sites in Hormone-Sensitive Lipase That Are Phosphorylated in Response to Isoproterenol and Govern Activation Properties in Vitro. J. Biol. Chem. 1998, 273, 215–221. [Google Scholar] [CrossRef]
- Su, C.-L.; Sztalryd, C.; Contreras, J.A.; Holm, C.; Kimmel, A.R.; Londos, C. Mutational Analysis of the Hormone-Sensitive Lipase Translocation Reaction in Adipocytes. J. Biol. Chem. 2003, 278, 43615–43619. [Google Scholar] [CrossRef] [PubMed]
- D’Eon, T.M.; Souza, S.C.; Aronovitz, M.; Obin, M.S.; Fried, S.K.; Greenberg, A.S. Estrogen Regulation of Adiposity and Fuel Partitioning. Evidence of Genomic and Non-genomic Regulation of Lipogenic and Oxidative Pathways. J. Biol. Chem. 2005, 280, 35983–35991. [Google Scholar] [CrossRef] [PubMed]
- Prossnitz, E.R.; Maggiolini, M. Mechanisms of Estrogen Signaling and Gene Expression via GPR30. Mol. Cell. Endocrinol. 2009, 308, 32–38. [Google Scholar] [CrossRef] [PubMed]
- Filardo, E.J.; Quinn, J.A.; Frackelton, A.R.; Bland, K.I. Estrogen Action Via the G Protein-Coupled Receptor, GPR30: Stimulation of Adenylyl Cyclase and cAMP-Mediated Attenuation of the Epidermal Growth Factor Receptor-to-MAPK Signaling Axis. Mol. Endocrinol. 2002, 16, 70–84. [Google Scholar] [CrossRef] [PubMed]
- Liao, R.S.; Ma, S.; Miao, L.; Li, R.; Yin, Y.; Raj, G.V. Androgen Receptor-Mediated Non-Genomic Regulation of Prostate Cancer Cell Proliferation. Transl. Androl. Urol. 2013, 2, 187–196. [Google Scholar]
- Lin, B.C.; Suzawa, M.; Blind, R.D.; Tobias, S.C.; Bulun, S.E.; Scanlan, T.S.; Ingraham, H.A. Stimulating the GPR30 Estrogen Receptor with a Novel Tamoxifen Analogue Activates SF-1 and Promotes Endometrial Cell Proliferation. Cancer Res. 2009, 69, 5415–5423. [Google Scholar] [CrossRef]
- Prossnitz, E.R.; Barton, M. The G-Protein-Coupled Estrogen Receptor GPER in Health and Disease. Nat. Rev. Endocrinol. 2011, 7, 715–726. [Google Scholar] [CrossRef] [PubMed]
- Prossnitz, E.R.; Barton, M. The G Protein-Coupled Oestrogen Receptor GPER in Health and Disease: An Update. Nat. Rev. Endocrinol. 2023, 19, 407–424. [Google Scholar] [CrossRef]
- Sharma, G.; Hu, C.; Brigman, J.L.; Zhu, G.; Hathaway, H.J.; Prossnitz, E.R. GPER Deficiency in Male Mice Results in Insulin Resistance, Dyslipidemia, and a Proinflammatory State. Endocrinology 2013, 154, 4136–4145. [Google Scholar] [CrossRef]
- Kwon, J.; Seo, S.; Heo, Y.-S.; Yue, S.; Cheng, J.-X.; Lee, K.; Kim, K.-H. Piceatannol, Natural Polyphenolic Stilbene, Inhibits Adipogenesis via Modulation of Mitotic Clonal Expansion and Insulin Receptor-dependent Insulin Signaling in Early Phase of Differentiation. J. Biol. Chem. 2012, 287, 11566–11578. [Google Scholar] [CrossRef]
- Prossnitz, E.R.; Arterburn, J.B. International Union of Basic and Clinical Pharmacology. XCVII. G Protein–Coupled Estrogen Receptor and Its Pharmacologic Modulators. Pharmacol. Rev. 2015, 67, 505–540. [Google Scholar] [CrossRef] [PubMed]
- Bligh, E.G.; Dyer, W.J. A Rapid Method of Total Lipid Extraction and Purification. Can. J. Biochem. Physiol. 1959, 37, 911–917. [Google Scholar] [CrossRef] [PubMed]
- McDonough, P.M.; Maciejewski-Lenoir, D.; Hartig, S.M.; Hanna, R.A.; Whittaker, R.; Heisel, A.; Nicoll, J.B.; Buehrer, B.M.; Christensen, K.; Mancini, M.G.; et al. Differential Phosphorylation of Perilipin 1A at the Initiation of Lipolysis Revealed by Novel Monoclonal Antibodies and High Content Analysis. PLoS ONE 2013, 8, e55511. [Google Scholar] [CrossRef] [PubMed]
- Bauzá-Thorbrügge, M.; Rodríguez-Cuenca, S.; Vidal-Puig, A.; Galmés-Pascual, B.M.; Sbert-Roig, M.; Gianotti, M.; Lladó, I.; Proenza, A.M. GPER and ERα Mediate Estradiol Enhancement of Mitochondrial Function in Inflamed Adipocytes through a PKA Dependent Mechanism. J. Steroid Biochem. Mol. Biol. 2019, 185, 256–267. [Google Scholar] [CrossRef] [PubMed]
- Dennis, M.K.; Burai, R.; Ramesh, C.; Petrie, W.K.; Alcon, S.N.; Nayak, T.K.; Bologa, C.G.; Leitao, A.; Brailoiu, E.; Deliu, E.; et al. In Vivo Effects of a GPR30 Antagonist. Nat. Chem. Biol. 2009, 5, 421–427. [Google Scholar] [CrossRef] [PubMed]
- Recazens, E.; Mouisel, E.; Langin, D. Hormone-Sensitive Lipase: Sixty Years Later. Prog. Lipid Res. 2021, 82, 101084. [Google Scholar] [CrossRef]
- Shimizu, H.; Shimomura, Y.; Nakanishi, Y.; Futawatari, T.; Ohtani, K.; Sato, N.; Mori, M. Estrogen Increases in Vivo Leptin Production in Rats and Human Subjects. J. Endocrinol. 1997, 154, 285–292. [Google Scholar] [CrossRef]
- Zidon, T.M.; Padilla, J.; Fritsche, K.L.; Welly, R.J.; McCabe, L.T.; Stricklin, O.E.; Frank, A.; Park, Y.; Clegg, D.J.; Lubahn, D.B.; et al. Effects of ERβ and ERα on OVX-Induced Changes in Adiposity and Insulin Resistance. J. Endocrinol. 2020, 245, 165–178. [Google Scholar] [CrossRef]
- Al-Qahtani, S.M.; Bryzgalova, G.; Valladolid-Acebes, I.; Korach-André, M.; Dahlman-Wright, K.; Efendić, S.; Berggren, P.-O.; Portwood, N. 17β-Estradiol Suppresses Visceral Adipogenesis and Activates Brown Adipose Tissue-Specific Gene Expression. Horm. Mol. Biol. Clin. Investig. 2016, 29, 13–26. [Google Scholar] [CrossRef]
- Palin, S.L.; McTernan, P.G.; Anderson, L.A.; Sturdee, D.W.; Barnett, A.H.; Kumar, S. 17β-Estradiol and Anti-Estrogen ICI: Compound 182,780 Regulate Expression of Lipoprotein Lipase and Hormone-Sensitive Lipase in Isolated Subcutaneous Abdominal Adipocytes. Metabolism 2003, 52, 383–388. [Google Scholar] [CrossRef]
- Filardo, E.J.; Quinn, J.A.; Bland, K.I.; Frackelton, A.R., Jr. Estrogen-Induced Activation of Erk-1 and Erk-2 Requires the G Protein-Coupled Receptor Homolog, GPR30, and Occurs via Trans-Activation of the Epidermal Growth Factor Receptor through Release of HB-EGF. Mol. Endocrinol. 2000, 14, 1649–1660. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.; Zhu, P.; Sham, K.W.Y.; Yuen, J.M.L.; Xie, C.; Zhang, Y.; Liu, Y.; Li, S.; Huang, X.; Cheng, C.H.K.; et al. Identification of a Membrane Estrogen Receptor in Zebrafish with Homology to Mammalian GPER and Its High Expression in Early Germ Cells of the Testis. Biol. Reprod. 2009, 80, 1253–1261. [Google Scholar] [CrossRef] [PubMed]
- Revankar, C.M.; Cimino, D.F.; Sklar, L.A.; Arterburn, J.B.; Prossnitz, E.R. A Transmembrane Intracellular Estrogen Receptor Mediates Rapid Cell Signaling. Science 2005, 307, 1625–1630. [Google Scholar] [CrossRef] [PubMed]
- Prossnitz, E.R.; Hathaway, H.J. What Have We Learned about GPER Function in Physiology and Disease from Knockout Mice? J. Steroid Biochem. Mol. Biol. 2015, 153, 114–126. [Google Scholar] [CrossRef] [PubMed]
- Zhu, P.; Yuen, J.M.L.; Sham, K.W.Y.; Cheng, C.H.K. GPER Mediates the Inhibitory Actions of Estrogen on Adipogenesis in 3T3-L1 Cells through Perturbation of Mitotic Clonal Expansion. Gen. Comp. Endocrinol. 2013, 193, 19–26. [Google Scholar] [CrossRef] [PubMed]
- Sharma, G.; Mauvais-Jarvis, F.; Prossnitz, E.R. Roles of G Protein-Coupled Estrogen Receptor GPER in Metabolic Regulation. J. Steroid Biochem. Mol. Biol. 2018, 176, 31–37. [Google Scholar] [CrossRef] [PubMed]
- Gehm, B.D.; Levenson, A.S.; Liu, H.; Lee, E.-J.; Amundsen, B.M.; Cushman, M.; Jordan, V.C.; Jameson, J.L. Estrogenic Effects of Resveratrol in Breast Cancer Cells Expressing Mutant and Wild-Type Estrogen Receptors: Role of AF-1 and AF-2. J. Steroid Biochem. Mol. Biol. 2004, 88, 223–234. [Google Scholar] [CrossRef]
- Wober, J.; Möller, F.; Richter, T.; Unger, C.; Weigt, C.; Jandausch, A.; Zierau, O.; Rettenberger, R.; Kaszkin-Bettag, M.; Vollmer, G. Activation of Estrogen Receptor-β by a Special Extract of Rheum Rhaponticum (ERr 731®), Its Aglycones and Structurally Related Compounds. J. Steroid Biochem. Mol. Biol. 2007, 107, 191–201. [Google Scholar] [CrossRef]
- Greenberg, A.S.; Shen, W.-J.; Muliro, K.; Patel, S.; Souza, S.C.; Roth, R.A.; Kraemer, F.B. Stimulation of Lipolysis and Hormone-Sensitive Lipase via the Extracellular Signal-Regulated Kinase Pathway. J. Biol. Chem. 2001, 276, 45456–45461. [Google Scholar] [CrossRef]
- Kim, N.; Kwon, J.; Shin, U.S.; Jung, J. Stimulatory Anticancer Effect of Resveratrol Mediated by G Protein-Coupled Estrogen Receptor in Colorectal Cancer. Biomol. Ther. 2023, 31, 655–660. [Google Scholar] [CrossRef]
- Carpéné, C.; Pejenaute, H.; del Moral, R.; Boulet, N.; Hijona, E.; Andrade, F.; Villanueva-Millán, M.; Aguirre, L.; Arbones-Mainar, J.M. The Dietary Antioxidant Piceatannol Inhibits Adipogenesis of Human Adipose Mesenchymal Stem Cells and Limits Glucose Transport and Lipogenic Activities in Adipocytes. Int. J. Mol. Sci. 2018, 19, 2081. [Google Scholar] [CrossRef] [PubMed]


Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Arisawa, K.; Matsuoka, A.; Ozawa, N.; Ishikawa, T.; Ichi, I.; Fujiwara, Y. GPER/PKA-Dependent Enhancement of Hormone-Sensitive Lipase Phosphorylation in 3T3-L1 Adipocytes by Piceatannol. Nutrients 2024, 16, 38. https://doi.org/10.3390/nu16010038
Arisawa K, Matsuoka A, Ozawa N, Ishikawa T, Ichi I, Fujiwara Y. GPER/PKA-Dependent Enhancement of Hormone-Sensitive Lipase Phosphorylation in 3T3-L1 Adipocytes by Piceatannol. Nutrients. 2024; 16(1):38. https://doi.org/10.3390/nu16010038
Chicago/Turabian StyleArisawa, Kotoko, Ayumi Matsuoka, Natsuki Ozawa, Tomoko Ishikawa, Ikuyo Ichi, and Yoko Fujiwara. 2024. "GPER/PKA-Dependent Enhancement of Hormone-Sensitive Lipase Phosphorylation in 3T3-L1 Adipocytes by Piceatannol" Nutrients 16, no. 1: 38. https://doi.org/10.3390/nu16010038