Microbiome Taxonomic and Functional Differences in C3H/HeJ Mice Fed a Long-Term High-Fat Diet with Beef Protein ± Ammonium Hydroxide Supplementation

 ,
,  and
and
Abstract
:1. Introduction
2. Methods
2.1. Mouse Study Design
2.2. Diet Preparation and Composition
2.3. Mouse Metrics
2.4. Fecal Collection and Processing
2.5. Metagenomic Assembly and Initial Taxonomic Analysis
2.6. Refined Taxonomic Analysis and Differential Abundance Tests
2.7. Functional Analysis and Gene Ontology Enrichment Tests
3. Results
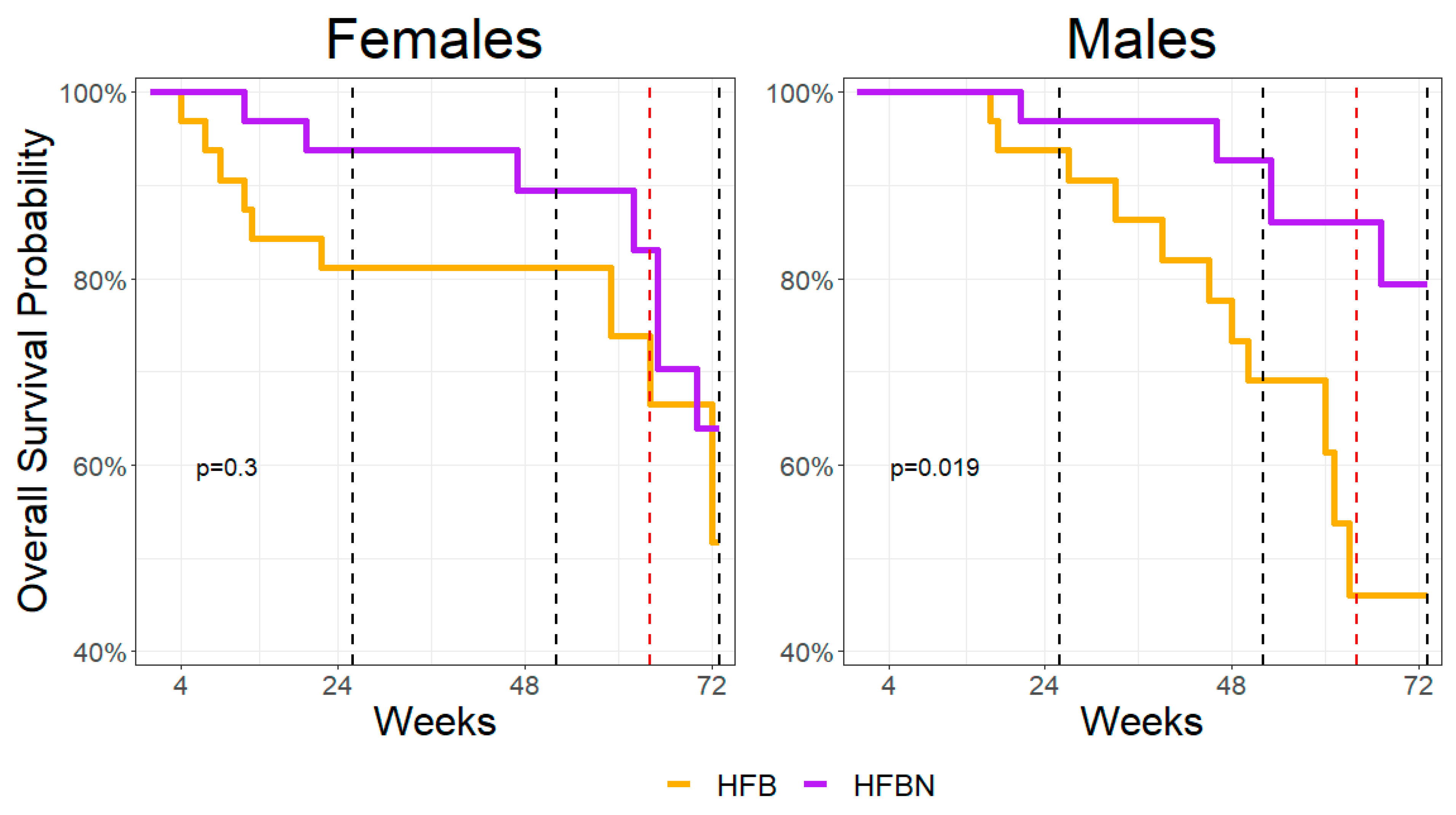
3.1. Survivability Assessment
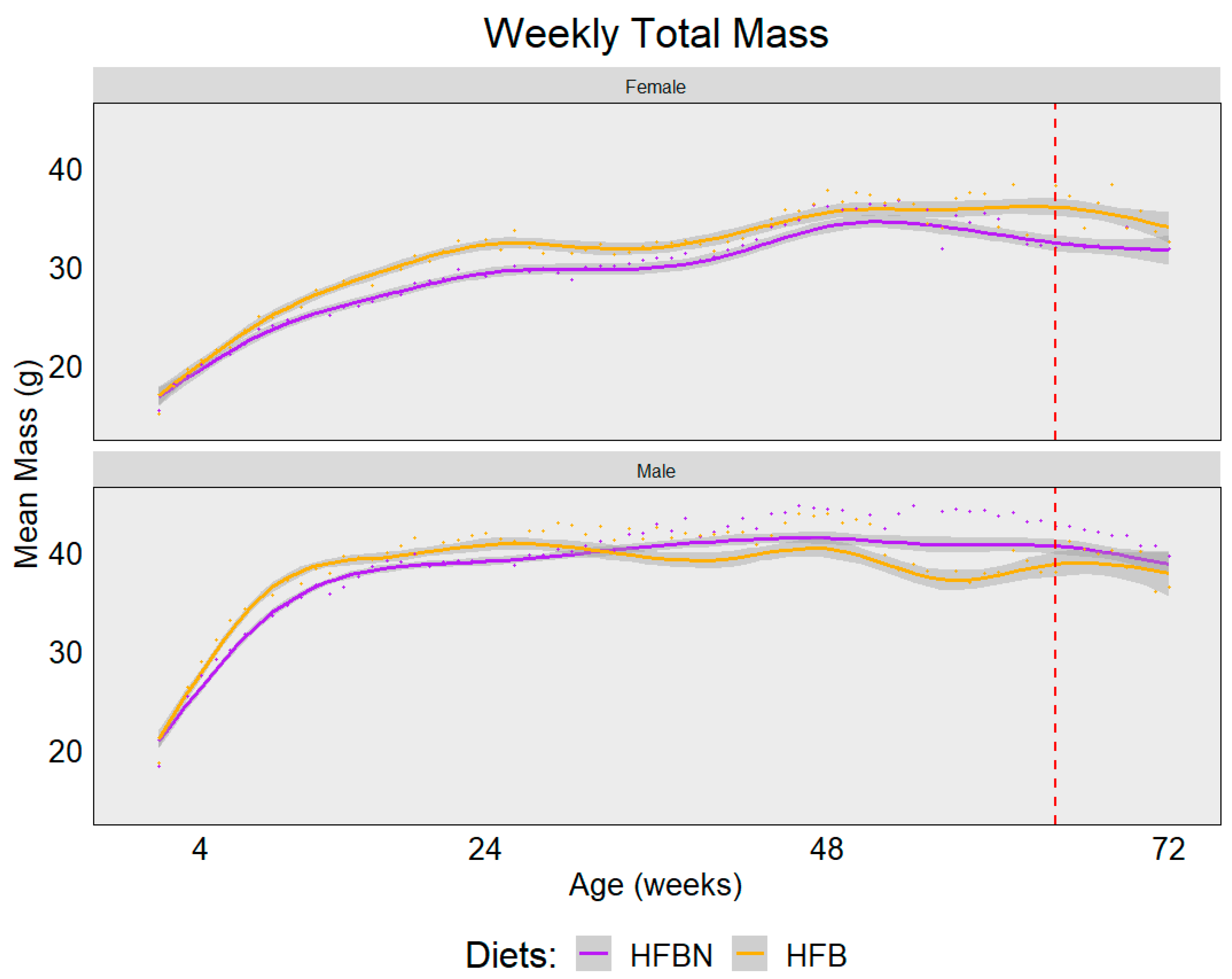
3.2. Food Consumption and Mass
3.3. Sequence Yield, Assembly Features, and Annotated Genes
3.4. Phylum-Level Taxonomic Composition of metaWRAP-Refined Bins
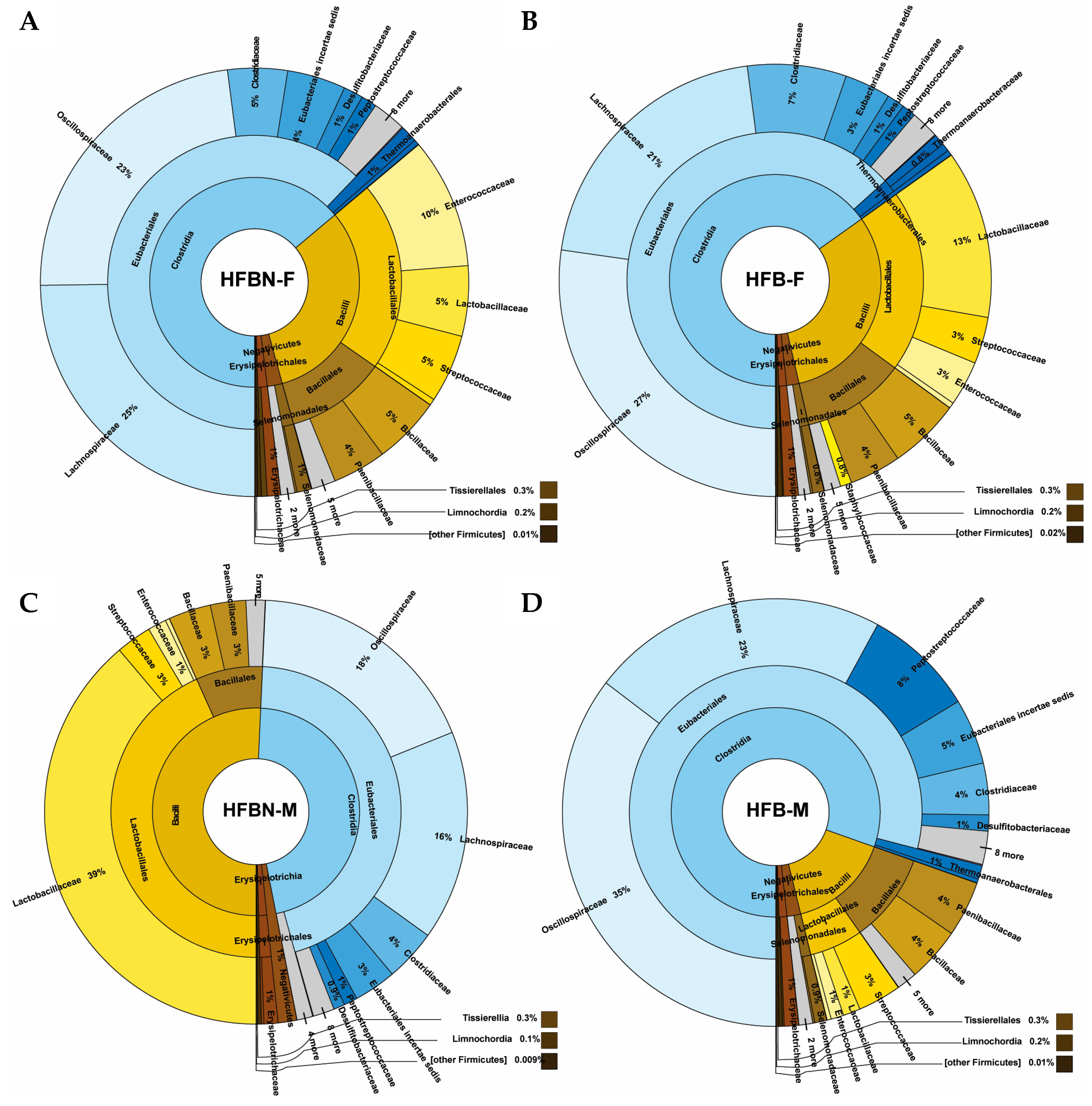
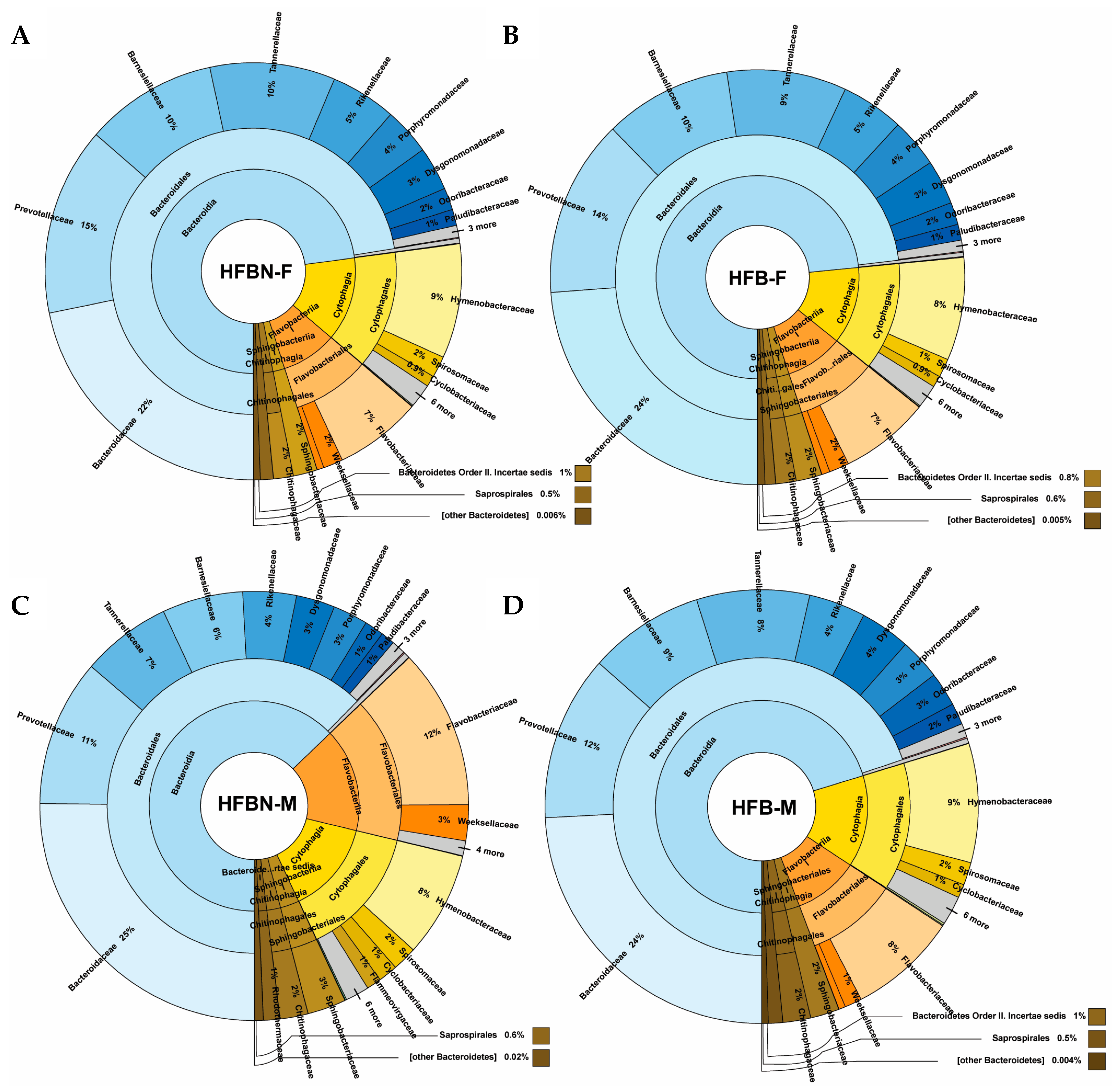
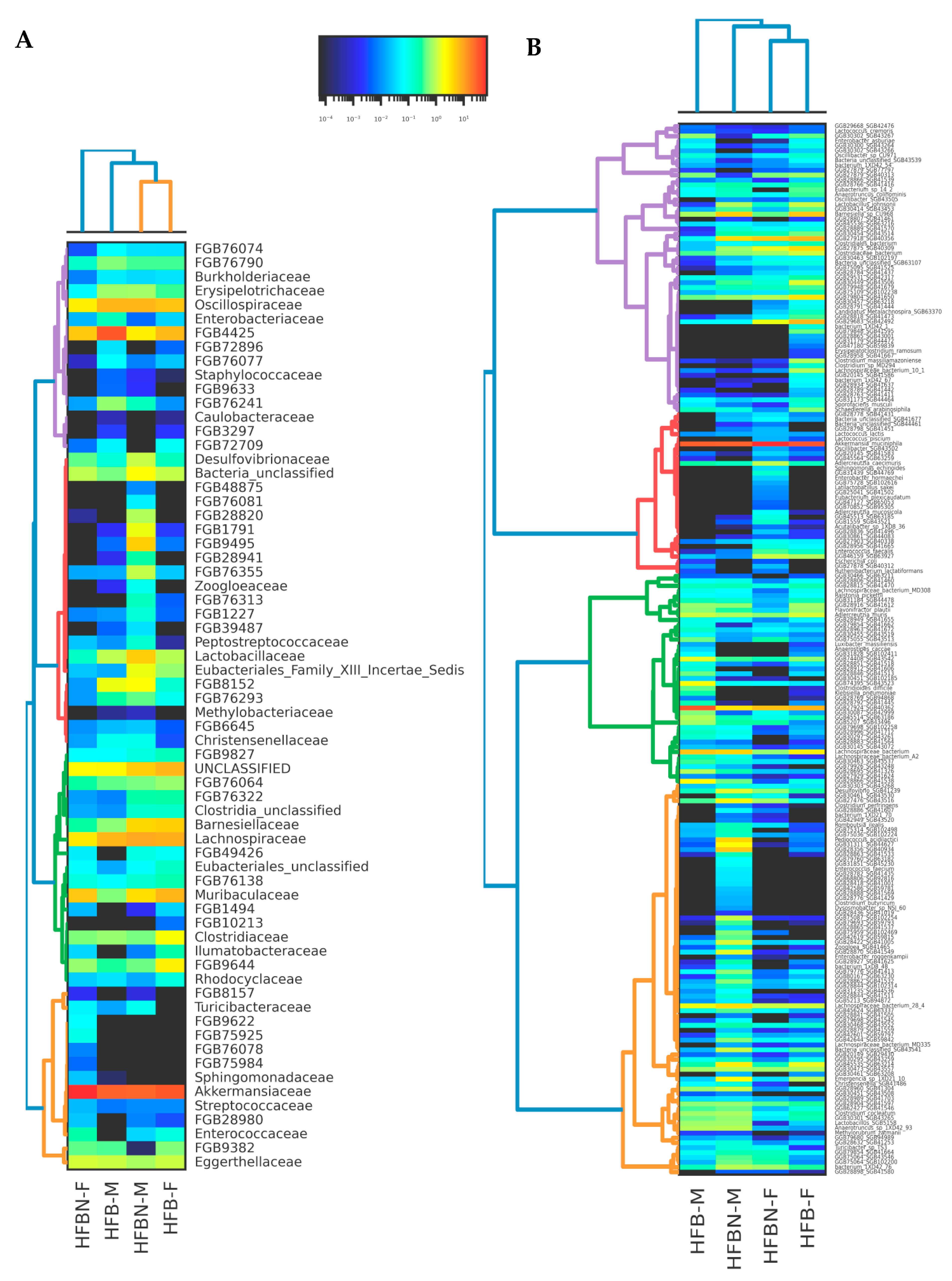
3.5. Lower-Level Taxonomic Classification of metaWRAP-Refined Bins
3.6. Refined Taxonomic Data Classification Using MetaPhlAn4
3.7. Statistical Tests of Differentially Abundant Taxa from ANCOM-BC2
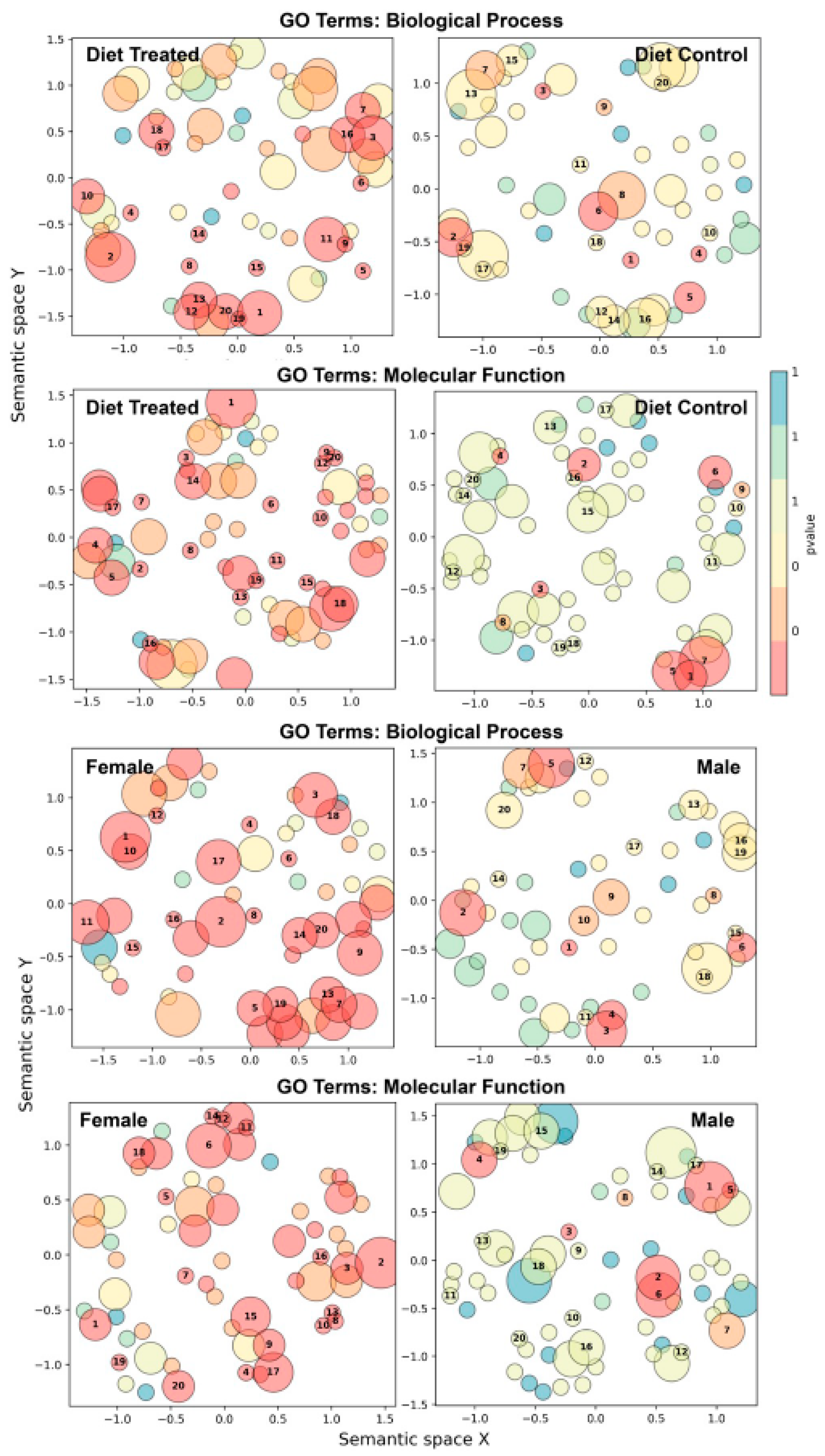
3.8. Metapangenomics and Gene Ontology (GO) Enrichment Analysis
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Kopp, W. How Western Diet and Lifestyle Drive the Pandemic of Obesity and Civilization Diseases. Diabetes Metab. Syndr. Obes. Targets Ther. 2019, 12, 2221–2236. [Google Scholar] [CrossRef]
- Williamson, M.; Moustaid-Moussa, N.; Gollahon, L. The Molecular Effects of Dietary Acid Load on Metabolic Disease (The Cellular PasaDoble: The Fast-Paced Dance of pH Regulation). Front. Mol. Med. 2021, 1, 777088. [Google Scholar] [CrossRef]
- Sanz, J.M.; Sergi, D.; Colombari, S.; Capatti, E.; Situlin, R.; Biolo, G.; Di Girolamo, F.G.; Lazzer, S.; Simunic, B.; Pisot, R.; et al. Dietary Acid Load but Not Mediterranean Diet Adherence Score is Associated with Metabolic and Cardiovascular Health State: A Population Observational Study from Northern Italy. Front. Nutr. 2022, 9, 828587. [Google Scholar] [CrossRef] [PubMed]
- Storz, M.A.; Ronco, A.L.; Hannibal, L. Observational and clinical evidence that plant-based nutrition reduces dietary acid load. J. Nutr. Sci. 2022, 11, e93. [Google Scholar] [CrossRef]
- Menikdiwela, K.; DesHotels, A.; Albracht-Schulte, K.; Scoggin, S.; Gollahon, L.; Loneragan, G.; Moustaid-Moussa, N. Metabolic Effects of pH Enhanced Ground Beef in Diet-induced Obese Mice. Curr. Dev. Nutr. 2021, 5, 1234. [Google Scholar] [CrossRef]
- Carnauba, R.A.; Baptistella, A.B.; Paschoal, V.; Hübscher, G.H. Diet-Induced Low-Grade Metabolic Acidosis and Clinical Outcomes: A Review. Nutrients 2017, 9, 538. [Google Scholar] [CrossRef] [PubMed]
- Osuna-Padilla, I.; Leal-Escobar, G.; Garza-García, C.; Rodríguez-Castellanos, F. Dietary acid load: Mechanisms and evidence of its health repercussions. Nefrologia 2019, 39, 343–354. [Google Scholar] [CrossRef] [PubMed]
- Robey, I.F. Examining the relationship between diet-induced acidosis and cancer. Nutr. Metab. 2012, 9, 72. [Google Scholar] [CrossRef]
- Williams, R.S.; Kozan, P.; Samocha-Bonet, D. The role of dietary acid load and mild metabolic acidosis in insulin resistance in humans. Biochimie 2016, 124, 171–177. [Google Scholar] [CrossRef]
- Matsushita, M.; Fujita, K.; Hatano, K.; Hayashi, T.; Kayama, H.; Motooka, D.; Hase, H.; Yamamoto, A.; Uemura, T.; Yamamichi, G.; et al. High-fat diet promotes prostate cancer growth through histamine signaling. Int. J. Cancer 2022, 151, 623–636. [Google Scholar] [CrossRef]
- Henning, S.M.; Yang, J.; Shao, P.; Lee, R.-P.; Huang, J.; Ly, A.; Hsu, M.; Lu, Q.-Y.; Thames, G.; Heber, D.; et al. Health benefit of vegetable/fruit juice-based diet: Role of microbiome. Sci. Rep. 2017, 7, 2167. [Google Scholar] [CrossRef] [PubMed]
- Maruvada, P.; Leone, V.; Kaplan, L.M.; Chang, E.B. The Human Microbiome and Obesity: Moving beyond Associations. Cell Host Microbe 2017, 22, 589–599. [Google Scholar] [CrossRef] [PubMed]
- Wilson, A.S.; Koller, K.R.; Ramaboli, M.C.; Nesengani, L.T.; Ocvirk, S.; Chen, C.; Flanagan, C.A.; Sapp, F.R.; Merritt, Z.T.; Bhatti, F.; et al. Diet and the Human Gut Microbiome: An International Review. Dig. Dis. Sci. 2020, 65, 723–740. [Google Scholar] [CrossRef] [PubMed]
- Saklayen, M.G. The global epidemic of the metabolic syndrome. Curr. Hypertens. Rep. 2018, 20, 12. [Google Scholar] [CrossRef] [PubMed]
- Ilhan, Z.E.; Marcus, A.K.; Kang, D.; Rittmann, B.E. pH-Mediated Microbial and Metabolic Interactions in Fecal Enrichment Cultures. mSphere 2017, 2, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Han, H.; Zhou, Y.; Liu, Q.; Wang, G.; Feng, J.; Zhang, M. Effects of Ammonia on Gut Microbiota and Growth Performance of Broiler Chickens. Animals 2021, 11, 1716. [Google Scholar] [CrossRef] [PubMed]
- Firrman, J.; Liu, L.; Mahalak, K.; Tanes, C.; Bittinger, K.; Tu, V.; Bobokalonov, J.; Mattei, L.; Zhang, H.; Abbeele, P.V.D. The impact of environmental pH on the gut microbiota community structure and short chain fatty acid production. FEMS Microbiol. Ecol. 2022, 98, fiac038. [Google Scholar] [CrossRef] [PubMed]
- Statovci, D.; Aguilera, M.; MacSharry, J.; Melgar, S. The Impact of Western Diet and Nutrients on the Microbiota and Immune Response at Mucosal Interfaces. Front. Immunol. 2017, 8, 838. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Jacobs, J.P.; Lagishetty, V.; Yuan, P.-Q.; Wu, S.V.; Million, M.; Reeve, J.R.; Pisegna, J.R.; Taché, Y.; Klingbeil, E.; et al. High-protein diet improves sensitivity to cholecystokinin and shifts the cecal microbiome without altering brain inflammation in diet-induced obesity in rats. Am. J. Physiol. Integr. Comp. Physiol. 2017, 313, R473–R486. [Google Scholar] [CrossRef]
- Albracht-Schulte, K.; Islam, T.; Johnson, P.; Moustaid-Moussa, N. Systematic review of beef protein effects on gut microbiota: Implications for health. Adv. Nutr. 2021, 12, 102–114. [Google Scholar] [CrossRef]
- Drouillard, J.S. Current situation and future trends for beef production in the United States of America—A review. Asian-Australas. J. Anim. Sci. 2018, 31, 1007–1016. [Google Scholar] [CrossRef]
- Tessler, M.; Neumann, J.S.; Afshinnekoo, E.; Pineda, M.; Hersch, R.; Velho, L.F.M.; Segovia, B.T.; Lansac-Toha, F.A.; Lemke, M.; DeSalle, R.; et al. Large-scale differences in microbial biodiversity discovery between 16S amplicon and shotgun sequencing. Sci. Rep. 2017, 7, 6589. [Google Scholar] [CrossRef]
- Planer, J.D.; Peng, Y.; Kau, A.L.; Blanton, L.V.; Ndao, I.M.; Tarr, P.I.; Warner, B.B.; Gordon, J.I. Development of the gut microbiota and mucosal IgA responses in twins and gnotobiotic mice. Nature 2016, 534, 263–266. [Google Scholar] [CrossRef]
- Liu, R.; Kang, J.D.; Sartor, R.B.; Sikaroodi, M.; Fagan, A.; Gavis, E.A.; Zhou, H.; Hylemon, P.B.; Herzog, J.W.; Li, X.; et al. Neuroinflammation in Murine Cirrhosis Is Dependent on the Gut Microbiome and Is Attenuated by Fecal Transplant. Hepatology 2019, 71, 611–626. [Google Scholar] [CrossRef] [PubMed]
- Koren, O.; Goodrich, J.K.; Cullender, T.C.; Spor, A.; Laitinen, K.; Bäckhed, H.K.; Gonzalez, A.; Werner, J.J.; Angenent, L.T.; Knight, R.; et al. Host Remodeling of the Gut Microbiome and Metabolic Changes during Pregnancy. Cell 2012, 150, 470–480. [Google Scholar] [CrossRef] [PubMed]
- Uritskiy, G.V.; DiRuggiero, J.; Taylor, J. MetaWRAP—A flexible pipeline for genome-resolved metagenomic data analysis. Microbiome 2018, 6, 158. [Google Scholar] [CrossRef]
- Zhang, J.; Kobert, K.; Flouri, T.; Stamatakis, A. PEAR: A fast and accurate Illumina Paired-End reAd mergeR. Bioinformatics 2013, 30, 614–620. [Google Scholar] [CrossRef]
- Bolger, A.M.; Lohse, M.; Usadel, B. Trimmomatic: A flexible trimmer for Illumina sequence data. Bioinformatics 2014, 30, 2114–2120. [Google Scholar] [CrossRef] [PubMed]
- Nurk, S.; Meleshko, D.; Korobeynikov, A.; Pevzner, P. metaSPAdes: A new versatile de novo metagenomics assembler. arXiv 2016, arXiv:1604.03071. [Google Scholar] [CrossRef]
- Alneberg, J.; Bjarnason, B.S.; de Bruijn, I.; Schirmer, M.; Quick, J.; Ijaz, U.Z.; Lahti, L.; Loman, N.J.; Andersson, A.F.; Quince, C. Binning metagenomic contigs by coverage and composition. Nat. Methods 2014, 11, 1144–1146. [Google Scholar] [CrossRef]
- Wu, Y.-W.; Simmons, B.A.; Singer, S.W. MaxBin 2.0: An automated binning algorithm to recover genomes from multiple metagenomic datasets. Bioinformatics 2015, 32, 605–607. [Google Scholar] [CrossRef] [PubMed]
- Kang, D.D.; Froula, J.; Egan, R.; Wang, Z. MetaBAT, an efficient tool for accurately reconstructing single genomes from complex microbial communities. PeerJ 2015, 3, e1165. [Google Scholar] [CrossRef] [PubMed]
- Bankevich, A.; Nurk, S.; Antipov, D.; Gurevich, A.A.; Dvorkin, M.; Kulikov, A.S.; Lesin, V.M.; Nikolenko, S.I.; Pham, S.; Prjibelski, A.D.; et al. SPAdes: A new genome assembly algorithm and its applications to single-cell sequencing. J. Comput. Biol. 2012, 19, 455–477. [Google Scholar] [CrossRef] [PubMed]
- Parks, D.H.; Imelfort, M.; Skennerton, C.T.; Hugenholtz, P.; Tyson, G.W. CheckM: Assessing the quality of microbial genomes recovered from isolates, single cells, and metagenomes. Genome Res. 2015, 25, 1043–1055. [Google Scholar] [CrossRef] [PubMed]
- Kim, D.; Song, L.; Breitwieser, F.P.; Salzberg, S.L. Centrifuge: Rapid and sensitive classification of metagenomic sequences. Genome Res. 2016, 26, 1721–1729. [Google Scholar] [CrossRef] [PubMed]
- Kumar, S.; Jones, M.; Koutsovoulos, G.; Clarke, M.; Blaxter, M. Blobology: Exploring raw genome data for contaminants, symbionts and parasites using taxon-annotated GC-coverage plots. Front. Genet. 2013, 4, 237. [Google Scholar] [CrossRef] [PubMed]
- Camacho, C.; Coulouris, G.; Avagyan, V.; Ma, N.; Papadopoulos, J.; Bealer, K.; Madden, T.L. BLAST+: Architecture and applications. BMC Bioinform. 2009, 10, 421. [Google Scholar] [CrossRef] [PubMed]
- Wickham, H.; Averick, M.; Bryan, J.; Chang, W.; McGowan, L.D.A.; François, R.; Grolemund, G.; Hayes, A.; Henry, L.; Hester, J.; et al. Welcome to the Tidyverse. J. Open Source Softw. 2019, 4, 1686. [Google Scholar] [CrossRef]
- Ondov, B.D.; Bergman, N.H.; Phillippy, A.M. Interactive metagenomic visualization in a Web browser. BMC Bioinform. 2011, 12, 385. [Google Scholar] [CrossRef]
- Segata, N.; Waldron, L.; Ballarini, A.; Narasimhan, V.; Jousson, O.; Huttenhower, C. Metagenomic microbial community profiling using unique clade-specific marker genes. Nat. Methods 2013, 9, 811–814. [Google Scholar] [CrossRef]
- Blanco-Míguez, A.; Beghini, F.; Cumbo, F.; McIver, L.J.; Thompson, K.N.; Zolfo, M.; Manghi, P.; Dubois, L.; Huang, K.D.; Thomas, A.M.; et al. Extending and improving metagenomic taxonomic profiling with uncharacterized species using MetaPhlAn 4. Nat. Biotechnol. 2023, 41, 1633–1644. [Google Scholar] [CrossRef] [PubMed]
- Manghi, P.; Blanco-Míguez, A.; Manara, S.; NabiNejad, A.; Cumbo, F.; Beghini, F.; Armanini, F.; Golzato, D.; Huang, K.D.; Thomas, A.M.; et al. MetaPhlAn 4 profiling of unknown species-level genome bins improves the characterization of diet-associated microbiome changes in mice. Cell Rep. 2023, 42, 112464. [Google Scholar] [CrossRef] [PubMed]
- McMurdie, P.J.; Holmes, S. phyloseq: An R package for reproducible interactive analysis and graphics of microbiome census data. PLoS ONE 2013, 8, e61217. [Google Scholar] [CrossRef] [PubMed]
- Li, S.; Vogtmann, E.; Graubard, B.I.; Gail, M.H.; Abnet, C.C.; Shi, J. fast.adonis: A computationally efficient non-parametric multivariate analysis of microbiome data for large-scale studies. Bioinform. Adv. 2022, 2, vbac044. [Google Scholar] [CrossRef] [PubMed]
- Mandal, S.; Van Treuren, W.; White, R.A.; Eggesbø, M.; Knight, R.T.; Peddada, S.D. Analysis of composition of microbiomes: A novel method for studying microbial composition. Microb. Ecol. Health Dis. 2015, 26, 27663. [Google Scholar] [CrossRef] [PubMed]
- Lin, H.; Das Peddada, S. Multigroup analysis of compositions of microbiomes with covariate adjustments and repeated measures. Nat. Methods 2023, 21, 83–91. [Google Scholar] [CrossRef] [PubMed]
- Seemann, T. Prokka: Rapid Prokaryotic Genome Annotation. Bioinformatics 2014, 30, 2068–2069. [Google Scholar] [CrossRef]
- Page, A.J.; Cummins, C.A.; Hunt, M.; Wong, V.K.; Reuter, S.; Holden, M.T.G.; Fookes, M.; Falush, D.; Keane, J.A.; Parkhill, J. Roary: Rapid large-scale prokaryote pan genome analysis. Bioinformatics 2015, 31, 3691–3693. [Google Scholar] [CrossRef] [PubMed]
- Alexa, A.; Rahnenfuhrer, J. topGO: Enrichment Analysis for Gene Ontology. R Package Version 2.40.0. 2020. Available online: https://bioconductor.org/packages/release/bioc/html/topGO.html (accessed on 14 May 2022).
- Reijnders, M.J.M.F.; Waterhouse, R.M. Summary Visualizations of Gene Ontology Terms With GO-Figure! Front. Bioinform. 2021, 1, 638255. [Google Scholar] [CrossRef]
- Org, E.; Mehrabian, M.; Parks, B.W.; Shipkova, P.; Liu, X.; Drake, T.A.; Lusis, A.J. Sex differences and hormonal effects on gut microbiota composition in mice. Gut Microbes 2016, 7, 313–322. [Google Scholar] [CrossRef]
- McGee, J.S.; Huttenhower, C. Of mice and men and women: Sexual dimorphism of the gut microbiome. Int. J. Women’s Dermatol. 2021, 7, 533–538. [Google Scholar] [CrossRef] [PubMed]
- Wu, Y.; Peng, X.; Li, X.; Li, D.; Tan, Z.; Yu, R. Sex hormones influence the intestinal microbiota composition in mice. Front. Microbiol. 2022, 13, 964847. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.J.; Kim, S.-E.; Kim, A.-R.; Kang, S.; Park, M.-Y.; Sung, M.-K. Dietary fat intake and age modulate the composition of the gut microbiota and colonic inflammation in C57BL/6J mice. BMC Microbiol. 2019, 19, 193. [Google Scholar] [CrossRef] [PubMed]
- Binda, C.; Lopetuso, L.R.; Rizzatti, G.; Gibiino, G.; Cennamo, V.; Gasbarrini, A. Actinobacteria: A relevant minority for the maintenance of gut homeostasis. Dig. Liver Dis. 2018, 50, 421–428. [Google Scholar] [CrossRef]
- Nitert, M.D.; Mousa, A.; Barrett, H.L.; Naderpoor, N.; de Courten, B. Altered Gut Microbiota Composition Is Associated With Back Pain in Overweight and Obese Individuals. Front. Endocrinol. 2020, 11, 514189. [Google Scholar] [CrossRef] [PubMed]
- Hexun, Z.Z.; Miyake, T.; Maekawa, T.; Mori, H.; Yasukawa, D.; Ohno, M.; Nishida, A.; Andoh, A.; Tani, M. High abundance of Lachnospiraceae in the human gut microbiome is related to high immunoscores in advanced colorectal cancer. Cancer Immunol. Immunother. 2023, 72, 315–326. [Google Scholar] [CrossRef] [PubMed]
- Vacca, M.; Celano, G.; Calabrese, F.M.; Portincasa, P.; Gobbetti, M.; De Angelis, M. The Controversial Role of Human Gut Lachnospiraceae. Microorganisms 2020, 8, 573. [Google Scholar] [CrossRef] [PubMed]
- Atzeni, A.; Bastiaanssen, T.; Cryan, J.; Tinahones, F.; Vioque, J.; Corella, D.; Fitó, M.; Vidal, J.; Moreno-Indias, I.; Gómez-Pérez, A.; et al. Taxonomic and Functional Fecal Microbiota Signatures Associated With Insulin Resistance in Non-Diabetic Subjects With Overweight/Obesity within the Frame of the PREDIMED-Plus Study. Front. Endocrinol. 2022, 13, 804455. [Google Scholar] [CrossRef] [PubMed]
- Lee, S.Y.; Kim, J.H.; Lee, D.Y.; Hur, S.J. Characterization of gut microbiota in mouse models of aging and sarcopenia. Microbiol. Res. 2023, 275, 127462. [Google Scholar] [CrossRef]
- Jeong, D.-Y.; Ryu, M.-S.; Yang, H.-J.; Jeong, S.-Y.; Zhang, T.; Yang, H.J.; Kim, M.J.; Park, S. Pediococcus acidilactici intake decreases the clinical severity of atopic dermatitis along with increasing mucin production and improving the gut microbiome in Nc/Nga mice. Biomed. Pharmacother. 2020, 129, 110488. [Google Scholar] [CrossRef]
- Qiao, Y.; Qiu, Z.; Tian, F.; Yu, L.; Zhao, J.; Zhang, H.; Zhai, Q.; Chen, W. Effect of bacteriocin-producing Pediococcus acidilactici strains on the immune system and intestinal flora of normal mice. Food Sci. Hum. Wellness 2021, 11, 238–246. [Google Scholar] [CrossRef]
- Gao, X.; Chang, S.; Liu, S.; Peng, L.; Xie, J.; Dong, W.; Tian, Y.; Sheng, J. Correlations between α-Linolenic Acid-Improved Multitissue Homeostasis and Gut Microbiota in Mice Fed a High-Fat Diet. mSystems 2020, 5, 1–17. [Google Scholar] [CrossRef]
- Lagkouvardos, I.; Lesker, T.R.; Hitch, T.C.A.; Galvez, E.J.C.; Smit, N.; Neuhaus, K.; Wang, J.; Baines, J.F.; Abt, B.; Stecher, B.; et al. Sequence and cultivation study of Muribaculaceae reveals novel species, host preference, and functional potential of this yet undescribed family. Microbiome 2019, 7, 28. [Google Scholar] [CrossRef] [PubMed]
- Paone, P.; Suriano, F.; Jian, C.; Korpela, K.; Delzenne, N.M.; Van Hul, M.; Salonen, A.; Cani, P.D. Prebiotic oligofructose protects against high-fat diet-induced obesity by changing the gut microbiota, intestinal mucus production, glycosylation and secretion. Gut Microbes 2022, 14, 2152307. [Google Scholar] [CrossRef]
- Togo, A.; Diop, A.; Dubourg, G.; Khelaifia, S.; Richez, M.; Armstrong, N.; Maraninchi, M.; Fournier, P.-E.; Raoult, D.; Million, M. Anaerotruncus massiliensis sp. nov., a succinate-producing bacterium isolated from human stool from an obese patient after bariatric surgery. New Microbes New Infect. 2019, 29, 100508. [Google Scholar] [CrossRef] [PubMed]
- Hases, L.; Stepanauskaite, L.; Birgersson, M.; Brusselaers, N.; Schuppe-Koistinen, I.; Archer, A.; Engstrand, L.; Williams, C. High-fat diet and estrogen modulate the gut microbiota in a sex-dependent manner in mice. Commun. Biol. 2023, 6, 20. [Google Scholar] [CrossRef] [PubMed]
- Cao, L.; Gil Lee, S.; Melough, M.M.; Sakaki, J.R.; Maas, K.R.; Koo, S.I.; Chun, O.K.; Cao, L.; Gil Lee, S.; Melough, M.M.; et al. Long-Term Blackcurrant Supplementation Modified Gut Microbiome Profiles in Mice in an Age-Dependent Manner: An Exploratory Study. Nutrients 2020, 12, 290. [Google Scholar] [CrossRef]
- Yang, M.; Bose, S.; Lim, S.; Seo, J.; Shin, J.; Lee, D.; Chung, W.-H.; Song, E.-J.; Nam, Y.-D.; Kim, H. Beneficial Effects of Newly Isolated Akkermansia muciniphila Strains from the Human Gut on Obesity and Metabolic Dysregulation. Microorganisms 2020, 8, 1413. [Google Scholar] [CrossRef]
- De Vos, W.M. Microbe Profile: Akkermansia muciniphila: A conserved intestinal symbiont that acts as the gatekeeper of our mucosa. Microbiology 2017, 163, 646–648. [Google Scholar] [CrossRef]
- Everard, A.; Belzer, C.; Geurts, L.; Ouwerkerk, J.P.; Druart, C.; Bindels, L.B.; Guiot, Y.; Derrien, M.; Muccioli, G.G.; Delzenne, N.M.; et al. Cross-talk between Akkermansia muciniphila and intestinal epithelium controls diet-induced obesity. Proc. Natl. Acad. Sci. USA 2013, 110, 9066–9071. [Google Scholar] [CrossRef]
- Karcher, N.; Nigro, E.; Puncochar, M.; Blanco-Miguez, A.; Ciciani, M.; Manghi, P.; Zolfo, M.; Cumbo, F.; Manara, S.; Golzato, D.; et al. Genomic diversity and ecology of human-associated Akkermansia species in the gut microbiome revealed by extensive metagenomic assembly. Genome Biol. 2021, 22, 209. [Google Scholar] [CrossRef] [PubMed]
- Xu, Y.; Wang, N.; Tan, H.-Y.; Li, S.; Zhang, C.; Feng, Y. Function of Akkermansia muciniphila in Obesity: Interactions With Lipid Metabolism, Immune Response and Gut Systems. Front. Microbiol. 2020, 11, 219. [Google Scholar] [CrossRef]
- Dubourg, G.; Lagier, J.-C.; Armougom, F.; Robert, C.; Audoly, G.; Papazian, L.; Raoult, D. High-level colonisation of the human gut by Verrucomicrobia following broad-spectrum antibiotic treatment. Int. J. Antimicrob. Agents 2013, 41, 149–155. [Google Scholar] [CrossRef]
- Zeng, S.-Y.; Liu, Y.-F.; Liu, J.-H.; Zeng, Z.-L.; Xie, H. Potential Effects of Akkermansia Muciniphila in Aging and Aging-Related Diseases: Current Evidence and Perspectives. Aging Dis. 2023, 14, 2015–2027. [Google Scholar] [CrossRef]
- Singh, H.; Torralba, M.G.; Moncera, K.J.; DiLello, L.; Petrini, J.; Nelson, K.E.; Pieper, R. Gastro-intestinal and oral microbiome signatures associated with healthy aging. GeroScience 2019, 41, 907–921. [Google Scholar] [CrossRef] [PubMed]
- Palmas, V.; Pisanu, S.; Madau, V.; Casula, E.; Deledda, A.; Cusano, R.; Uva, P.; Loviselli, A.; Velluzzi, F.; Manzin, A. Gut Microbiota Markers and Dietary Habits Associated with Extreme Longevity in Healthy Sardinian Centenarians. Nutrients 2022, 14, 2436. [Google Scholar] [CrossRef] [PubMed]
- Barcena, C.; Valdes-Mas, R.; Mayoral, P.; Garabaya, C.; Durand, S.; Rodriguez, F.; Fernandez-Garcia, M.T.; Salazar, N.; Nogacka, A.M.; Garatachea, N.; et al. Healthspan and lifespan extension by fecal microbiota transplantation into progeroid mice. Nat. Med. 2019, 25, 1234–1242. [Google Scholar] [CrossRef] [PubMed]
- Zhang, N.; Zhang, Y.; Wang, Z.; Pan, F.; Ren, R.; Li, Z.; Zhao, H.; Luo, X.; Li, Z.; Wang, L.; et al. Regular fecal microbiota transplantation to Senescence Accelerated Mouse-Prone 8 (SAMP8) mice delayed the aging of locomotor and exploration ability by rejuvenating the gut microbiota. Front. Aging Neurosci. 2022, 14, 991157. [Google Scholar] [CrossRef]
- Ijaz, M.U.; Ahmed, M.I.; Zou, X.; Hussain, M.; Zhang, M.; Zhao, F.; Xu, X.; Zhou, G.; Li, C. Beef, Casein, and Soy Proteins Differentially Affect Lipid Metabolism, Triglycerides Accumulation and Gut Microbiota of High-Fat Diet-Fed C57BL/6J Mice. Front. Microbiol. 2018, 9, 2200. [Google Scholar] [CrossRef]
- Ma, Z. Testing the Anna Karenina Principle in Human Microbiome-Associated Diseases. iScience 2020, 23, 101007. [Google Scholar] [CrossRef]
- Zaneveld, J.R.; McMinds, R.; Thurber, R.V. Stress and stability: Applying the Anna Karenina principle to animal microbiomes. Nat. Microbiol. 2017, 2, 17121. [Google Scholar] [CrossRef] [PubMed]
- Liu, M.; Liu, H.; Shi, M.; Jiang, M.; Li, L.; Zheng, Y. Microbial production of ectoine and hydroxyectoine as high-value chemicals. Microb. Cell Factories 2021, 20, 76. [Google Scholar] [CrossRef] [PubMed]
- Koistinen, V.M.; Kärkkäinen, O.; Borewicz, K.; Zarei, I.; Jokkala, J.; Micard, V.; Rosa-Sibakov, N.; Auriola, S.; Aura, A.-M.; Smidt, H.; et al. Contribution of gut microbiota to metabolism of dietary glycine betaine in mice and in vitro colonic fermentation. Microbiome 2019, 7, 103. [Google Scholar] [CrossRef] [PubMed]
- Ticak, T.; Kountz, D.J.; Girosky, K.E.; Krzycki, J.A.; Ferguson, D.J. A nonpyrrolysine member of the widely distributed trimethylamine methyltransferase family is a glycine betaine methyltransferase. Proc. Natl. Acad. Sci. USA 2014, 111, E4668–E4676. [Google Scholar] [CrossRef] [PubMed]
- Rampelli, S.; Soverini, M.; D’Amico, F.; Barone, M.; Tavella, T.; Monti, D.; Capri, M.; Astolfi, A.; Brigidi, P.; Biagi, E.; et al. Shotgun Metagenomics of Human Gut Microbiota Up to Extreme Longevity and the Increasing Role of Xenobiotics Degradation. SSRN Electron. J. 2019, 5, 1–21. [Google Scholar] [CrossRef]
- Mach, N.; Clark, A. Micronutrient Deficiencies and the Human Gut Microbiota. Trends Microbiol. 2017, 25, 607–610. [Google Scholar] [CrossRef] [PubMed]
- Rosier, B.T.; Buetas, E.; Moya-Gonzalvez, E.M.; Artacho, A.; Mira, A.; Rosier, B.T.; Buetas, E.; Moya-Gonzalvez, E.M.; Artacho, A.; Mira, A. Nitrate as a potential prebiotic for the oral microbiome. Sci. Rep. 2020, 10, 12895. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.-Y.; Tsolis, R.M.; Bäumler, A.J. The microbiome and gut homeostasis. Science 2022, 377, eabp9960. [Google Scholar] [CrossRef] [PubMed]
- Ma, H.-D.; Wang, Y.-H.; Chang, C.; Gershwin, M.E.; Lian, Z.-X. The intestinal microbiota and microenvironment in liver. Autoimmun. Rev. 2014, 14, 183–191. [Google Scholar] [CrossRef]
- Zaiatz-Bittencourt, V.; Jones, F.; Tosetto, M.; Scaife, C.; Cagney, G.; Jones, E.; Doherty, G.A.; Ryan, E.J. Butyrate limits human natural killer cell effector function. Sci. Rep. 2023, 13, 2715. [Google Scholar] [CrossRef]
- Barbosa, P.; Melnyk, S.; Bennuri, S.C.; Delhey, L.; Reis, A.; Moura, G.R.; Borsheim, E.; Rose, S.; Carvalho, E. Redox Imbalance and Methylation Disturbances in Early Childhood Obesity. Oxidative Med. Cell. Longev. 2021, 2021, 2207125. [Google Scholar] [CrossRef] [PubMed]
- Qiao, Y.; Sun, J.; Ding, Y.; Le, G.; Shi, Y. Alterations of the gut microbiota in high-fat diet mice is strongly linked to oxidative stress. Appl. Microbiol. Biotechnol. 2012, 97, 1689–1697. [Google Scholar] [CrossRef] [PubMed]
- Fouesnard, M.; Zoppi, J.; Petera, M.; Le Gleau, L.; Migne, C.; Devime, F.; Durand, S.; Benani, A.; Chaffron, S.; Douard, V.; et al. Dietary switch to Western diet induces hypothalamic adaptation associated with gut microbiota dysbiosis in rats. Int. J. Obes. 2021, 45, 1271–1283. [Google Scholar] [CrossRef]
- Schwalm, N.D.; Groisman, E.A. Navigating the Gut Buffet: Control of Polysaccharide Utilization in Bacteroides spp. Trends Microbiol. 2017, 25, 1005–1015. [Google Scholar] [CrossRef] [PubMed]
- Mathlouthi, N.E.H.; Kriaa, A.; Keskes, L.A.; Rhimi, M.; Gdoura, R. Virulence Factors in Colorectal Cancer Metagenomes and Association of Microbial Siderophores with Advanced Stages. Microorganisms 2022, 10, 2365. [Google Scholar] [CrossRef]
- Vidulin, V.; Šmuc, T.; Džeroski, S.; Supek, F. The evolutionary signal in metagenome phyletic profiles predicts many gene functions. Microbiome 2018, 6, 129. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Ingredient | HFBN Dietary Components | HFB Dietary Components |
|---|---|---|
| Ammonium-supplemented beef (cooked, freeze-dried) | 352.42 g | 0 g |
| Unsupplemented beef (cooked, freeze-dried) | 0 g | 293.64 g |
| L-cystine | 3 g | 3 g |
| Corn starch | 68.68 g | 68.46 g |
| Maltodextrin 10 | 100 g | 100 g |
| Sucrose | 175.21 g | 175.21 g |
| Cellulose | 45.07 g | 48.53 g |
| Soybean oil | 25 g | 25 g |
| Beef fat, Bunge | 23.28 g | 78.76 g |
| Mineral mix S10026A (No Ca, P, K, Na, Cl) | 5 g | 5 g |
| Dicalcium phosphate | 13 g | 13 g |
| Calcium carbonate | 5.5 g | 5.5 g |
| Potassium citrate, 1 H2O | 16.5 g | 16.5 g |
| Sodium chloride | 1.088 g | 0.87 g |
| Vitamin mix V10001 | 10 g | 10 g |
| Choline bitartrate | 2 g | 2 g |
| Cholesterol | 0 g | 0.02 g |
| Yellow dye #5 FD&C | 0 g | 0.025 g |
| Red dye #40 FD&C | 0.025 g | 0.025 g |
| Blue dye #1 FD&C | 0.025 g | 0 g |
| TOTAL MASS | 845.798 g | 845.54 g |
| Protein | 18% of Kcal | 18% of Kcal |
| Carbohydrates | 36% of Kcal | 36% of Kcal |
| Fats | 46% of Kcal | 46% of Kcal |
| TOTAL Kcal% | 100% of Kcal | 100% of Kcal |
| Average Food Consumed per Mouse (g) | Standard Error | |
|---|---|---|
| HFBN-F | 15.3 | ±0.02 |
| HFBN-M | 21.4 | ±0.04 |
| HFB-F | 16.8 | ±0.03 |
| HFB-M | 19.8 | ±0.03 |
| Raw Reads (Millions) | Number of Contigs | Maximum Contig Length | Total Contig Length (Mbp) | Assembly N50 | Number of Genes Annotated | Unique Gene Orthologs with Known Function | |
|---|---|---|---|---|---|---|---|
| HFBN-F | 76.80 | 180,626 | 457,219 | 173.05 | 2355 | 176,033 | 3931 |
| HFBN-M | 66.90 | 342,839 | 447,818 | 322.73 | 2837 | 325,674 | 4319 |
| HFB-F | 62.73 | 219,182 | 684,229 | 246.30 | 2575 | 213,531 | 4214 |
| HFB-M | 62.32 | 250,995 | 447,727 | 257.89 | 3141 | 239,786 | 4359 |
| Ammonium Beef Diet (HFBN) | p-Value | Control Diet (HFB) | p-Value |
|---|---|---|---|
| Comparison 1 | |||
| GO Biological Process: | GO Biological Process: | ||
| glycine betaine transport | 0.0042 | glutathione metabolic process | 0.0041 |
| xenobiotic detoxification by transmembrane export across the plasma membrane | 0.0092 | response to oxidative stress | 0.0075 |
| defense response to bacterium | 0.0198 | ion transport | 0.0185 |
| nitrate assimilation | 0.0371 | respiratory electron transport chain | 0.0331 |
| anaerobic respiration | 0.0371 | organic phosphonate metabolic process | 0.0451 |
| polysaccharide transport | 0.0457 | ||
| sucrose metabolic process | 0.0457 | ||
| dormancy process | 0.0457 | ||
| aromatic compound catabolic process | 0.0472 | ||
| GO Molecular Function: | GO Molecular Function: | ||
| phosphorelay sensor kinase activity | 0.00068 | transmembrane transporter activity | 0.0023 |
| phosphopantetheine binding | 0.00882 | glutathione transferase activity | 0.0036 |
| lipoic acid binding | 0.01292 | porin activity | 0.0066 |
| catalytic activity | 0.02998 | copper ion binding | 0.0162 |
| toxin activity | 0.03361 | carbohydrate:proton symporter activity | 0.024 |
| protein serine/threonine kinase activity | 0.03361 | glucose transmembrane transporter activity | 0.0469 |
| flavin adenine dinucleotide binding | 0.0487 | ||
| GO Cellular Component: | GO Cellular Component: | ||
| integral component of membrane | 0.019 | pore complex | 0.00089 |
| cell outer membrane | 0.00107 | ||
| periplasmic space | 0.00349 | ||
| ATP-binding cassette (ABC) transporter complex | 0.01176 | ||
| integral component of membrane | 0.0157 | ||
| Comparison 2 | |||
| Females | p-Value | Males | p-Value |
| GO Biological Process: | GO Biological Process: | ||
| response to toxic substance | 0.012 | glutathione metabolic process | 0.004 |
| aerobic electron transport chain | 0.013 | ion transport | 0.018 |
| nitrate assimilation | 0.023 | glycine betaine transport | 0.031 |
| carotenoid biosynthetic process | 0.024 | response to oxidative stress | 0.036 |
| heme metabolic process | 0.031 | organic phosphonate metabolic process | 0.044 |
| polysaccharide transport | 0.045 | ||
| dormancy process | 0.045 | ||
| cell adhesion | 0.049 | ||
| GO Molecular Function: | GO Molecular Function: | ||
| FAD binding | 0.032 | phosphopantetheine binding | 0.0011 |
| fatty acid binding | 0.032 | glutathione transferase activity | 0.0035 |
| N-acetylmuramoyl-l-alanine amidase activity | 0.039 | l-phosphoserine phosphatase activity | 0.0035 |
| porin activity | 0.0065 | ||
| flavin adenine dinucleotide binding | 0.0168 | ||
| 3-oxoacyl-[acyl-carrier-protein] synthase activity | 0.0236 | ||
| phosphorelay sensor kinase activity | 0.0375 | ||
| anti-sigma factor antagonist activity | 0.0463 | ||
| GO Cellular Component: | |||
| pore complex | 0.00091 | ||
| cell outer membrane | 0.00425 | ||
| periplasmic space | 0.01042 | ||
| integral component of membrane | 0.0165 | ||
| outer membrane | 0.04496 | ||
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Garrison, E.C.; Brown, A.M.V.; Salazar, M.M.; Barr, B.; Moustaid-Moussa, N.; Gollahon, L.S. Microbiome Taxonomic and Functional Differences in C3H/HeJ Mice Fed a Long-Term High-Fat Diet with Beef Protein ± Ammonium Hydroxide Supplementation. Nutrients 2024, 16, 1613. https://doi.org/10.3390/nu16111613
Garrison EC, Brown AMV, Salazar MM, Barr B, Moustaid-Moussa N, Gollahon LS. Microbiome Taxonomic and Functional Differences in C3H/HeJ Mice Fed a Long-Term High-Fat Diet with Beef Protein ± Ammonium Hydroxide Supplementation. Nutrients. 2024; 16(11):1613. https://doi.org/10.3390/nu16111613
Chicago/Turabian StyleGarrison, Emily C., Amanda M. V. Brown, McKinlee M. Salazar, Benjamin Barr, Naima Moustaid-Moussa, and Lauren S. Gollahon. 2024. "Microbiome Taxonomic and Functional Differences in C3H/HeJ Mice Fed a Long-Term High-Fat Diet with Beef Protein ± Ammonium Hydroxide Supplementation" Nutrients 16, no. 11: 1613. https://doi.org/10.3390/nu16111613