
Variation in the Conservation of Species-Specific Gene Sets for HMO Degradation and Its Effects on HMO Utilization in Bifidobacteria


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Bacterial Strains and Growth Conditions
2.2. Growth on Carbohydrate Sources
2.3. Genome Sequencing, Assembly, Annotation, and GH Identification
2.4. Homology Searches for HMO Utilization Genes
3. Results and Discussion
3.1. Genome Sequencing
3.2. Common HMO Degradation Machinery of Human Bifidobacteria
3.2.1. GHs Involved in Degradation of Neutral Type I and II Chain HMOs
3.2.2. Lnp Locus
3.2.3. Fucosyllactose–Fucose Utilization Loci
3.3. Species-Specific HMO Degradation Patterns and Genetic Machinery
3.3.1. B. breve
Results
Discussion
3.3.2. B. longum subsp. infantis
Results
Discussion
3.3.3. B. longum subsp. longum
Results
Discussion
3.3.4. B. bifidum
Results
Discussion
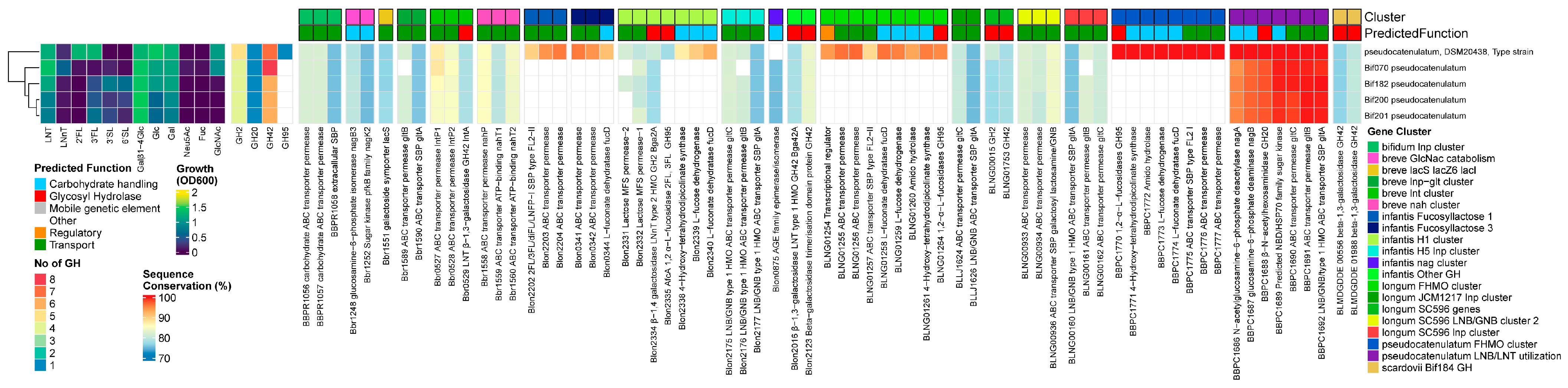
3.3.5. B. pseudocatenulatum
Results
Discussion
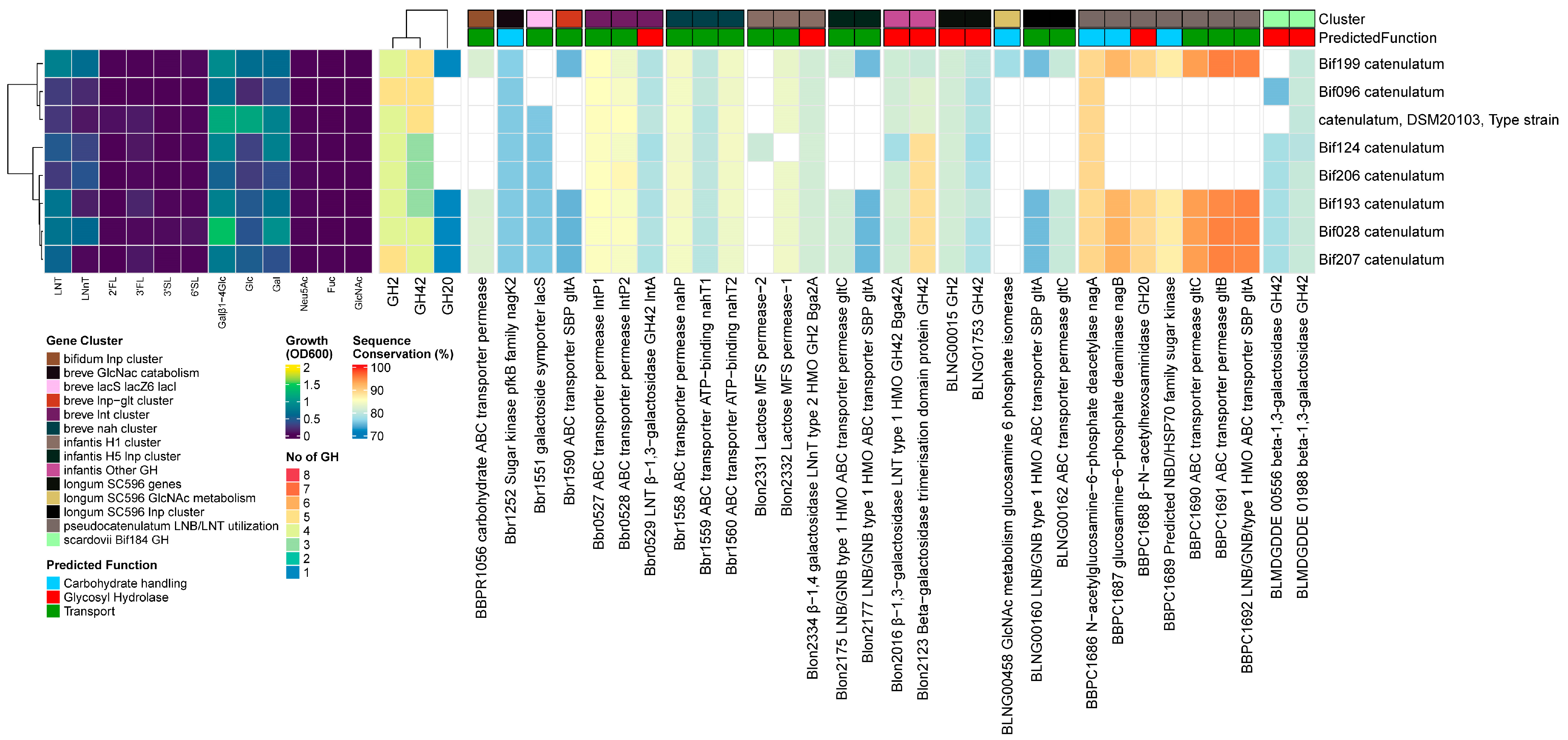
3.3.6. B. catenulatum
Results
Discussion
3.3.7. B. adolescentis
Results
Discussion
3.3.8. B. scardovii
Results
Discussion
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Wampach, L.; Heintz-Buschart, A.; Fritz, J.V.; Ramiro-Garcia, J.; Habier, J.; Herold, M.; Narayanasamy, S.; Kaysen, A.; Hogan, A.H.; Bindl, L.; et al. Birth Mode Is Associated with Earliest Strain-Conferred Gut Microbiome Functions and Immunostimulatory Potential. Nat. Commun. 2018, 9, 5091. [Google Scholar] [CrossRef] [PubMed]
- Tamburini, S.; Shen, N.; Wu, H.C.; Clemente, J.C. The Microbiome in Early Life: Implications for Health Outcomes. Nat. Med. 2016, 22, 713–722. [Google Scholar] [CrossRef] [PubMed]
- UNICEF; WHO. Capture the Moment—Early Initiation of Breastfeeding: The Best Start for Every Newborn; UNICEF: New York, NY, USA, 2018; ISBN 9789280649765. Available online: https://www.unicef.org/eca/media/4256/file/Capture-the-moment-EIBF-report.pdf (accessed on 12 May 2024).
- Bokulich, N.A.; Chung, J.; Battaglia, T.; Henderson, N.; Jay, M.; Li, H.; Lieber, A.D.; Wu, F.; Perez-Perez, G.I.; Chen, Y.; et al. Antibiotics, Birth Mode, and Diet Shape Microbiome Maturation during Early Life. Sci. Transl. Med. 2016, 8, 343ra82. [Google Scholar] [CrossRef] [PubMed]
- Hermes, G.D.A.; Eckermann, H.A.; de Vos, W.M.; de Weerth, C. Does Entry to Center-Based Childcare Affect Gut Microbial Colonization in Young Infants? Sci. Rep. 2020, 10, 10235. [Google Scholar] [CrossRef] [PubMed]
- Van Daele, E.; Kamphorst, K.; Vlieger, A.M.; Hermes, G.; Milani, C.; Ventura, M.; Belzer, C.; Smidt, H.; van Elburg, R.M.; Knol, J. Effect of Antibiotics in the First Week of Life on Faecal Microbiota Development. Arch. Dis. Child. Fetal Neonatal Ed. 2022, 107, 603–610. [Google Scholar] [CrossRef] [PubMed]
- Rothschild, D.; Weissbrod, O.; Barkan, E.; Kurilshikov, A.; Korem, T.; Zeevi, D.; Costea, P.I.; Godneva, A.; Kalka, I.N.; Bar, N.; et al. Environment Dominates over Host Genetics in Shaping Human Gut Microbiota. Nature 2018, 555, 210–215. [Google Scholar] [CrossRef]
- Bogaert, D.; van Beveren, G.J.; de Koff, E.M.; Lusarreta Parga, P.; Balcazar Lopez, C.E.; Koppensteiner, L.; Clerc, M.; Hasrat, R.; Arp, K.; Chu, M.L.J.N.; et al. Mother-to-Infant Microbiota Transmission and Infant Microbiota Development across Multiple Body Sites. Cell Host Microbe 2023, 31, 447–460.e6. [Google Scholar] [CrossRef]
- Melsaether, C.; Høtoft, D.; Wellejus, A.; Hermes, G.D.A.; Damholt, A. Seeding the Infant Gut in Early Life-Effects of Maternal and Infant Seeding with Probiotics on Strain Transfer, Microbiota, and Gastrointestinal Symptoms in Healthy Breastfed Infants. Nutrients 2023, 15, 4000. [Google Scholar] [CrossRef] [PubMed]
- De Agüero, M.G.; Ganal-Vonarburg, S.C.; Fuhrer, T.; Rupp, S.; Uchimura, Y.; Li, H.; Steinert, A.; Heikenwalder, M.; Hapfelmeier, S.; Sauer, U.; et al. The Maternal Microbiota Drives Early Postnatal Innate Immune Development. Science 2016, 351, 1296–1302. [Google Scholar] [CrossRef]
- Ventura, M.; Canchaya, C.; Tauch, A.; Chandra, G.; Fitzgerald, G.F.; Chater, K.F.; van Sinderen, D. Genomics of Actinobacteria: Tracing the Evolutionary History of an Ancient Phylum. Microbiol. Mol. Biol. Rev. 2007, 71, 495–548. [Google Scholar] [CrossRef]
- Turroni, F.; Foroni, E.; Pizzetti, P.; Giubellini, V.; Ribbera, A.; Merusi, P.; Cagnasso, P.; Bizzarri, B.; De’Angelis, G.L.; Shanahan, F.; et al. Exploring the Diversity of the Bifidobacterial Population in the Human Intestinal Tract. Appl. Environ. Microbiol. 2009, 75, 1534–1545. [Google Scholar] [CrossRef] [PubMed]
- Avershina, E.; Storrø, O.; Øien, T.; Johnsen, R.; Wilson, R.; Egeland, T.; Rudia, K. Bifidobacterial Succession and Correlation Networks in a Large Unselected Cohort of Mothers and Their Children. Appl. Environ. Microbiol. 2013, 79, 497–507. [Google Scholar] [CrossRef] [PubMed]
- Kujawska, M.; La Rosa, S.L.; Roger, L.C.; Pope, P.B.; Hoyles, L.; McCartney, A.L.; Hall, L.J. Succession of Bifidobacterium Longum Strains in Response to a Changing Early Life Nutritional Environment Reveals Dietary Substrate Adaptations. iScience 2020, 23, 101368. [Google Scholar] [CrossRef] [PubMed]
- Arboleya, S.; Bottacini, F.; O’Connell-Motherway, M.; Ryan, C.A.; Ross, R.P.; van Sinderen, D.; Stanton, C. Gene-Trait Matching across the Bifidobacterium Longum Pan-Genome Reveals Considerable Diversity in Carbohydrate Catabolism among Human Infant Strains. BMC Genom. 2018, 19, 33. [Google Scholar] [CrossRef] [PubMed]
- Chung The, H.; Nguyen Ngoc Minh, C.; Tran Thi Hong, C.; Nguyen Thi Nguyen, T.; Pike, L.J.; Zellmer, C.; Pham Duc, T.; Tran, T.-A.; Ha Thanh, T.; Van, M.P.; et al. Exploring the Genomic Diversity and Antimicrobial Susceptibility of Bifidobacterium Pseudocatenulatum in a Vietnamese Population. Microbiol. Spectr. 2021, 9, e00526-21. [Google Scholar] [CrossRef] [PubMed]
- Bunesova, V.; Lacroix, C.; Schwab, C. Fucosyllactose and L-Fucose Utilization of Infant Bifidobacterium Longum and Bifidobacterium Kashiwanohense. BMC Microbiol. 2016, 16, 248. [Google Scholar] [CrossRef] [PubMed]
- Lawson, M.A.E.; O’Neill, I.J.; Kujawska, M.; Gowrinadh Javvadi, S.; Wijeyesekera, A.; Flegg, Z.; Chalklen, L.; Hall, L.J. Breast Milk-Derived Human Milk Oligosaccharides Promote Bifidobacterium Interactions within a Single Ecosystem. ISME J. 2019, 14, 635–648. [Google Scholar] [CrossRef]
- Shani, G.; Hoeflinger, J.L.; Heiss, B.E.; Masarweh, C.F.; Larke, J.A.; Jensen, N.M.; Wickramasinghe, S.; Davis, J.C.; Goonatilleke, E.; El-Hawiet, A.; et al. Fucosylated Human Milk Oligosaccharide Foraging within the Species Bifidobacterium Pseudocatenulatum Is Driven by Glycosyl Hydrolase Content and Specificity. Appl. Environ. Microbiol. 2022, 88, e01707-21. [Google Scholar] [CrossRef] [PubMed]
- Ruiz-Moyano, S.; Totten, S.M.; Garrido, D.A.; Smilowitz, J.T.; Bruce German, J.; Lebrilla, C.B.; Mills, D.A. Variation in Consumption of Human Milk Oligosaccharides by Infant Gut-Associated Strains of Bifidobacterium Breve. Appl. Environ. Microbiol. 2013, 79, 6040–6049. [Google Scholar] [CrossRef]
- Matsuki, T.; Yahagi, K.; Mori, H.; Matsumoto, H.; Hara, T.; Tajima, S.; Ogawa, E.; Kodama, H.; Yamamoto, K.; Yamada, T.; et al. A Key Genetic Factor for Fucosyllactose Utilization Affects Infant Gut Microbiota Development. Nat. Commun. 2016, 7, 11939. [Google Scholar] [CrossRef]
- Engfer, M.B.; Stahl, B.; Finke, B.; Sawatzki, G.; Daniel, H. Human Milk Oligosaccharides Are Resistant to Enzymatic Hydrolysis in the Upper Gastrointestinal Tract. Am. J. Clin. Nutr. 2000, 71, 1589–1596. [Google Scholar] [CrossRef] [PubMed]
- Parschat, K.; Melsaether, C.; Jäpelt, K.R.; Jennewein, S. Clinical Evaluation of 16-Week Supplementation with 5HMO-Mix in Healthy-Term Human Infants to Determine Tolerability, Safety, and Effect on Growth. Nutrients 2021, 13, 2871. [Google Scholar] [CrossRef] [PubMed]
- Erney, R.M.; Malone, W.T.; Skelding, M.B.; Marcon, A.A.; Kleman–Leyer, K.M.; O’Ryan, M.L.; Ruiz–Palacios, G.; Hilty, M.D.; Pickering, L.K.; Prieto, P.A. Variability of Human Milk Neutral Oligosaccharides in a Diverse Population. J. Pediatr. Gastroenterol. Nutr. 2000, 30, 181–192. [Google Scholar] [PubMed]
- McGuire, M.K.; Meehan, C.L.; McGuire, M.A.; Williams, J.E.; Foster, J.; Sellen, D.W.; Kamau-Mbuthia, E.W.; Kamundia, E.W.; Mbugua, S.; Moore, S.E.; et al. What’s Normal? Oligosaccharide Concentrations and Profiles in Milk Produced by Healthy Women Vary Geographically. Am. J. Clin. Nutr. 2017, 105, 1086. [Google Scholar] [CrossRef] [PubMed]
- Duar, R.M.; Casaburi, G.; Mitchell, R.D.; Scofield, L.N.C.; Ortega Ramirez, C.A.; Barile, D.; Henrick, B.M.; Frese, S.A. Comparative Genome Analysis of Bifidobacterium Longum Subsp. Infantis Strains Reveals Variation in Human Milk Oligosaccharide Utilization Genes among Commercial Probiotics. Nutrients 2020, 12, 3247. [Google Scholar] [CrossRef] [PubMed]
- Zabel, B.E.; Gerdes, S.; Evans, K.C.; Nedveck, D.; Singles, S.K.; Volk, B.; Budinoff, C. Strain-Specific Strategies of 2′-Fucosyllactose, 3-Fucosyllactose, and Difucosyllactose Assimilation by Bifidobacterium Longum Subsp. Infantis Bi-26 and ATCC 15697. Sci. Rep. 2020, 10, 15919. [Google Scholar] [CrossRef] [PubMed]
- Sela, D.A.; Garrido, D.; Lerno, L.; Wu, S.; Tan, K.; Eom, H.J.; Joachimiak, A.; Lebrilla, C.B.; Mills, D.A. Bifidobacterium Longum Subsp. Infantis ATCC 15697 α-Fucosidases Are Active on Fucosylated Human Milk Oligosaccharides. Appl. Environ. Microbiol. 2012, 78, 795–803. [Google Scholar] [CrossRef] [PubMed]
- Ward, R.E.; Niñonuevo, M.; Mills, D.A.; Lebrilla, C.B.; German, J.B. In Vitro Fermentability of Human Milk Oligosaccharides by Several Strains of Bifidobacteria. Mol. Nutr. Food Res. 2007, 51, 1398–1405. [Google Scholar] [CrossRef] [PubMed]
- Garrido, D.; Ruiz-Moyano, S.; Lemay, D.G.; Sela, D.A.; German, J.B.; Mills, D.A. Comparative Transcriptomics Reveals Key Differences in the Response to Milk Oligosaccharides of Infant Gut-Associated Bifidobacteria. Sci. Rep. 2015, 5, 13517. [Google Scholar] [CrossRef] [PubMed]
- Garrido, D.; Ruiz-Moyano, S.; Kirmiz, N.; Davis, J.C.; Totten, S.M.; Lemay, D.G.; Ugalde, J.A.; German, J.B.; Lebrilla, C.B.; Mills, D.A. A Novel Gene Cluster Allows Preferential Utilization of Fucosylated Milk Oligosaccharides in Bifidobacterium Longum Subsp. Longum SC596. Sci. Rep. 2016, 6, 35045. [Google Scholar] [CrossRef]
- James, K.; Motherway, M.O.C.; Bottacini, F.; Van Sinderen, D. Bifidobacterium Breve UCC2003 Metabolises the Human Milk Oligosaccharides Lacto-N-Tetraose and Lacto-N-Neo-Tetraose through Overlapping, yet Distinct Pathways. Sci. Rep. 2016, 6, 38560. [Google Scholar] [CrossRef] [PubMed]
- Özcan, E.; Sela, D.A. Inefficient Metabolism of the Human Milk Oligosaccharides Lacto-N-Tetraose and Lacto-N-Neotetraose Shifts Bifidobacterium Longum Subsp. Infantis Physiology. Front. Nutr. 2018, 5, 46. [Google Scholar] [CrossRef] [PubMed]
- Nishiyama, K.; Nagai, A.; Uribayashi, K.; Yamamoto, Y.; Mukai, T.; Okada, N. Two Extracellular Sialidases from Bifidobacterium Bifidum Promote the Degradation of Sialyl-Oligosaccharides and Support the Growth of Bifidobacterium Breve. Anaerobe 2018, 52, 22–28. [Google Scholar] [CrossRef] [PubMed]
- Wong, C.B.; Odamaki, T.; Xiao, J.Z. Insights into the Reason of Human-Residential Bifidobacteria (HRB) Being the Natural Inhabitants of the Human Gut and Their Potential Health-Promoting Benefits. FEMS Microbiol. Rev. 2020, 44, 369–385. [Google Scholar] [CrossRef] [PubMed]
- Katayama, T.; Sakuma, A.; Kimura, T.; Makimura, Y.; Hiratake, J.; Sakata, K.; Yamanoi, T.; Kumagai, H.; Yamamoto, K. Molecular Cloning and Characterization of Bifidobacterium Bifidum 1,2-α-L-Fucosidase (AfcA), a Novel Inverting Glycosidase (Glycoside Hydrolase Family 95). J. Bacteriol. 2004, 186, 4885–4893. [Google Scholar] [CrossRef] [PubMed]
- Yoshida, E.; Sakurama, H.; Kiyohara, M.; Nakajima, M.; Kitaoka, M.; Ashida, H.; Hirose, J.; Katayama, T.; Yamamoto, K.; Kumagai, H. Bifidobacterium Longum Subsp. Infantis Uses Two Different β-Galactosidases for Selectively Degrading Type-1 and Type-2 Human Milk Oligosaccharides. Glycobiology 2012, 22, 361–368. [Google Scholar] [CrossRef] [PubMed]
- Kitaoka, M.; Tian, J.; Nishimoto, M. Novel Putative Galactose Operon Involving Lacto-N-Biose Phosphorylase in Bifidobacterium Longum. Appl. Environ. Microbiol. 2005, 71, 3158–3162. [Google Scholar] [CrossRef] [PubMed]
- Fujita, K.; Oura, F.; Nagamine, N.; Katayama, T.; Hiratake, J.; Sakata, K.; Kumagai, H.; Yamamoto, K. Identification and Molecular Cloning of a Novel Glycoside Hydrolase Family of Core 1 Type O-Glycan-Specific Endo-Alpha-N-Acetylgalactosaminidase from Bifidobacterium Longum. J. Biol. Chem. 2005, 280, 37415–37422. [Google Scholar] [CrossRef] [PubMed]
- Nagae, M.; Tsuchiya, A.; Katayama, T.; Yamamoto, K.; Wakatsuki, S.; Kato, R. Structural Basis of the Catalytic Reaction Mechanism of Novel 1,2-Alpha-L-Fucosidase from Bifidobacterium Bifidum. J. Biol. Chem. 2007, 282, 18497–18509. [Google Scholar] [CrossRef]
- Wada, J.; Ando, T.; Kiyohara, M.; Ashida, H.; Kitaoka, M.; Yamaguchi, M.; Kumagai, H.; Katayama, T.; Yamamoto, K. Bifidobacterium Bifidum Lacto-N-Biosidase, a Critical Enzyme for the Degradation of Human Milk Oligosaccharides with a Type 1 Structure. Appl. Environ. Microbiol. 2008, 74, 3996–4004. [Google Scholar] [CrossRef]
- Goulas, T.; Goulas, A.; Tzortzis, G.; Gibson, G.R. Comparative Analysis of Four β-Galactosidases from Bifidobacterium Bifidum NCIMB41171: Purification and Biochemical Characterisation. Appl. Microbiol. Biotechnol. 2009, 82, 1079–1088. [Google Scholar] [CrossRef] [PubMed]
- Ashida, H.; Miyake, A.; Kiyohara, M.; Wada, J.; Yoshida, E.; Kumagai, H.; Katayama, T.; Yamamoto, K. Two Distinct α-L-Fucosidases from Bifidobacterium Bifidum Are Essential for the Utilization of Fucosylated Milk Oligosaccharides and Glycoconjugates. Glycobiology 2009, 19, 1010–1017. [Google Scholar] [CrossRef]
- Miwa, M.; Horimoto, T.; Kiyohara, M.; Katayama, T.; Kitaoka, M.; Ashida, H.; Yamamoto, K. Cooperation of β-Galactosidase and β-N-Acetylhexosaminidase from Bifidobacteria in Assimilation of Human Milk Oligosaccharides with Type 2 Structure. Glycobiology 2010, 20, 1402–1409. [Google Scholar] [CrossRef]
- Garrido, D.; Ruiz-Moyano, S.; Mills, D.A. Release and Utilization of N-Acetyl-d-Glucosamine from Human Milk Oligosaccharides by Bifidobacterium Longum Subsp. Infantis. Anaerobe 2012, 18, 430–435. [Google Scholar] [CrossRef]
- Kiyohara, M.; Tanigawa, K.; Chaiwangsri, T.; Katayama, T.; Ashida, H.; Yamamoto, K. An Exo—Sialidase from Bifidobacteria Involved in the Degradation of Sialyloligosaccharides in Human Milk and Intestinal Glycoconjugates. Glycobiology 2011, 21, 437–447. [Google Scholar] [CrossRef] [PubMed]
- Hidaka, M.; Nishimoto, M.; Kitaoka, M.; Wakagi, T.; Shoun, H.; Fushinobu, S. The Crystal Structure of Galacto-N-Biose/Lacto-N-Biose I Phosphorylase. A Large Deformation of a Tim Barrel Scaffold. J. Biol. Chem. 2009, 284, 7273–7283. [Google Scholar] [CrossRef]
- Turroni, F.; Bottacini, F.; Foroni, E.; Mulder, I.; Kim, J.H.; Zomer, A.; Sánchez, B.; Bidossi, A.; Ferrarini, A.; Giubellini, V.; et al. Genome Analysis of Bifidobacterium Bifidum PRL2010 Reveals Metabolic Pathways for Host-Derived Glycan Foraging. Proc. Natl. Acad. Sci. USA 2010, 107, 19514–19519. [Google Scholar] [CrossRef] [PubMed]
- Sakanaka, M.; Gotoh, A.; Yoshida, K.; Odamaki, T.; Koguchi, H.; Xiao, J.Z.; Kitaoka, M.; Katayama, T. Varied Pathways of Infant Gut-Associated Bifidobacterium to Assimilate Human Milk Oligosaccharides: Prevalence of the Gene Set and Its Correlation with Bifidobacteria-Rich Microbiota Formation. Nutrients 2020, 12, 71. [Google Scholar] [CrossRef]
- Garrido, D.; Kim, J.H.; German, J.B.; Raybould, H.E.; Mills, D.A. Oligosaccharide Binding Proteins from Bifidobacterium Longum Subsp. Infantis Reveal a Preference for Host Glycans. PLoS ONE 2011, 6, e17315. [Google Scholar] [CrossRef]
- Bankevich, A.; Nurk, S.; Antipov, D.; Gurevich, A.A.; Dvorkin, M.; Kulikov, A.S.; Lesin, V.M.; Nikolenko, S.I.; Pham, S.; Prjibelski, A.D.; et al. SPAdes: A New Genome Assembly Algorithm and Its Applications to Single-Cell Sequencing. J. Comput. Biol. 2012, 19, 455. [Google Scholar] [CrossRef]
- Seemann, T. Prokka: Rapid Prokaryotic Genome Annotation. Bioinformatics 2014, 30, 2068–2069. [Google Scholar] [CrossRef] [PubMed]
- Chklovski, A.; Parks, D.H.; Woodcroft, B.J.; Tyson, G.W. CheckM2: A Rapid, Scalable and Accurate Tool for Assessing Microbial Genome Quality Using Machine Learning. Nat. Methods 2023, 20, 1203–1212. [Google Scholar] [CrossRef]
- Drula, E.; Garron, M.L.; Dogan, S.; Lombard, V.; Henrissat, B.; Terrapon, N. The Carbohydrate-Active Enzyme Database: Functions and Literature. Nucleic Acids Res. 2022, 50, D571–D577. [Google Scholar] [CrossRef] [PubMed]
- Bortolaia, V.; Kaas, R.S.; Ruppe, E.; Roberts, M.C.; Schwarz, S.; Cattoir, V.; Philippon, A.; Allesoe, R.L.; Rebelo, A.R.; Florensa, A.F.; et al. ResFinder 4.0 for Predictions of Phenotypes from Genotypes. J. Antimicrob. Chemother. 2020, 75, 3491–3500. [Google Scholar] [CrossRef]
- R Core Team. R: A Language and Environment for Statistical Computing; R Core Team: Vienna, Austria, 2022. [Google Scholar]
- Gu, Z.; Eils, R.; Schlesner, M. Complex Heatmaps Reveal Patterns and Correlations in Multidimensional Genomic Data. Bioinformatics 2016, 32, 2847–2849. [Google Scholar] [CrossRef] [PubMed]
- Bottacini, F.; Morrissey, R.; Esteban-Torres, M.; James, K.; Van Breen, J.; Dikareva, E.; Egan, M.; Lambert, J.; Van Limpt, K.; Knol, J.; et al. Comparative Genomics and Genotype-Phenotype Associations in Bifidobacterium Breve. Sci. Rep. 2018, 8, 10633. [Google Scholar] [CrossRef] [PubMed]
- Duranti, S.; Milani, C.; Lugli, G.A.; Mancabelli, L.; Turroni, F.; Ferrario, C.; Mangifesta, M.; Viappiani, A.; Sanchez, B.; Margolles, A.; et al. Evaluation of Genetic Diversity among Strains of the Human Gut Commensal Bifidobacterium Adolescentis. Sci. Rep. 2016, 6, 23971. [Google Scholar] [CrossRef] [PubMed]
- Lin, G.; Liu, Q.; Wang, L.; Li, H.; Zhao, J.; Zhang, H.; Wang, G.; Chen, W. The Comparative Analysis of Genomic Diversity and Genes Involved in Carbohydrate Metabolism of Eighty-Eight Bifidobacterium Pseudocatenulatum Isolates from Different Niches of China. Nutrients 2022, 14, 2347. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Li, W.; Yao, C.; Yu, J.; Zhang, H. Comparative Genomic Analysis Revealed Genetic Divergence between Bifidobacterium Catenulatum Subspecies Present in Infant versus Adult Guts. BMC Microbiol. 2022, 22, 158. [Google Scholar] [CrossRef]
- Ambrogi, V.; Bottacini, F.; O’Sullivan, J.; O’Connell Motherway, M.; Linqiu, C.; Schoemaker, B.; Schoterman, M.; van Sinderen, D. Characterization of GH2 and GH42 β-Galactosidases Derived from Bifidobacterial Infant Isolates. AMB Express 2019, 9, 9. [Google Scholar] [CrossRef]
- O’Connell, K.J.; Motherway, M.O.C.; O’Callaghan, J.; Fitzgerald, G.F.; Paul Ross, R.; Ventura, M.; Stanton, C.; van Sinderen, D. Metabolism of Four α-Glycosidic Linkage-Containing Oligosaccharides by Bifidobacterium Breve UCC2003. Appl. Environ. Microbiol. 2013, 79, 6280–6292. [Google Scholar] [CrossRef] [PubMed]
- Nishimoto, M.; Kitaoka, M. Identification of the Putative Proton Donor Residue of Lacto-N-Biose Phosphorylase (EC 2.4.1.211). Biosci. Biotechnol. Biochem. 2007, 71, 1587–1591. [Google Scholar] [CrossRef]
- Arzamasov, A.A.; Nakajima, A.; Sakanaka, M.; Ojima, M.N.; Katayama, T.; Rodionov, D.A.; Osterman, A.L. Human Milk Oligosaccharide Utilization in Intestinal Bifidobacteria Is Governed by Global Transcriptional Regulator NagR. mSystems 2022, 7, e00343-22. [Google Scholar] [CrossRef] [PubMed]
- Odamaki, T.; Horigome, A.; Sugahara, H.; Hashikura, N.; Minami, J.; Xiao, J.Z.; Abe, F. Comparative Genomics Revealed Genetic Diversity and Species/Strain-Level Differences in Carbohydrate Metabolism of Three Probiotic Bifidobacterial Species. Int. J. Genomics 2015, 2015, 567809. [Google Scholar] [CrossRef] [PubMed]
- Garrido, D.; Ruiz-Moyano, S.; Jimenez-Espinoza, R.; Eom, H.J.; Block, D.E.; Mills, D.A. Utilization of Galactooligosaccharides by Bifidobacterium Longum Subsp. Infantis Isolates. Food Microbiol. 2013, 33, 262–270. [Google Scholar] [CrossRef] [PubMed]
- Katoh, T.; Ojima, M.N.; Sakanaka, M.; Ashida, H.; Gotoh, A.; Katayama, T. Enzymatic Adaptation of Bifidobacterium Bifidum to Host Glycans, Viewed from Glycoside Hydrolyases and Carbohydrate-Binding Modules. Microorganisms 2020, 8, 481. [Google Scholar] [CrossRef]
- Zabel, B.; Yde, C.C.; Roos, P.; Marcussen, J.; Jensen, H.M.; Salli, K.; Hirvonen, J.; Ouwehand, A.C.; Morovic, W. Novel Genes and Metabolite Trends in Bifidobacterium Longum Subsp. Infantis Bi-26 Metabolism of Human Milk Oligosaccharide 2′-Fucosyllactose. Sci. Rep. 2019, 9, 7983. [Google Scholar] [CrossRef]
- Sela, D.A.; Chapman, J.; Adeuya, A.; Kim, J.H.; Chen, F.; Whitehead, T.R.; Lapidus, A.; Rokhsar, D.S.; Lebrilla, C.B.; German, J.B.; et al. The Genome Sequence of Bifidobacterium Longum Subsp. Infantis Reveals Adaptations for Milk Utilization within the Infant Microbiome. Proc. Natl. Acad. Sci. USA 2008, 105, 18964–18969. [Google Scholar] [CrossRef] [PubMed]
- LoCascio, R.G.; Desai, P.; Sela, D.A.; Weimer, B.; Mills, D.A. Broad Conservation of Milk Utilization Genes in Bifidobacterium Longum Subsp. Infantis as Revealed by Comparative Genomic Hybridization. Appl. Environ. Microbiol. 2010, 76, 7373–7381. [Google Scholar] [CrossRef]
- Sela, D.A.; Li, Y.; Lerno, L.; Wu, S.; Marcobal, A.M.; Bruce German, J.; Chen, X.; Lebrilla, C.B.; Mills, D.A. An Infant-Associated Bacterial Commensal Utilizes Breast Milk Sialyloligosaccharides. J. Biol. Chem. 2011, 286, 11909–11918. [Google Scholar] [CrossRef]
- Suzuki, R.; Wada, J.; Katayama, T.; Fushinobu, S.; Wakagi, T.; Shoun, H.; Sugimoto, H.; Tanaka, A.; Kumagai, H.; Ashida, H.; et al. Structural and Thermodynamic Analyses of Solute-Binding Protein from Bifidobacterium Longum Specific for Core 1 Disaccharide and Lacto-N-Biose I. J. Biol. Chem. 2008, 283, 13165–13173. [Google Scholar] [CrossRef] [PubMed]
- Barratt, M.J.; Nuzhat, S.; Ahsan, K.; Frese, S.A.; Arzamasov, A.A.; Sarker, S.A.; Munirul Islam, M.; Palit, P.; Islam, M.R.; Hibberd, M.C.; et al. Bifidobacterium Infantis Treatment Promotes Weight Gain in Bangladeshi Infants with Severe Acute Malnutrition. Sci. Transl. Med. 2022, 14, 1107. [Google Scholar] [CrossRef] [PubMed]
- Kim, T.B.; Song, S.H.; Kang, S.C.; Oh, D.K. Quantitative Comparison of Lactose and Glucose Utilization in Bifidobacterium Longum Cultures. Biotechnol. Prog. 2003, 19, 672–675. [Google Scholar] [CrossRef] [PubMed]
- Parche, S.; Beleut, M.; Rezzonico, E.; Jacobs, D.; Arigoni, F.; Titgemeyer, F.; Jankovic, I. Lactose-over-Glucose Preference in Bifidobacterium Longum NCC2705: GlcP, Encoding a Glucose Transporter, Is Subject to Lactose Repression. J. Bacteriol. 2006, 188, 1260–1265. [Google Scholar] [CrossRef] [PubMed]
- Lanigan, N.; Kelly, E.; Arzamasov, A.A.; Stanton, C.; Rodionov, D.A.; van Sinderen, D. Transcriptional Control of Central Carbon Metabolic Flux in Bifidobacteria by Two Functionally Similar, yet Distinct LacI-Type Regulators. Sci. Rep. 2019, 9, 17851. [Google Scholar] [CrossRef]
- Schwab, C.; Ruscheweyh, H.J.; Bunesova, V.; Pham, V.T.; Beerenwinkel, N.; Lacroix, C. Trophic Interactions of Infant Bifidobacteria and Eubacterium Hallii during L-Fucose and Fucosyllactose Degradation. Front. Microbiol. 2017, 8, 244135. [Google Scholar] [CrossRef] [PubMed]
- Honda, Y.; Nishimoto, M.; Katayama, T.; Kitaoka, M. Characterization of the Cytosolic β-N-Acetylglucosaminidase from Bifidobacterium Longum Subsp. Longum. J. Appl. Glycosci. 2013, 60, 141–146. [Google Scholar] [CrossRef] [PubMed]
- Sakurama, H.; Kiyohara, M.; Wada, J.; Honda, Y.; Yamaguchi, M.; Fukiya, S.; Yokota, A.; Ashida, H.; Kumagai, H.; Kitaoka, M.; et al. Lacto-N-Biosidase Encoded by a Novel Gene of Bifidobacterium Longum Subspecies Longum Shows Unique Substrate Specificity and Requires a Designated Chaperone for Its Active Expression. J. Biol. Chem. 2013, 288, 25194–25206. [Google Scholar] [CrossRef]
- Oki, K.; Akiyama, T.; Matsuda, K.; Gawad, A.; Makino, H.; Ishikawa, E.; Oishi, K.; Kushiro, A.; Fujimoto, J. Long-Term Colonization Exceeding Six Years from Early Infancy of Bifidobacterium Longum Subsp. Longum in Human Gut. BMC Microbiol. 2018, 18, 209. [Google Scholar] [CrossRef]
- Yamada, C.; Gotoh, A.; Sakanaka, M.; Hattie, M.; Stubbs, K.A.; Katayama-Ikegami, A.; Hirose, J.; Kurihara, S.; Arakawa, T.; Kitaoka, M.; et al. Molecular Insight into Evolution of Symbiosis between Breast-Fed Infants and a Member of the Human Gut Microbiome Bifidobacterium Longum. Cell Chem. Biol. 2017, 24, 515–524.e5. [Google Scholar] [CrossRef]
- Sakanaka, M.; Hansen, M.E.; Gotoh, A.; Katoh, T.; Yoshida, K.; Odamaki, T.; Yachi, H.; Sugiyama, Y.; Kurihara, S.; Hirose, J.; et al. Evolutionary Adaptation in Fucosyllactose Uptake Systems Supports Bifidobacteria-Infant Symbiosis. Sci. Adv. 2019, 5, eaaw7696. [Google Scholar] [CrossRef] [PubMed]
- Asakuma, S.; Hatakeyama, E.; Urashima, T.; Yoshida, E.; Katayama, T.; Yamamoto, K.; Kumagai, H.; Ashida, H.; Hirose, J.; Kitaoka, M. Physiology of Consumption of Human Milk Oligosaccharides by Infant Gut-Associated Bifidobacteria. J. Biol. Chem. 2011, 286, 34583–34592. [Google Scholar] [CrossRef] [PubMed]
- Gotoh, A.; Katoh, T.; Sakanaka, M.; Ling, Y.; Yamada, C.; Asakuma, S.; Urashima, T.; Tomabechi, Y.; Katayama-Ikegami, A.; Kurihara, S.; et al. Sharing of Human Milk Oligosaccharides Degradants within Bifidobacterial Communities in Faecal Cultures Supplemented with Bifidobacterium Bifidum. Sci. Rep. 2018, 8, 13958. [Google Scholar] [CrossRef] [PubMed]
- Turroni, F.; Strati, F.; Foroni, E.; Serafini, F.; Duranti, S.; van Sinderen, D.; Ventura, M. Analysis of Predicted Carbohydrate Transport Systems Encoded by Bifidobacterium Bifidum PRL2010. Appl. Environ. Microbiol. 2012, 78, 5002–5012. [Google Scholar] [CrossRef] [PubMed]
- Hakomori, S. itiroh Structure and Function of Glycosphingolipids and Sphingolipids: Recollections and Future Trends. Biochim. Biophys. Acta Gen. Subj. 2008, 1780, 325–346. [Google Scholar] [CrossRef] [PubMed]
- Nishimoto, M.; Kitaoka, M. Identification of N-Acetylhexosamine 1-Kinase in the Complete Lacto-N-Biose I/Galacto-N-Biose Metabolic Pathway in Bifidobacterium Longum. Appl. Environ. Microbiol. 2007, 73, 6444–6449. [Google Scholar] [CrossRef] [PubMed]
- Junick, J.; Blaut, M. Quantification of Human Fecal Bifidobacterium Species by Use of Quantitative Real-Time PCR Analysis Targeting the GroEL Gene. Appl. Environ. Microbiol. 2012, 78, 2613–2622. [Google Scholar] [CrossRef] [PubMed]
- Wu, G.; Zhang, C.; Wu, H.; Wang, R.; Shen, J.; Wang, L.; Zhao, Y.; Pang, X.; Zhang, X.; Zhao, L.; et al. Genomic Microdiversity of Bifidobacterium Pseudocatenulatum Underlying Differential Strain-Level Responses to Dietary Carbohydrate Intervention. MBio 2017, 8, 10-1128. [Google Scholar] [CrossRef] [PubMed]
- Matsuki, T.; Watanabe, K.; Fujimoto, J.; Kado, Y.; Takada, T.; Matsumoto, K.; Tanaka, R. Quantitative PCR with 16S RRNA-Gene-Targeted Species-Specific Primers for Analysis of Human Intestinal Bifidobacteria. Appl. Environ. Microbiol. 2004, 70, 167–173. [Google Scholar] [CrossRef]
- Turroni, F.; Peano, C.; Pass, D.A.; Foroni, E.; Severgnini, M.; Claesson, M.J.; Kerr, C.; Hourihane, J.; Murray, D.; Fuligni, F.; et al. Diversity of Bifidobacteria within the Infant Gut Microbiota. PLoS ONE 2012, 7, e36957. [Google Scholar] [CrossRef]
- Kiyohara, M.; Tachizawa, A.; Nishimoto, M.; Kitaoka, M.; Ashida, H.; Yamamot, K. Prebiotic Effect of Lacto-n-Biose i on Bifidobacterial Growth. Biosci. Biotechnol. Biochem. 2009, 73, 1175–1179. [Google Scholar] [CrossRef] [PubMed]






Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hermes, G.D.A.; Rasmussen, C.; Wellejus, A. Variation in the Conservation of Species-Specific Gene Sets for HMO Degradation and Its Effects on HMO Utilization in Bifidobacteria. Nutrients 2024, 16, 1893. https://doi.org/10.3390/nu16121893
Hermes GDA, Rasmussen C, Wellejus A. Variation in the Conservation of Species-Specific Gene Sets for HMO Degradation and Its Effects on HMO Utilization in Bifidobacteria. Nutrients. 2024; 16(12):1893. https://doi.org/10.3390/nu16121893
Chicago/Turabian StyleHermes, Gerben D. A., Christine Rasmussen, and Anja Wellejus. 2024. "Variation in the Conservation of Species-Specific Gene Sets for HMO Degradation and Its Effects on HMO Utilization in Bifidobacteria" Nutrients 16, no. 12: 1893. https://doi.org/10.3390/nu16121893