
The “Sunshine Vitamin” and Its Antioxidant Benefits for Enhancing Muscle Function


{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
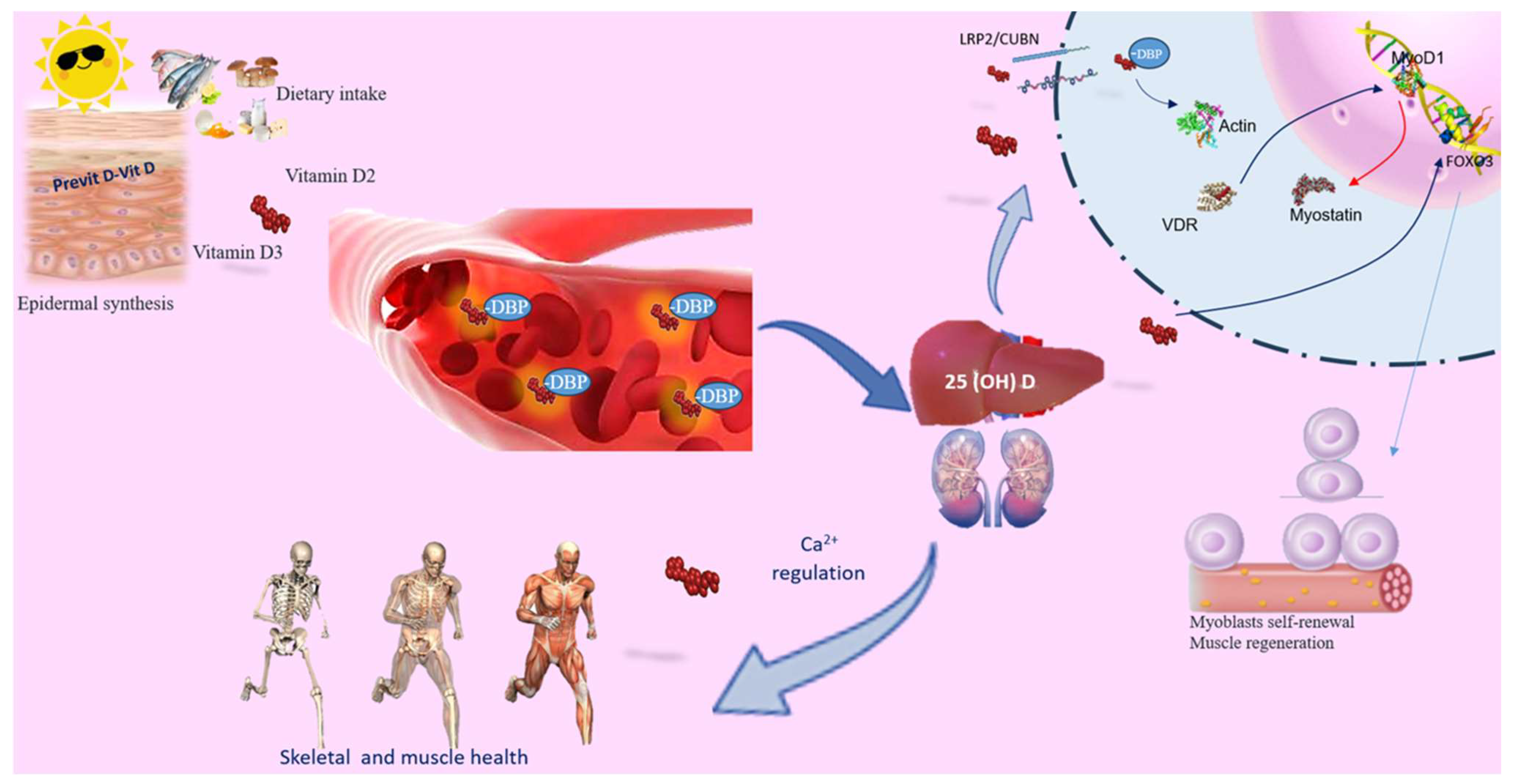
2. Understanding the Biological Influence of Vitamin D on Muscle Performance
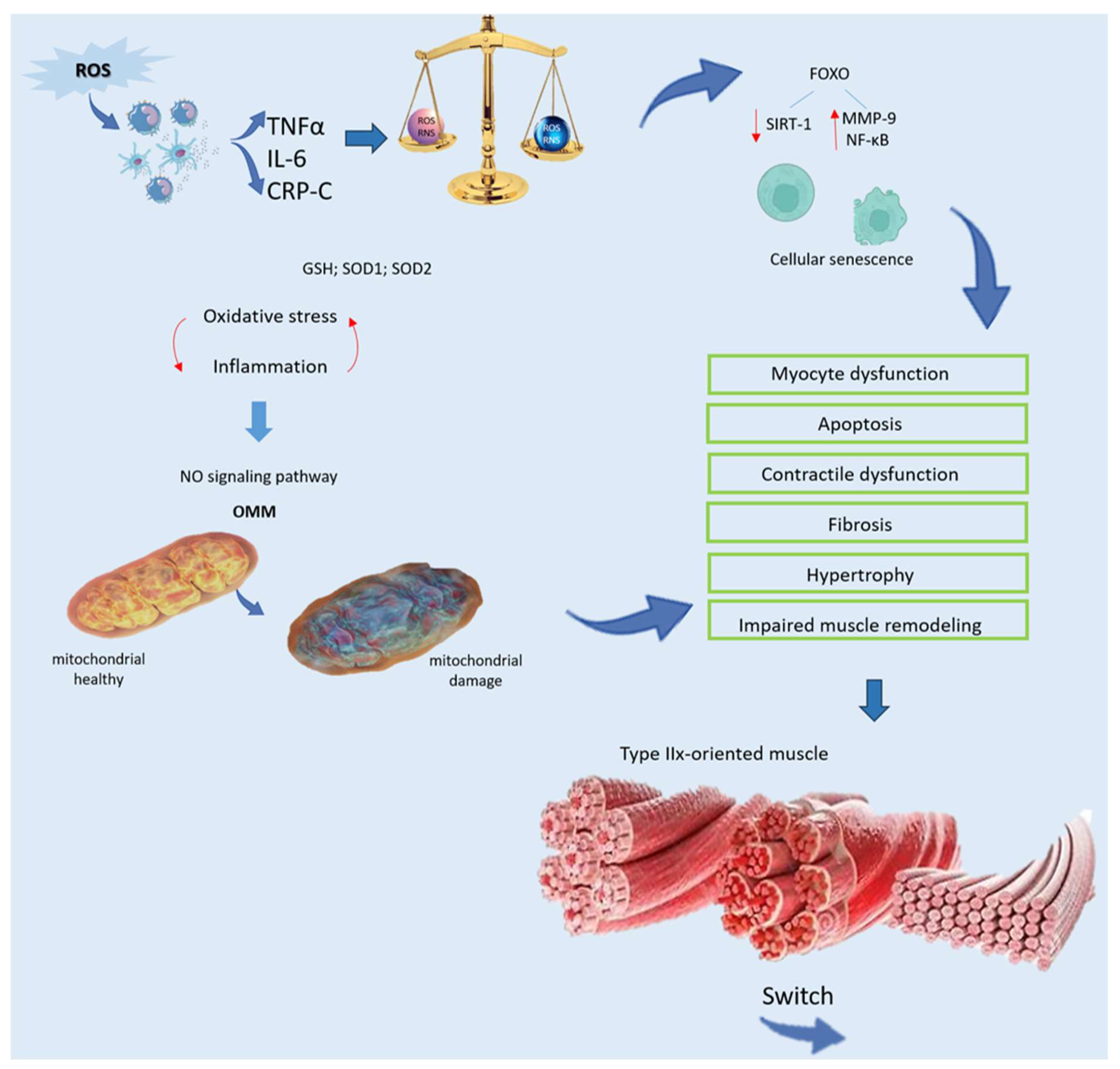
3. Exploring the Role of Oxidative Stress on Muscle Weakness
4. Unveiling the Molecular Pathways of Vitamin D in Musculoskeletal Balance
5. Vitamin D Deficiency and Muscle Dysfunction
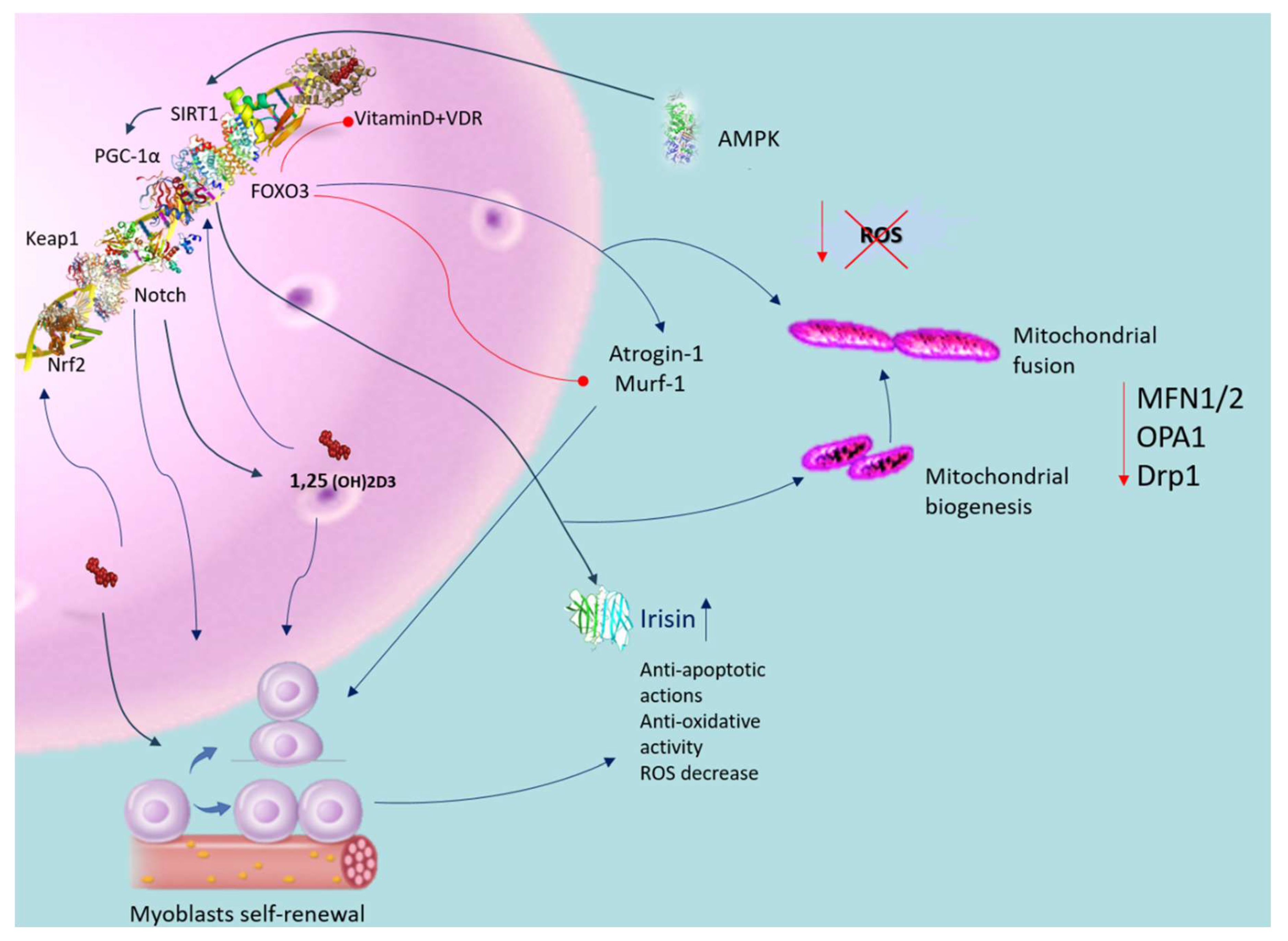
6. The Antioxidative Capacity of Vitamin D in Muscle Dysfunction
7. Examining the Role of Mitochondrial Function in Muscle Weakness
8. Modulating Mitochondrial Functionality through Vitamin D Regulation
9. Conclusions
Author Contributions
Funding
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Rai, M.; Demontis, F. Systemic Nutrient and Stress Signaling via Myokines and Myometabolites. Annu. Rev. Physiol. 2016, 78, 85–107. [Google Scholar] [CrossRef] [PubMed]
- Blaauw, B.; Schiaffino, S.; Reggiani, C. Mechanisms modulating skeletal muscle phenotype. Compr. Physiol. 2013, 3, 1645–1687. [Google Scholar] [CrossRef] [PubMed]
- Srikanthan, P.; Karlamangla, A.S. Muscle mass index as a predictor of longevity in older adults. Am. J. Med. 2014, 127, 547–553. [Google Scholar] [CrossRef]
- Casabona, A.; Valle, M.S.; Laudani, L.; Crimi, C.; Russo, C.; Malaguarnera, L.; Crimi, N.; Cioni, M. Is the Power Spectrum of Electromyography Signal a Feasible Tool to Estimate Muscle Fiber Composition in Patients with COPD? J. Clin. Med. 2021, 10, 3815. [Google Scholar] [CrossRef] [PubMed]
- Derbré, F.; Gratas-Delamarche, A.; Gómez-Cabrera, M.C.; Viña, J. Inactivity-induced oxidative stress: A central role in age-related sarcopenia? Eur. J. Sport Sci. 2014, 1, S98–S108. [Google Scholar] [CrossRef] [PubMed]
- Cao, R.Y.; Li, J.; Dai, Q.; Li, Q.; Yang, J. Muscle Atrophy: Present and Future. Adv. Exp. Med. Biol. 2018, 1088, 605–624. [Google Scholar] [CrossRef]
- Valle, M.S.; Casabona, A.; Di Fazio, E.; Crimi, C.; Russo, C.; Malaguarnera, L.; Crimi, N.; Cioni, M. Impact of chronic obstructive pulmonary disease on passive viscoelastic components of the musculoarticular system. Sci. Rep. 2021, 11, 18077. [Google Scholar] [CrossRef]
- Russo, C.; Valle, M.S.; Casabona, A.; Spicuzza, L.; Sambataro, G.; Malaguarnera, L. Vitamin D Impacts on Skeletal Muscle Dysfunction in Patients with COPD Promoting Mitochondrial Health. Biomedicines 2022, 10, 898. [Google Scholar] [CrossRef]
- Patergnani, S.; Bouhamida, E.; Leo, S.; Pinton, P.; Rimessi, A. Mitochondrial Oxidative Stress and “Mito-Inflammation”: Actors in the Diseases. Biomedicines 2021, 9, 216. [Google Scholar] [CrossRef]
- Sartori, R.; Romanello, V.; Sandri, M. Mechanisms of muscle atrophy and hypertrophy: Implications in health and disease. Nat. Commun. 2021, 12, 330. [Google Scholar] [CrossRef]
- Romanello, V.; Sandri, M. The connection between the dynamic remodeling of the mitochondrial network and the regulation of muscle mass. Cell Mol. Life Sci. 2021, 78, 1305–1328. [Google Scholar] [CrossRef] [PubMed]
- Bollen, S.E.; Bass, J.J.; Fujita, S.; Wilkinson, D.; Hewison, M.; Atherton, P.J. The Vitamin D/Vitamin D receptor (VDR) axis in muscle atrophy and sarcopenia. Cell Signal. 2022, 96, 110355. [Google Scholar] [CrossRef]
- Valle, M.S.; Russo, C.; Casabona, A.; Crimi, N.; Crimi, C.; Colaianni, V.; Cioni, M.; Malaguarnera, L. Anti-inflammatory role of vitamin D in muscle dysfunctions of patients with chronic obstructive pulmonary disease: A comprehensive review. Minerva Med. 2023, 114, 357–371. [Google Scholar] [CrossRef] [PubMed]
- Laird, E.; Ward, M.; McSorley, E.; Strain, J.J.; Wallace, J. Vitamin D and bone health: Potential mechanisms. Nutrients 2010, 2, 693–724. [Google Scholar] [CrossRef]
- Ceglia, L. Vitamin D and skeletal muscle tissue and function. Mol. Aspects Med. 2008, 29, 407–414. [Google Scholar] [CrossRef]
- Bischoff, H.A.; Borchers, M.; Gudat, F.; Duermueller, U.; Theiler, R.; Stähelin, H.B.; Dick, W. In situ detection of 1,25-dihydroxyvitamin D3 receptor in human skeletal muscle tissue. Histochem. J. 2001, 33, 19–24. [Google Scholar] [CrossRef] [PubMed]
- Garcia, L.A.; King, K.K.; Ferrini, M.G.; Norris, K.C.; Artaza, J.N. 1,25(OH)2 vitamin D3 stimulates myogenic differentiation by inhibiting cell proliferation and modulating the expression of promyogenic growth factors and myostatin in C2C12 skeletal muscle cells. Endocrinology 2011, 152, 2976–2986. [Google Scholar] [CrossRef]
- Mannino, G.; Russo, C.; Maugeri, G.; Musumeci, G.; Vicario, N.; Tibullo, D.; Giuffrida, R.; Parenti, R.; Lo Furno, D. Adult stem cell niches for tissue homeostasis. J. Cell. Physiol. 2022, 237, 239–257. [Google Scholar] [CrossRef]
- Srikuea, R.; Hirunsai, M.; Charoenphandhu, N. Regulation of vitamin D system in skeletal muscle and resident myogenic stem cell during development, maturation, and ageing. Sci. Rep. 2020, 10, 8239. [Google Scholar] [CrossRef]
- Dalton, T.P.; Shertzer, H.G.; Puga, A. Regulation of gene expression by reactive oxygen. Annu. Rev. Pharmacol. Toxicol. 1999, 39, 67–101. [Google Scholar] [CrossRef]
- Sies, H.; Belousov, V.V.; Chandel, N.S.; Davies, M.J.; Jones, D.P.; Mann, G.E.; Murphy, M.P.; Yamamoto, M.; Winterbourn, C. Defining roles of specific reactive oxygen species (ROS) in cell biology and physiology. Nat. Rev. Mol. Cell Biol. 2022, 23, 499–515. [Google Scholar] [CrossRef] [PubMed]
- Piacenza, L.; Zeida, A.; Trujillo, M.; Radi, R. The superoxide radical switch in the biology of nitric oxide and peroxynitrite. Physiol. Rev. 2022, 102, 1881–1906. [Google Scholar] [CrossRef] [PubMed]
- Warraich, U.E.; Hussain, F.; Kayani, H.U.R. Aging-Oxidative stress, antioxidants and computational modeling. Heliyon 2020, 6, e04107. [Google Scholar] [CrossRef] [PubMed]
- Ji, Y.; Li, M.; Chang, M.; Liu, R.; Qiu, J.; Wang, K.; Deng, C.; Shen, Y.; Zhu, J.; Wang, W.; et al. Inflammation: Roles in Skeletal Muscle Atrophy. Antioxidants 2022, 11, 1686. [Google Scholar] [CrossRef] [PubMed]
- Andrade, B.; Jara-Gutiérrez, C.; Paz-Araos, M.; Vázquez, M.C.; Díaz, P.; Murgas, P. The Relationship between Reactive Oxygen Species and the cGAS/STING Signaling Pathway in the Inflammaging Process. Int. J. Mol. Sci. 2022, 23, 15182. [Google Scholar] [CrossRef] [PubMed]
- Grivennikova, V.G.; Vinogradov, A.D. Mitochondrial production of reactive oxygen species. Biochemistry 2013, 78, 1490–1511. [Google Scholar] [CrossRef] [PubMed]
- Turrens, J.F. Mitochondrial formation of reactive oxygen species. J. Physiol. 2003, 552 Pt 2, 335–344. [Google Scholar] [CrossRef] [PubMed]
- Cinelli, M.A.; Do, H.T.; Miley, G.P.; Silverman, R.B. Inducible nitric oxide synthase: Regulation, structure, and inhibition. Med. Res. Rev. 2020, 40, 158–189. [Google Scholar] [CrossRef] [PubMed]
- Magnani, F.; Mattevi, A. Structure and mechanisms of ROS generation by NADPH oxidases. Curr. Opin. Struct. Biol. 2019, 59, 91–97. [Google Scholar] [CrossRef]
- Miller, A.F. Superoxide dismutases: Ancient enzymes and new insights. FEBS Lett. 2012, 586, 585–595. [Google Scholar] [CrossRef]
- Cantin, A.M.; Larivée, P.; Bégin, R.O. Extracellular glutathione suppresses human lung fibroblast proliferation. Am. J. Respir. Cell Mol. Biol. 1990, 3, 79–85. [Google Scholar] [CrossRef] [PubMed]
- Akira, S.; Kishimoto, A. NF-IL6 and NF-kB in cytokine gene regulation. Immunol. Adv. 1997, 65, 1–46. [Google Scholar]
- Gilmore, T.D. Introduction to NF-kappa B: Players, paths, perspectives. Oncogene 2006, 25, 6680–6684. [Google Scholar] [CrossRef] [PubMed]
- Hirota, K.; Murata, M.; Sachi, Y.; Nakamura, H.; Takeuchi, J.; Mori, K.; Yodoi, J. Distinct roles of thioredoxin in the cytoplasm and in the nucleus. A two-step redox regulation mechanism of NF-kappaB transcription factor. J. Biol. Chem. 1999, 274, 27891–27897. [Google Scholar] [CrossRef] [PubMed]
- Barnes, P.J.; Baker, J.; Donnelly, L.E. Cellular Senescence as a Mechanism and Target in Chronic Lung Diseases. Am. J. Respir. Crit. Care Med. 2019, 200, 556–564. [Google Scholar] [CrossRef] [PubMed]
- Gambini, J.; Stromsnes, K. Oxidative Stress and Inflammation: From Mechanisms to Therapeutic Approaches. Biomedicines 2022, 10, 753. [Google Scholar] [CrossRef] [PubMed]
- Bienaimé, F.; Prié, D.; Friedlander, G.; Souberbielle, J.C. Metabolism and activity of vitamin D in the parathyroid gland. Mol. Cell. Endocrinol. 2011, 347, 30–41. [Google Scholar] [CrossRef] [PubMed]
- Ainbinder, A.; Boncompagni, S.; Protasi, F.; Dirksen, R.T. Role of Mitofusin-2 in mitochondrial localization and calcium uptake in skeletal muscle. Cell Calcium 2015, 57, 14–24. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Gu, Y.; Huang, J.; Wu, H.; Meng, G.; Zhang, Q.; Liu, L.; Zhang, S.; Wang, X.; Zhang, J.; et al. Serum vitamin D status and circulating irisin levels in older adults with sarcopenia. Front. Nutr. 2022, 9, 1051870. [Google Scholar] [CrossRef]
- Cannon, B.; Nedergaard, J. Brown adipose tissue: Function and physiological significance. Physiol. Rev. 2004, 84, 277–359. [Google Scholar] [CrossRef]
- Halling, J.F.; Pilegaard, H. PGC-1α-mediated regulation of mitochondrial function and physiological implications. Appl. Physiol. Nutr. Metab. 2020, 45, 927–936. [Google Scholar] [CrossRef] [PubMed]
- Adamovich, Y.; Shlomai, A.; Tsvetkov, P.; Umansky, K.B.; Reuven, N.; Estall, J.L.; Spiegelman, B.M.; Shaul, Y. The protein level of PGC-1α, a key metabolic regulator, is controlled by NADH-NQO1. Mol. Cell Biol. 2013, 33, 2603–2613. [Google Scholar] [CrossRef] [PubMed]
- Korta, P.; Pocheć, E.; Mazur-Biały, A. Irisin as a Multifunctional Protein: Implications for Health and Certain Diseases. Medicina 2019, 55, 485. [Google Scholar] [CrossRef]
- Askari, H.; Rajani, S.F.; Poorebrahim, M.; Haghi-Aminjan, H.; Raeis-Abdollahi, E.; Abdollahi, M. A glance at the therapeutic potential of irisin against diseases involving inflammation, oxidative stress, and apoptosis: An introductory review. Pharmacol. Res. 2018, 129, 44–55. [Google Scholar] [CrossRef] [PubMed]
- Xin, C.; Liu, J.; Zhang, J.; Zhu, D.; Wang, H.; Xiong, L.; Lee, Y.; Ye, J.; Lian, K.; Xu, C.; et al. Irisin improves fatty acid oxidation and glucose utilization in type 2 diabetes by regulating the AMPK signaling pathway. Int. J. Obes. 2016, 40, 443–451. [Google Scholar] [CrossRef] [PubMed]
- Stavenuiter, A.W.; Arcidiacono, M.V.; Ferrantelli, E.; Keuning, E.D.; Vila Cuenca, M.; Wee, P.M.; Beelen, R.H.; Vervloet, M.G.; Dusso, A.S. A novel rat model of vitamin D deficiency: Safe and rapid induction of vitamin D and calcitriol deficiency without hyperparathyroidism. Biomed. Res. Int. 2015, 2015, 604275. [Google Scholar] [CrossRef] [PubMed]
- Nadimi, H.; Djazayery, A.; Javanbakht, M.H.; Dehpour, A.; Ghaedi, E.; Derakhshanian, H.; Mohammadi, H.; Zarei, M.; Djalali, M. The Effect of Vitamin D Supplementation on Serum and Muscle Irisin Levels, and FNDC5 Expression in Diabetic Rats. Rep. Biochem. Mol. Biol. 2019, 8, 236–243. [Google Scholar] [PubMed]
- Faienza, M.F.; Brunetti, G.; Sanesi, L.; Colaianni, G.; Celi, M.; Piacente, L.; D’Amato, G.; Schipani, E.; Colucci, S.; Grano, M. High irisin levels are associated with better glycemic control and bone health in children with Type 1 diabetes. Diabetes Res. Clin. Pract. 2018, 141, 10–17. [Google Scholar] [CrossRef] [PubMed]
- Colaianni, G.; Oranger, A.; Dicarlo, M.; Lovero, R.; Storlino, G.; Pignataro, P.; Fontana, A.; Di Serio, F.; Ingravallo, A.; Caputo, G.; et al. Irisin Serum Levels and Skeletal Muscle Assessment in a Cohort of Charcot-Marie-Tooth Patients. Front. Endocrinol. (Lausanne) 2022, 13, 886243. [Google Scholar] [CrossRef]
- Chang, E. 1,25-Dihydroxyvitamin D Decreases Tertiary Butyl-Hydrogen Peroxide-Induced Oxidative Stress and Increases AMPK/SIRT1 Activation in C2C12 Muscle Cells. Molecules 2019, 24, 3903. [Google Scholar] [CrossRef]
- Cantó, C.; Jiang, L.Q.; Deshmukh, A.S.; Mataki, C.; Coste, A.; Lagouge, M.; Zierath, J.R.; Auwerx, J. Interdependence of AMPK and SIRT1 for metabolic adaptation to fasting and exercise in skeletal muscle. Cell Metab. 2010, 11, 213–219. [Google Scholar] [CrossRef]
- Wiciński, M.; Adamkiewicz, D.; Adamkiewicz, M.; Śniegocki, M.; Podhorecka, M.; Szychta, P.; Malinowski, B. Impact of Vitamin D on Physical Efficiency and Exercise Performance-A Review. Nutrients 2019, 11, 2826. [Google Scholar] [CrossRef] [PubMed]
- Wimalawansa, S.J. Vitamin D Deficiency: Effects on Oxidative Stress, Epigenetics, Gene Regulation, and Aging. Biology 2019, 8, 30. [Google Scholar] [CrossRef] [PubMed]
- Prokopidis, K.; Giannos, P.; Katsikas Triantafyllidis, K.; Kechagias, K.S.; Mesinovic, J.; Witard, O.C.; Scott, D. Effect of vitamin D monotherapy on indices of sarcopenia in community-dwelling older adults: A systematic review and meta-analysis. J. Cachexia Sarcopenia Muscle 2022, 13, 1642–1652. [Google Scholar] [CrossRef]
- Ceglia, L.; Niramitmahapanya, S.; da Silva Morais, M.; Rivas, D.A.; Harris, S.S.; Bischoff-Ferrari, H.; Fielding, R.A.; Dawson-Hughes, B. A randomized study on the effect of vitamin D3 supplementation on skeletal muscle morphology and vitamin D receptor concentration in older women. J. Clin. Endocrinol. Metab. 2013, 98, E1927–E1935. [Google Scholar] [CrossRef]
- Pfeifer, M.; Begerow, B.; Minne, H.W.; Suppan, K.; Fahrleitner-Pammer, A.; Dobnig, H. Effects of a long-term vitamin D and calcium supplementation on falls and parameters of muscle function in community-dwelling older individuals. Osteoporos. Int. 2009, 20, 315–322. [Google Scholar] [CrossRef]
- Hirose, Y.; Onishi, T.; Miura, S.; Hatazawa, Y.; Kamei, Y. Vitamin D Attenuates FOXO1-Target Atrophy Gene Expression in C2C12 Muscle Cells. J. Nutr. Sci. Vitaminol. 2018, 64, 229–232. [Google Scholar] [CrossRef] [PubMed]
- Endo, I.; Inoue, D.; Mitsui, T.; Umaki, Y.; Akaike, M.; Yoshizawa, T.; Kato, S.; Matsumoto, T. Deletion of vitamin D receptor gene in mice results in abnormal skeletal muscle development with deregulated expression of myoregulatory transcription factors. Endocrinology 2003, 144, 5138–5144. [Google Scholar] [CrossRef]
- Valle, M.S.; Russo, C.; Malaguarnera, L. Protective role of vitamin D against oxidative stress in diabetic retinopathy. Diabetes Metab. Res. Rev. 2021, 37, e3447. [Google Scholar] [CrossRef]
- Rossi, A.; Pizzo, P.; Filadi, R. Calcium, mitochondria and cell metabolism: A functional triangle in bioenergetics. Biochim. Biophys. Acta Mol. Cell Res. 2019, 1866, 1068–1078. [Google Scholar] [CrossRef]
- Modesti, L.; Danese, A.; Vitto, V.A.M.; Ramaccini, D.; Aguiari, G.; Gafà, R.; Lanza, G.; Giorgi, C.; Pinton, P. Mitochondrial Ca2+ Signaling in Health, Disease and Therapy. Cells 2021, 10, 1317. [Google Scholar] [CrossRef] [PubMed]
- van der Meijden, K.; Bravenboer, N.; Dirks, N.F.; Heijboer, A.C.; den Heijer, M.; de Wit, G.M.; Offringa, C.; Lips, P.; Jaspers, R.T. Effects of 1,25(OH)2D3 and 25(OH)D3 on C2C12 Myoblast Proliferation, Differentiation, and Myotube Hypertrophy. J. Cell Physiol. 2016, 231, 2517–2528. [Google Scholar] [CrossRef] [PubMed]
- Latham, C.M.; Brightwell, C.R.; Keeble, A.R.; Munson, B.D.; Thomas, N.T.; Zagzoog, A.M.; Fry, C.S.; Fry, J.L. Vitamin D Promotes Skeletal Muscle Regeneration and Mitochondrial Health. Front. Physiol. 2021, 12, 660498. [Google Scholar] [CrossRef] [PubMed]
- Bhat, M.; Ismail, A. Vitamin D treatment protects against and reverses oxidative stress induced muscle proteolysis. J. Steroid Biochem. Mol. Biol. 2015, 152, 171–179. [Google Scholar] [CrossRef] [PubMed]
- Ryan, Z.C.; Craig, T.A.; Folmes, C.D.; Wang, X.; Lanza, I.R.; Schaible, N.S.; Salisbury, J.L.; Nair, K.S.; Terzic, A.; Sieck, G.C.; et al. 1α,25-Dihydroxyvitamin D3 Regulates Mitochondrial Oxygen Consumption and Dynamics in Human Skeletal Muscle Cells. J. Biol. Chem. 2016, 291, 1514–1528. [Google Scholar] [CrossRef] [PubMed]
- Seldeen, K.L.; Berman, R.N.; Pang, M.; Lasky, G.; Weiss, C.; MacDonald, B.A.; Thiyagarajan, R.; Redae, Y.; Troen, B.R. Vitamin D Insufficiency Reduces Grip Strength, Grip Endurance and Increases Frailty in Aged C57Bl/6J Mice. Nutrients 2020, 12, 3005. [Google Scholar] [CrossRef] [PubMed]
- Ke, C.Y.; Yang, F.L.; Wu, W.T.; Chung, C.H.; Lee, R.P.; Yang, W.T.; Subeq, Y.M.; Liao, K.W. Vitamin D3 Reduces Tissue Damage and Oxidative Stress Caused by Exhaustive Exercise. Int. J. Med. Sci. 2016, 13, 147–153. [Google Scholar] [CrossRef]
- Chen, L.; Yang, R.; Qiao, W.; Zhang, W.; Chen, J.; Mao, L.; Goltzman, D.; Miao, D. 1,25-Dihydroxyvitamin D exerts an antiaging role by activation of Nrf2-antioxidant signaling and inactivation of p16/p53-senescence signaling. Aging Cell 2019, 18, e12951. [Google Scholar] [CrossRef] [PubMed]
- Xiang, Y.; Fu, L.; Xiang, H.X.; Zheng, L.; Tan, Z.X.; Wang, L.X.; Cao, W.; Xu, D.X.; Zhao, H. Correlations among Pulmonary DJ-1, VDR and Nrf-2 in patients with Chronic Obstructive Pulmonary Disease: A Case-control Study. Int. J. Med. Sci. 2021, 18, 2449–2456. [Google Scholar] [CrossRef]
- Mathyssen, C.; Aelbrecht, C.; Serré, J.; Everaerts, S.; Maes, K.; Gayan-Ramirez, G.; Vanaudenaerde, B.; Janssens, W. Local expression profiles of vitamin D-related genes in airways of COPD patients. Respir. Res. 2020, 21, 137. [Google Scholar] [CrossRef]
- Montecinos-Franjola, F.; Ramachandran, R. Imaging Dynamin-Related Protein 1 (Drp1)-Mediated Mitochondrial Fission in Living Cells. Methods Mol. Biol. 2020, 2159, 205–217. [Google Scholar] [CrossRef] [PubMed]
- Zsurka, G.; Peeva, V.; Kotlyar, A.; Kunz, W.S. Is There Still Any Role for Oxidative Stress in Mitochondrial DNA-Dependent Aging? Genes 2018, 9, 175. [Google Scholar] [CrossRef]
- Wang, J.; Aung, L.H.; Prabhakar, B.S.; Li, P. The mitochondrial ubiquitin ligase plays an anti-apoptotic role in cardiomyocytes by regulating mitochondrial fission. J. Cell Mol. Med. 2016, 20, 2278–2288. [Google Scholar] [CrossRef]
- Tengan, C.H.; Moraes, C.T. NO control of mitochondrial function in normal and transformed cells. Biochim. Biophys. Acta Bioenerg. 2017, 1858, 573–581. [Google Scholar] [CrossRef] [PubMed]
- Boyd, C.S.; Cadenas, E. Nitric oxide and cell signaling pathways in mitochondrial-dependent apoptosis. Biol. Chem. 2002, 383, 411–423. [Google Scholar] [CrossRef]
- Parameswaran, N.; Patial, S. Tumor necrosis factor-α signaling in macrophages. Crit. Rev. Eukaryot. Gene Expr. 2010, 20, 87–103. [Google Scholar] [CrossRef] [PubMed]
- Remels, A.H.; Langen, R.C.; Schrauwen, P.; Schaart, G.; Schols, A.M.; Gosker, H.R. Regulation of mitochondrial biogenesis during myogenesis. Mol. Cell Endocrinol. 2010, 315, 113–120. [Google Scholar] [CrossRef]
- Tang, K.; Murano, G.; Wagner, H.; Nogueira, L.; Wagner, P.D.; Tang, A.; Dalton, N.D.; Gu, Y.; Peterson, K.L.; Breen, E.C. Impaired exercise capacity and skeletal muscle function in a mouse model of pulmonary inflammation. J. Appl. Physiol. (1985) 2013, 114, 1340–1350. [Google Scholar] [CrossRef]
- De la Fuente, M.; Miquel, J. An update of the oxidation-inflammation theory of aging: The involvement of the immune system in oxi-inflamm-aging. Curr. Pharm. Des. 2009, 15, 3003–3026. [Google Scholar] [CrossRef]
- Rosa, M.D.; Distefano, G.; Gagliano, C.; Rusciano, D.; Malaguarnera, L. Autophagy in Diabetic Retinopathy. Curr. Neuropharmacol. 2016, 14, 810–825. [Google Scholar] [CrossRef]
- St-Pierre, J.; Drori, S.; Uldry, M.; Silvaggi, J.M.; Rhee, J.; Jäger, S.; Handschin, C.; Zheng, K.; Lin, J.; Yang, W.; et al. Suppression of reactive oxygen species and neurodegeneration by the PGC-1 transcriptional coactivators. Cell 2006, 127, 397–408. [Google Scholar] [CrossRef]
- Salles, J.; Chanet, A.; Giraudet, C.; Patrac, V.; Pierre, P.; Jourdan, M.; Luiking, Y.C.; Verlaan, S.; Migné, C.; Boirie, Y.; et al. 1,25(OH)2-vitamin D3 enhances the stimulating effect of leucine and insulin on protein synthesis rate through Akt/PKB and mTOR mediated pathways in murine C2C12 skeletal myotubes. Mol. Nutr. Food Res. 2013, 57, 2137–2146. [Google Scholar] [CrossRef] [PubMed]
- Sandri, M.; Sandri, C.; Gilbert, A.; Skurk, C.; Calabria, E.; Picard, A.; Walsh, K.; Schiaffino, S.; Lecker, S.H.; Goldberg, A.L. Foxo transcription factors induce the atrophy-related ubiquitin ligase atrogin-1 and cause skeletal muscle atrophy. Cell 2004, 117, 399–412. [Google Scholar] [CrossRef]
- Tran, H.; Brunet, A.; Griffith, E.C.; Greenberg, M.E. The many forks in FOXO’s road. Sci. STKE 2003, 2003, RE5. [Google Scholar] [CrossRef] [PubMed]
- Chen, S.; Villalta, S.A.; Agrawal, D.K. FOXO1 Mediates Vitamin D Deficiency-Induced Insulin Resistance in Skeletal Muscle. J. Bone Miner. Res. 2016, 31, 585–595. [Google Scholar] [CrossRef] [PubMed]
- Kitajima, Y.; Yoshioka, K.; Suzuki, N. The ubiquitin-proteasome system in regulation of the skeletal muscle homeostasis and atrophy: From basic science to disorders. J. Physiol. Sci. 2020, 70, 40. [Google Scholar] [CrossRef] [PubMed]
- Saline, M.; Badertscher, L.; Wolter, M.; Lau, R.; Gunnarsson, A.; Jacso, T.; Norris, T.; Ottmann, C.; Snijder, A. AMPK and AKT protein kinases hierarchically phosphorylate the N-terminus of the FOXO1 transcription factor, modulating interactions with 14-3-3 proteins. J. Biol. Chem. 2019, 294, 13106–13116. [Google Scholar] [CrossRef]
- Brenkman, A.B.; van den Broek, N.J.; de Keizer, P.L.; van Gent, D.C.; Burgering, B.M. The DNA damage repair protein Ku70 interacts with FOXO4 to coordinate a conserved cellular stress response. FASEB J. 2010, 24, 4271–4280. [Google Scholar] [CrossRef] [PubMed]
- DeLuca, G.C.; Kimball, S.M.; Kolasinski, J.; Ramagopalan, S.V.; Ebers, G.C. Review: The role of vitamin D in nervous system health and disease. Neuropathol. Appl. Neurobiol. 2013, 39, 458–484. [Google Scholar] [CrossRef]
- Smolders, J.; Menheere, P.; Thewissen, M.; Peelen, E.; Cohen Tervaert, J.W.; Hupperts, R.; Damoiseaux, J. Regulatory T cell function correlates with serum 25-hydroxyvitamin D, but not with 1, 25-dihydroxyvita-min D, parathyroid hormone and calcium levels in patients with relapsing remitting multiple sclerosis. J. Steroid Biochem. Mol. Biol. 2010, 121, 243–246. [Google Scholar] [CrossRef]
- Mahon, B.D.; Gordon, S.A.; Cruz, J.; Cosman, F.; Cantorna, M.T. Cytokine profile in patients with multiple sclerosis following vitamin D supplementation. J. Neuroimmunol. 2003, 134, 128–132. [Google Scholar] [CrossRef] [PubMed]
- Usategui-Martín, R.; De Luis-Román, D.A.; Fernández-Gómez, J.M.; Ruiz-Mambrilla, M.; Pérez-Castrillón, J.L. Vitamin D Receptor (VDR) Gene Polymorphisms Modify the Response to Vitamin D Supplementation: A Systematic Review and Meta-Analysis. Nutrients 2022, 14, 360. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Russo, C.; Santangelo, R.; Malaguarnera, L.; Valle, M.S. The “Sunshine Vitamin” and Its Antioxidant Benefits for Enhancing Muscle Function. Nutrients 2024, 16, 2195. https://doi.org/10.3390/nu16142195
Russo C, Santangelo R, Malaguarnera L, Valle MS. The “Sunshine Vitamin” and Its Antioxidant Benefits for Enhancing Muscle Function. Nutrients. 2024; 16(14):2195. https://doi.org/10.3390/nu16142195
Chicago/Turabian StyleRusso, Cristina, Rosa Santangelo, Lucia Malaguarnera, and Maria Stella Valle. 2024. "The “Sunshine Vitamin” and Its Antioxidant Benefits for Enhancing Muscle Function" Nutrients 16, no. 14: 2195. https://doi.org/10.3390/nu16142195
APA StyleRusso, C., Santangelo, R., Malaguarnera, L., & Valle, M. S. (2024). The “Sunshine Vitamin” and Its Antioxidant Benefits for Enhancing Muscle Function. Nutrients, 16(14), 2195. https://doi.org/10.3390/nu16142195