
Neurodevelopment Is Dependent on Maternal Diet: Placenta and Brain Glucose Transporters GLUT1 and GLUT3

Abstract
:
1. Introduction
2. GLUT3
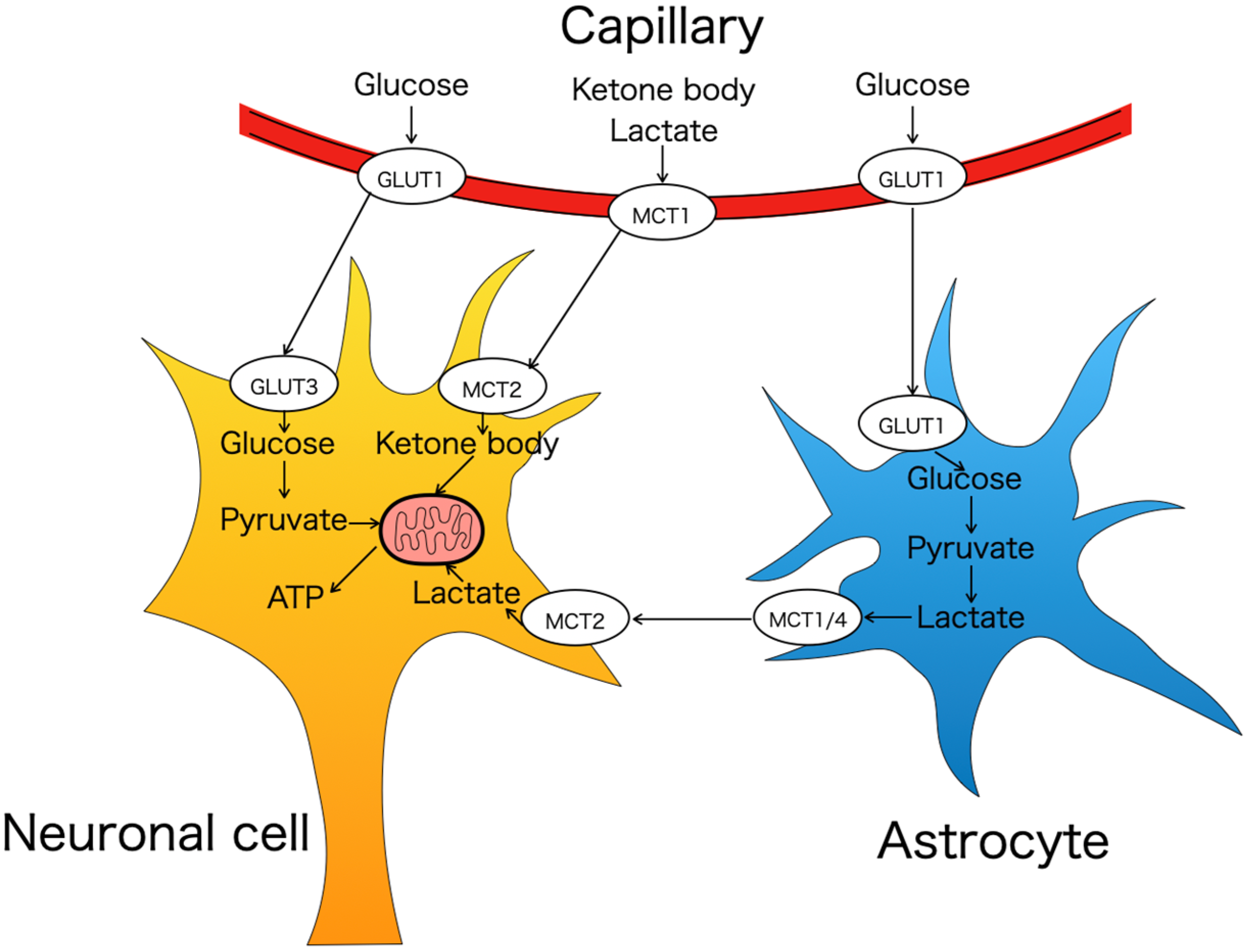
2.1. Glucose Transporters (GLUTs and SGLTs Family)

{kind=link}
{kind=link}
| Classification | Transporters | Km (mM) | Tissue Distribution | Substrate | Function in Brain |
|---|---|---|---|---|---|
| GLUT family class I | GLUT1 | 6.9 | Fibroblasts, erythrocytes, brain (endothelial cells, astrocytic processes and oligodendrocytes) >skeletal and cardiac muscle, white adipose tissue, liver | Glucose/Galactose | Transport glucose through BBB and into astrocyte |
| GLUT2 | 11.2 | Liver, pancreas, kidney, small intestine, brain (brainstem, thalamus, cortex, hypothalamus, hippocampus) [39,40,43] | Glucose/Galactose /Fructose | Regulation of food and glucose intake | |
| GLUT3 | 1.4 | Brain (hippocampus, temporal neocortex, gyrus) >testis, placenta, white blood cells, platelets [31] | Glucose/Galactose | Transport glucose to neurons | |
| GLUT4 | 4.6 | White adipose tissue, skeletal and cardiac muscle cells > brain (basal ganglia, neocortex, hypothalamus, hippocampus) | Glucose | Insulin-dependent regulation of active neuronal circuits | |
| GLUT14 | - | Testis | - | - | |
| GLUT family class II | GLUT5 | 5 | Small intestine >kidney, skeletal muscle, adipose tissue [63,64] | Fructose | - |
| GLUT7 | 0.3 | Small intestine, colon [65] | Glucose/Fructose | - | |
| GLUT9 | 0.3 | GLUT9a: liver, kidney, lung, placenta, leukocytes GLUT9b: kidney, placenta [31,52] | Glucose/Fructose | - | |
| GLUT11 | 0.2 | Heart, skeletal muscle, pancreas, kidney [66,67] | Glucose/Fructose | - | |
| GLUT family class III | GLUT6 | White adipose, spleen, brain (median eminence), peripheral leucocytes | Glucose | Regulation of glucohomeostasis | |
| GLUT8 | 2.4 | Testis, skeletal muscle, heart, small intestine >brain (amygdala, primary olfactory cortex, dentate gyrus, dorsal hypothalamic area, pituitary stark, posterior pituitary, dentate gyrus, hippocampus) | Glucose | Energy supply for neurons in hippocampus | |
| GLUT10 | 0.3 | Liver, pancreas > placenta, heart, lung skeletal muscle, kidney, adipose tissue [31] | Glucose/Galactose | - | |
| GLUT12 | 4–5 | Skeletal muscle, adipose tissue, small intestine [55] | Glucose/Galactose/Fructose | - | |
| HMIT GLUT13 | 0.1 | Brain (hippocampus, hypothalamus, cerebellum) >white and brown adipose tissues, kidney [68] | Myoinositol | Control of myo-inositol metabolism in brain | |
| SGLT family | SGLT1 | 0.5 | Intestine, Trachea, kidney, heart, testis, prostate, brain (cortical neurons, hippocampal pyramidal cell, Purkinje cells, BBB) | Glucose/Galactose | Removal of glucose from brain interstitium |
| SGLT2 | 5.0 | Kidney, brain (cerebellum), liver, thyroid, muscle, heart | Glucose | Not determined | |
| SGLT3 | 20 | Skeletal muscle, testis, uterus, small intestine, brain (hypothalamus), thyroid | Glucose | Activation of glucosensitive neurons | |
| SGLT4 | 2.0 | Small intestine, skeletal muscle [56,57] | Glucose/Mannose | - | |
| SGLT5 | - | Kidney [56,57] | Glucose/Galactose | - | |
| SGLT6 | - | Small intestine, brain (hypothalamus and substantia nigra) | Inositol | Recognize nutrients to control food intake and reward processing |
2.2. GLUT3—Structure and Kinetics
2.3. GLUT3—Expression/Investigations in Brain and Placenta
2.4. GLUT3 Is Potentially Associated with Neurodevelopmental and Neurodegenerative Disorders
3. Maternal and Postnatal Dietary Exposures Affect Offspring’s GLUTs
3.1. High-Fat Diet
3.2. Ketogenic Diet
3.3. Energy Restriction
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Conflicts of Interest
References
- Vannucci, S.J.; Clark, R.R.; Koehler-Stec, E.; Li, K.; Smith, C.B.; Davies, P.; Maher, F.; Simpson, I.A. Glucose transporter expression in brain: Relationship to cerebral glucose utilization. Dev. Neurosci. 1998, 20, 369–379. [Google Scholar] [CrossRef] [PubMed]
- Leino, R.L.; Gerhart, D.Z.; van Bueren, A.M.; McCall, A.L.; Drewes, L.R. Ultrastructural localization of GLUT 1 and GLUT 3 glucose transporters in rat brain. J. Neurosci. Res. 1997, 49, 617–626. [Google Scholar] [CrossRef]
- Maher, F.; Simpson, I.A. The GLUT3 glucose transporter is the predominant isoform in primary cultured neurons: Assessment by biosynthetic and photoaffinity labelling. Biochem. J. 1994, 301 Pt 2, 379–384. [Google Scholar] [CrossRef] [PubMed]
- Simpson, I.A.; Dwyer, D.; Malide, D.; Moley, K.H.; Travis, A.; Vannucci, S.J. The facilitative glucose transporter GLUT3: 20 years of distinction. Am. J. Physiol. Endocrinol. Metab. 2008, 295, E242–E253. [Google Scholar] [CrossRef] [PubMed]
- Nagamatsu, S.; Kornhauser, J.M.; Burant, C.F.; Seino, S.; Mayo, K.E.; Bell, G.I. Glucose transporter expression in brain. cDNA sequence of mouse GLUT3, the brain facilitative glucose transporter isoform, and identification of sites of expression by in situ hybridization. J. Biol. Chem. 1992, 267, 467–472. [Google Scholar] [CrossRef] [PubMed]
- Lesch, K.P.; Selch, S.; Renner, T.J.; Jacob, C.; Nguyen, T.T.; Hahn, T.; Romanos, M.; Walitza, S.; Shoichet, S.; Dempfle, A.; et al. Genome-wide copy number variation analysis in attention-deficit/hyperactivity disorder: Association with neuropeptide Y gene dosage in an extended pedigree. Mol. Psychiatry 2011, 16, 491–503. [Google Scholar] [CrossRef] [PubMed]
- Peng, W.; Tan, C.; Mo, L.; Jiang, J.; Zhou, W.; Du, J.; Zhou, X.; Liu, X.; Chen, L. Glucose transporter 3 in neuronal glucose metabolism: Health and diseases. Metabolism 2021, 123, 154869. [Google Scholar] [CrossRef] [PubMed]
- Roeske, D.; Ludwig, K.U.; Neuhoff, N.; Becker, J.; Bartling, J.; Bruder, J.; Brockschmidt, F.F.; Warnke, A.; Remschmidt, H.; Hoffmann, P.; et al. First genome-wide association scan on neurophysiological endophenotypes points to trans-regulation effects on SLC2A3 in dyslexic children. Mol. Psychiatry 2011, 16, 97–107. [Google Scholar] [CrossRef] [PubMed]
- Tomi, M.; Zhao, Y.; Thamotharan, S.; Shin, B.C.; Devaskar, S.U. Early life nutrient restriction impairs blood-brain metabolic profile and neurobehavior predisposing to Alzheimer’s disease with aging. Brain Res. 2013, 1495, 61–75. [Google Scholar] [CrossRef]
- Zhao, Y.; Fung, C.; Shin, D.; Shin, B.C.; Thamotharan, S.; Sankar, R.; Ehninger, D.; Silva, A.; Devaskar, S.U. Neuronal glucose transporter isoform 3 deficient mice demonstrate features of autism spectrum disorders. Mol. Psychiatry 2010, 15, 286–299. [Google Scholar] [CrossRef]
- Wu, A.; Lee, D.; Xiong, W.C. Lactate Metabolism, Signaling, and Function in Brain Development, Synaptic Plasticity, Angiogenesis, and Neurodegenerative Diseases. Int. J. Mol. Sci. 2023, 24, 13398. [Google Scholar] [CrossRef] [PubMed]
- Simpson, I.A.; Carruthers, A.; Vannucci, S.J. Supply and demand in cerebral energy metabolism: The role of nutrient transporters. J. Cereb. Blood Flow Metab. 2007, 27, 1766–1791. [Google Scholar] [CrossRef] [PubMed]
- Rho, J.M.; Boison, D. The metabolic basis of epilepsy. Nat. Rev. Neurol. 2022, 18, 333–347. [Google Scholar] [CrossRef] [PubMed]
- Aghayan, M.; Rao, L.V.; Smith, R.M.; Jarett, L.; Charron, M.J.; Thorens, B.; Heyner, S. Developmental expression and cellular localization of glucose transporter molecules during mouse preimplantation development. Development 1992, 115, 305–312. [Google Scholar] [CrossRef] [PubMed]
- Ganguly, A.; McKnight, R.A.; Raychaudhuri, S.; Shin, B.C.; Ma, Z.; Moley, K.; Devaskar, S.U. Glucose transporter isoform-3 mutations cause early pregnancy loss and fetal growth restriction. Am. J. Physiol. Endocrinol. Metab. 2007, 292, E1241–E1255. [Google Scholar] [CrossRef] [PubMed]
- Heilig, C.W.; Saunders, T.; Brosius, F.C., 3rd; Moley, K.; Heilig, K.; Baggs, R.; Guo, L.; Conner, D. Glucose transporter-1-deficient mice exhibit impaired development and deformities that are similar to diabetic embryopathy. Proc. Natl. Acad. Sci. USA 2003, 100, 15613–15618. [Google Scholar] [CrossRef] [PubMed]
- Ohtsuki, S.; Kikkawa, T.; Hori, S.; Terasaki, T. Modulation and compensation of the mRNA expression of energy related transporters in the brain of glucose transporter 1-deficient mice. Biol. Pharm. Bull. 2006, 29, 1587–1591. [Google Scholar] [CrossRef] [PubMed]
- Adastra, K.L.; Frolova, A.I.; Chi, M.M.; Cusumano, D.; Bade, M.; Carayannopoulos, M.O.; Moley, K.H. Slc2a8 deficiency in mice results in reproductive and growth impairments. Biol. Reprod. 2012, 87, 49. [Google Scholar] [CrossRef] [PubMed]
- Smith, D.E.; Gridley, T. Differential screening of a PCR-generated mouse embryo cDNA library: Glucose transporters are differentially expressed in early postimplantation mouse embryos. Development 1992, 116, 555–561. [Google Scholar] [CrossRef]
- Pantaleon, M.; Kaye, P.L. Glucose transporters in preimplantation development. Rev. Reprod. 1998, 3, 77–81. [Google Scholar] [CrossRef]
- Matsumoto, K.; Akazawa, S.; Ishibashi, M.; Trocino, R.A.; Matsuo, H.; Yamasaki, H.; Yamaguchi, Y.; Nagamatsu, S.; Nagataki, S. Abundant expression of GLUT1 and GLUT3 in rat embryo during the early organogenesis period. Biochem. Biophys. Res. Commun. 1995, 209, 95–102. [Google Scholar] [CrossRef] [PubMed]
- Khan, J.Y.; Rajakumar, R.A.; McKnight, R.A.; Devaskar, U.P.; Devaskar, S.U. Developmental regulation of genes mediating murine brain glucose uptake. Am. J. Physiol. 1999, 276, R892–R900. [Google Scholar] [CrossRef] [PubMed]
- Brown, K.; Heller, D.S.; Zamudio, S.; Illsley, N.P. Glucose transporter 3 (GLUT3) protein expression in human placenta across gestation. Placenta 2011, 32, 1041–1049. [Google Scholar] [CrossRef] [PubMed]
- Kamei, Y.; Tsutsumi, O.; Yamakawa, A.; Oka, Y.; Taketani, Y.; Imaki, J. Maternal epidermal growth factor deficiency causes fetal hypoglycemia and intrauterine growth retardation in mice: Possible involvement of placental glucose transporter GLUT3 expression. Endocrinology 1999, 140, 4236–4243. [Google Scholar] [CrossRef] [PubMed]
- Shin, B.C.; Fujikura, K.; Suzuki, T.; Tanaka, S.; Takata, K. Glucose transporter GLUT3 in the rat placental barrier: A possible machinery for the transplacental transfer of glucose. Endocrinology 1997, 138, 3997–4004. [Google Scholar] [CrossRef] [PubMed]
- Dawson, P.A.; Mychaleckyj, J.C.; Fossey, S.C.; Mihic, S.J.; Craddock, A.L.; Bowden, D.W. Sequence and functional analysis of GLUT10: A glucose transporter in the Type 2 diabetes-linked region of chromosome 20q12-13.1. Mol. Genet. Metab. 2001, 74, 186–199. [Google Scholar] [CrossRef] [PubMed]
- Janzen, C.; Lei, M.Y.Y.; Jeong, I.S.D.; Ganguly, A.; Sullivan, P.; Paharkova, V.; Capodanno, G.; Nakamura, H.; Perry, A.; Shin, B.C.; et al. Humanin (HN) and glucose transporter 8 (GLUT8) in pregnancies complicated by intrauterine growth restriction. PLoS ONE 2018, 13, e0193583. [Google Scholar] [CrossRef]
- Limesand, S.W.; Regnault, T.R.; Hay, W.W., Jr. Characterization of glucose transporter 8 (GLUT8) in the ovine placenta of normal and growth restricted fetuses. Placenta 2004, 25, 70–77. [Google Scholar] [CrossRef] [PubMed]
- Devaskar, S.U.; Chu, A. Intrauterine Growth Restriction: Hungry for an Answer. Physiology 2016, 31, 131–146. [Google Scholar] [CrossRef]
- Sullivan, E.L.; Smith, M.S.; Grove, K.L. Perinatal exposure to high-fat diet programs energy balance, metabolism and behavior in adulthood. Neuroendocrinology 2011, 93, 1–8. [Google Scholar] [CrossRef]
- Mueckler, M.; Thorens, B. The SLC2 (GLUT) family of membrane transporters. Mol. Aspects Med. 2013, 34, 121–138. [Google Scholar] [CrossRef] [PubMed]
- Scheepers, A.; Joost, H.G.; Schurmann, A. The glucose transporter families SGLT and GLUT: Molecular basis of normal and aberrant function. JPEN J. Parenter. Enteral Nutr. 2004, 28, 364–371. [Google Scholar] [CrossRef]
- Manolescu, A.R.; Witkowska, K.; Kinnaird, A.; Cessford, T.; Cheeseman, C. Facilitated hexose transporters: New perspectives on form and function. Physiology 2007, 22, 234–240. [Google Scholar] [CrossRef] [PubMed]
- Joost, H.G.; Thorens, B. The extended GLUT-family of sugar/polyol transport facilitators: Nomenclature, sequence characteristics, and potential function of its novel members (review). Mol. Membr. Biol. 2001, 18, 247–256. [Google Scholar] [CrossRef]
- Holman, G.D. Structure, function and regulation of mammalian glucose transporters of the SLC2 family. Pflug. Arch. 2020, 472, 1155–1175. [Google Scholar] [CrossRef] [PubMed]
- Kasahara, M.; Hinkle, P.C. Reconstitution and purification of the D-glucose transporter from human erythrocytes. J. Biol. Chem. 1977, 252, 7384–7390. [Google Scholar] [CrossRef]
- Mann, G.E.; Yudilevich, D.L.; Sobrevia, L. Regulation of amino acid and glucose transporters in endothelial and smooth muscle cells. Physiol. Rev. 2003, 83, 183–252. [Google Scholar] [CrossRef] [PubMed]
- Mueckler, M.; Caruso, C.; Baldwin, S.A.; Panico, M.; Blench, I.; Morris, H.R.; Allard, W.J.; Lienhard, G.E.; Lodish, H.F. Sequence and structure of a human glucose transporter. Science 1985, 229, 941–945. [Google Scholar] [CrossRef]
- Fukumoto, H.; Seino, S.; Imura, H.; Seino, Y.; Eddy, R.L.; Fukushima, Y.; Byers, M.G.; Shows, T.B.; Bell, G.I. Sequence, tissue distribution, and chromosomal localization of mRNA encoding a human glucose transporter-like protein. Proc. Natl. Acad. Sci. USA 1988, 85, 5434–5438. [Google Scholar] [CrossRef]
- Kayano, T.; Fukumoto, H.; Eddy, R.L.; Fan, Y.S.; Byers, M.G.; Shows, T.B.; Bell, G.I. Evidence for a family of human glucose transporter-like proteins. Sequence and gene localization of a protein expressed in fetal skeletal muscle and other tissues. J. Biol. Chem. 1988, 263, 15245–15248. [Google Scholar] [CrossRef]
- Koepsell, H. Glucose transporters in brain in health and disease. Pflug. Arch. 2020, 472, 1299–1343. [Google Scholar] [CrossRef] [PubMed]
- Martinez, F.; Cifuentes, M.; Tapia, J.C.; Nualart, F. The median eminence as the hypothalamic area involved in rapid transfer of glucose to the brain: Functional and cellular mechanisms. J. Mol. Med. 2019, 97, 1085–1097. [Google Scholar] [CrossRef] [PubMed]
- Thorens, B.; Mueckler, M. Glucose transporters in the 21st Century. Am. J. Physiol. Endocrinol. Metab. 2010, 298, E141–E145. [Google Scholar] [CrossRef] [PubMed]
- Cushman, S.W.; Wardzala, L.J. Potential mechanism of insulin action on glucose transport in the isolated rat adipose cell. Apparent translocation of intracellular transport systems to the plasma membrane. J. Biol. Chem. 1980, 255, 4758–4762. [Google Scholar] [CrossRef] [PubMed]
- Suzuki, K.; Kono, T. Evidence that insulin causes translocation of glucose transport activity to the plasma membrane from an intracellular storage site. Proc. Natl. Acad. Sci. USA 1980, 77, 2542–2545. [Google Scholar] [CrossRef] [PubMed]
- Wallberg-Henriksson, H.; Zierath, J.R. GLUT4: A key player regulating glucose homeostasis? Insights from transgenic and knockout mice (review). Mol. Membr. Biol. 2001, 18, 205–211. [Google Scholar] [CrossRef] [PubMed]
- Doege, H.; Bocianski, A.; Joost, H.G.; Schurmann, A. Activity and genomic organization of human glucose transporter 9 (GLUT9), a novel member of the family of sugar-transport facilitators predominantly expressed in brain and leucocytes. Biochem. J. 2000, 350 Pt 3, 771–776. [Google Scholar] [CrossRef] [PubMed]
- Lisinski, I.; Schurmann, A.; Joost, H.G.; Cushman, S.W.; Al-Hasani, H. Targeting of GLUT6 (formerly GLUT9) and GLUT8 in rat adipose cells. Biochem. J. 2001, 358, 517–522. [Google Scholar] [CrossRef] [PubMed]
- Ibberson, M.; Riederer, B.M.; Uldry, M.; Guhl, B.; Roth, J.; Thorens, B. Immunolocalization of GLUTX1 in the testis and to specific brain areas and vasopressin-containing neurons. Endocrinology 2002, 143, 276–284. [Google Scholar] [CrossRef]
- Scheepers, A.; Doege, H.; Joost, H.G.; Schurmann, A. Mouse GLUT8: Genomic organization and regulation of expression in 3T3-L1 adipocytes by glucose. Biochem. Biophys. Res. Commun. 2001, 288, 969–974. [Google Scholar] [CrossRef]
- Schmidt, S.; Gawlik, V.; Holter, S.M.; Augustin, R.; Scheepers, A.; Behrens, M.; Wurst, W.; Gailus-Durner, V.; Fuchs, H.; Hrabe de Angelis, M.; et al. Deletion of glucose transporter GLUT8 in mice increases locomotor activity. Behav. Genet. 2008, 38, 396–406. [Google Scholar] [CrossRef] [PubMed]
- Augustin, R.; Carayannopoulos, M.O.; Dowd, L.O.; Phay, J.E.; Moley, J.F.; Moley, K.H. Identification and characterization of human glucose transporter-like protein-9 (GLUT9): Alternative splicing alters trafficking. J. Biol. Chem. 2004, 279, 16229–16236. [Google Scholar] [CrossRef] [PubMed]
- McVie-Wylie, A.J.; Lamson, D.R.; Chen, Y.T. Molecular cloning of a novel member of the GLUT family of transporters, SLC2a10 (GLUT10), localized on chromosome 20q13.1: A candidate gene for NIDDM susceptibility. Genomics 2001, 72, 113–117. [Google Scholar] [CrossRef] [PubMed]
- Segade, F.; Allred, D.C.; Bowden, D.W. Functional characterization of the promoter of the human glucose transporter 10 gene. Biochim. Biophys. Acta 2005, 1730, 147–158. [Google Scholar] [CrossRef] [PubMed]
- Rogers, S.; Macheda, M.L.; Docherty, S.E.; Carty, M.D.; Henderson, M.A.; Soeller, W.C.; Gibbs, E.M.; James, D.E.; Best, J.D. Identification of a novel glucose transporter-like protein-GLUT-12. Am. J. Physiol. Endocrinol. Metab. 2002, 282, E733–E738. [Google Scholar] [CrossRef] [PubMed]
- Wright, E.M.; Loo, D.D.; Hirayama, B.A. Biology of human sodium glucose transporters. Physiol. Rev. 2011, 91, 733–794. [Google Scholar] [CrossRef]
- Chen, J.; Williams, S.; Ho, S.; Loraine, H.; Hagan, D.; Whaley, J.M.; Feder, J.N. Quantitative PCR tissue expression profiling of the human SGLT2 gene and related family members. Diabetes Ther. 2010, 1, 57–92. [Google Scholar] [CrossRef] [PubMed]
- Ferrannini, E.; Solini, A. SGLT2 inhibition in diabetes mellitus: Rationale and clinical prospects. Nat. Rev. Endocrinol. 2012, 8, 495–502. [Google Scholar] [CrossRef] [PubMed]
- Wright, E.M. Renal Na(+)-glucose cotransporters. Am. J. Physiol. Ren. Physiol. 2001, 280, F10–F18. [Google Scholar] [CrossRef]
- Diez-Sampedro, A.; Hirayama, B.A.; Osswald, C.; Gorboulev, V.; Baumgarten, K.; Volk, C.; Wright, E.M.; Koepsell, H. A glucose sensor hiding in a family of transporters. Proc. Natl. Acad. Sci. USA 2003, 100, 11753–11758. [Google Scholar] [CrossRef]
- Baader-Pagler, T.; Eckhardt, M.; Himmelsbach, F.; Sauer, A.; Stierstorfer, B.E.; Hamilton, B.S. SGLT6—A pharmacological target for the treatment of obesity? Adipocyte 2018, 7, 277–284. [Google Scholar] [CrossRef] [PubMed]
- Lin, X.; Ma, L.; Fitzgerald, R.L.; Ostlund, R.E., Jr. Human sodium/inositol cotransporter 2 (SMIT2) transports inositols but not glucose in L6 cells. Arch. Biochem. Biophys. 2009, 481, 197–201. [Google Scholar] [CrossRef] [PubMed]
- Burant, C.F.; Takeda, J.; Brot-Laroche, E.; Bell, G.I.; Davidson, N.O. Fructose transporter in human spermatozoa and small intestine is GLUT5. J. Biol. Chem. 1992, 267, 14523–14526. [Google Scholar] [CrossRef] [PubMed]
- Kayano, T.; Burant, C.F.; Fukumoto, H.; Gould, G.W.; Fan, Y.S.; Eddy, R.L.; Byers, M.G.; Shows, T.B.; Seino, S.; Bell, G.I. Human facilitative glucose transporters. Isolation, functional characterization, and gene localization of cDNAs encoding an isoform (GLUT5) expressed in small intestine, kidney, muscle, and adipose tissue and an unusual glucose transporter pseudogene-like sequence (GLUT6). J. Biol. Chem. 1990, 265, 13276–13282. [Google Scholar] [PubMed]
- Cheeseman, C. GLUT7: A new intestinal facilitated hexose transporter. Am. J. Physiol. Endocrinol. Metab. 2008, 295, E238–E241. [Google Scholar] [CrossRef] [PubMed]
- Wu, X.; Li, W.; Sharma, V.; Godzik, A.; Freeze, H.H. Cloning and characterization of glucose transporter 11, a novel sugar transporter that is alternatively spliced in various tissues. Mol. Genet. Metab. 2002, 76, 37–45. [Google Scholar] [CrossRef] [PubMed]
- Doege, H.; Bocianski, A.; Scheepers, A.; Axer, H.; Eckel, J.; Joost, H.G.; Schurmann, A. Characterization of human glucose transporter (GLUT) 11 (encoded by SLC2A11), a novel sugar-transport facilitator specifically expressed in heart and skeletal muscle. Biochem. J. 2001, 359, 443–449. [Google Scholar] [CrossRef]
- Uldry, M.; Ibberson, M.; Horisberger, J.D.; Chatton, J.Y.; Riederer, B.M.; Thorens, B. Identification of a mammalian H(+)-myo-inositol symporter expressed predominantly in the brain. EMBO J. 2001, 20, 4467–4477. [Google Scholar] [CrossRef] [PubMed]
- Carruthers, A.; DeZutter, J.; Ganguly, A.; Devaskar, S.U. Will the original glucose transporter isoform please stand up! Am. J. Physiol. Endocrinol. Metab. 2009, 297, E836–E848. [Google Scholar] [CrossRef]
- Watts, A.G.; Donovan, C.M. Sweet talk in the brain: Glucosensing, neural networks, and hypoglycemic counterregulation. Front. Neuroendocrinol. 2010, 31, 32–43. [Google Scholar] [CrossRef]
- Haber, R.S.; Weinstein, S.P.; O’Boyle, E.; Morgello, S. Tissue distribution of the human GLUT3 glucose transporter. Endocrinology 1993, 132, 2538–2543. [Google Scholar] [CrossRef] [PubMed]
- Burant, C.F.; Davidson, N.O. GLUT3 glucose transporter isoform in rat testis: Localization, effect of diabetes mellitus, and comparison to human testis. Am. J. Physiol. 1994, 267, R1488–R1495. [Google Scholar] [CrossRef] [PubMed]
- Devaskar, S.U.; Devaskar, U.P.; Schroeder, R.E.; deMello, D.; Fiedorek, F.T., Jr.; Mueckler, M. Expression of genes involved in placental glucose uptake and transport in the nonobese diabetic mouse pregnancy. Am. J. Obstet. Gynecol. 1994, 171, 1316–1323. [Google Scholar] [CrossRef] [PubMed]
- Zhou, J.; Bondy, C.A. Placental glucose transporter gene expression and metabolism in the rat. J. Clin. Investig. 1993, 91, 845–852. [Google Scholar] [CrossRef]
- Mantych, G.J.; James, D.E.; Chung, H.D.; Devaskar, S.U. Cellular localization and characterization of Glut 3 glucose transporter isoform in human brain. Endocrinology 1992, 131, 1270–1278. [Google Scholar] [CrossRef] [PubMed]
- Fung, C.; Evans, E.; Shin, D.; Shin, B.C.; Zhao, Y.; Sankar, R.; Chaudhuri, G.; Devaskar, S.U. Hypoxic-ischemic brain injury exacerbates neuronal apoptosis and precipitates spontaneous seizures in glucose transporter isoform 3 heterozygous null mice. J. Neurosci. Res. 2010, 88, 3386–3398. [Google Scholar] [CrossRef] [PubMed]
- Choeiri, C.; Staines, W.; Messier, C. Immunohistochemical localization and quantification of glucose transporters in the mouse brain. Neuroscience 2002, 111, 19–34. [Google Scholar] [CrossRef] [PubMed]
- Sankar, R.; Thamotharan, S.; Shin, D.; Moley, K.H.; Devaskar, S.U. Insulin-responsive glucose transporters-GLUT8 and GLUT4 are expressed in the developing mammalian brain. Brain Res. Mol. Brain Res. 2002, 107, 157–165. [Google Scholar] [CrossRef] [PubMed]
- Shin, B.C.; Cepeda, C.; Eghbali, M.; Byun, S.Y.; Levine, M.S.; Devaskar, S.U. Adult glut3 homozygous null mice survive to demonstrate neural excitability and altered neurobehavioral responses reminiscent of neurodevelopmental disorders. Exp. Neurol. 2021, 338, 113603. [Google Scholar] [CrossRef]
- Shin, B.C.; Cepeda, C.; Estrada-Sanchez, A.M.; Levine, M.S.; Hodaei, L.; Dai, Y.; Jung, J.; Ganguly, A.; Clark, P.; Devaskar, S.U. Neural Deletion of Glucose Transporter Isoform 3 Creates Distinct Postnatal and Adult Neurobehavioral Phenotypes. J. Neurosci. 2018, 38, 9579–9599. [Google Scholar] [CrossRef]
- Carayannopoulos, M.O.; Xiong, F.; Jensen, P.; Rios-Galdamez, Y.; Huang, H.; Lin, S.; Devaskar, S.U. GLUT3 gene expression is critical for embryonic growth, brain development and survival. Mol. Genet. Metab. 2014, 111, 477–483. [Google Scholar] [CrossRef] [PubMed]
- Stanirowski, P.J.; Lipa, M.; Bomba-Opoń, D.; Wielgoś, M. Expression of placental glucose transporter proteins in pregnancies complicated by fetal growth disorders. Adv. Protein Chem. Struct. Biol. 2021, 123, 95–131. [Google Scholar] [CrossRef]
- Khan, J.Y.; Rajakumar, R.A.; Devaskar, U.P.; Weissfeld, L.A.; Devaskar, S.U. Effect of primary congenital hypothyroidism upon expression of genes mediating murine brain glucose uptake. Pediatr. Res. 1999, 45, 718–725. [Google Scholar] [CrossRef]
- Rajakumar, A.; Thamotharan, S.; Raychaudhuri, N.; Menon, R.K.; Devaskar, S.U. Trans-activators regulating neuronal glucose transporter isoform-3 gene expression in mammalian neurons. J. Biol. Chem. 2004, 279, 26768–26779. [Google Scholar] [CrossRef] [PubMed]
- Rajakumar, R.A.; Thamotharan, S.; Menon, R.K.; Devaskar, S.U. Sp1 and Sp3 regulate transcriptional activity of the facilitative glucose transporter isoform-3 gene in mammalian neuroblasts and trophoblasts. J. Biol. Chem. 1998, 273, 27474–27483. [Google Scholar] [CrossRef]
- Boado, R.J. Post-transcription modulation of the blood-brain barrier GLUT1 glucose transporter by brain-derived factors. J. Neural Transm. Suppl. 2000, 59, 255–261. [Google Scholar] [CrossRef]
- Thamotharan, S.; Stout, D.; Shin, B.C.; Devaskar, S.U. Temporal and spatial distribution of murine placental and brain GLUT3-luciferase transgene as a readout of in vivo transcription. Am. J. Physiol. Endocrinol. Metab. 2013, 304, E254–E266. [Google Scholar] [CrossRef] [PubMed]
- Plassmeyer, S.P.; Florian, C.P.; Kasper, M.J.; Chase, R.; Mueller, S.; Liu, Y.; White, K.M.; Jungers, C.F.; Djuranovic, S.P.; Djuranovic, S.; et al. A Massively Parallel Screen of 5′UTR Mutations Identifies Variants Impacting Translation and Protein Production in Neurodevelopmental Disorder Genes. medRxiv 2023. [Google Scholar] [CrossRef]
- Vannucci, S.J.; Reinhart, R.; Maher, F.; Bondy, C.A.; Lee, W.H.; Vannucci, R.C.; Simpson, I.A. Alterations in GLUT1 and GLUT3 glucose transporter gene expression following unilateral hypoxia-ischemia in the immature rat brain. Brain Res. Dev. Brain Res. 1998, 107, 255–264. [Google Scholar] [CrossRef]
- Gronlund, K.M.; Gerhart, D.Z.; Leino, R.L.; McCall, A.L.; Drewes, L.R. Chronic seizures increase glucose transporter abundance in rat brain. J. Neuropathol. Exp. Neurol. 1996, 55, 832–840. [Google Scholar] [CrossRef]
- Leroy, C.; Pierre, K.; Simpson, I.A.; Pellerin, L.; Vannucci, S.J.; Nehlig, A. Temporal changes in mRNA expression of the brain nutrient transporters in the lithium-pilocarpine model of epilepsy in the immature and adult rat. Neurobiol. Dis. 2011, 43, 588–597. [Google Scholar] [CrossRef]
- McClory, H.; Williams, D.; Sapp, E.; Gatune, L.W.; Wang, P.; DiFiglia, M.; Li, X. Glucose transporter 3 is a rab11-dependent trafficking cargo and its transport to the cell surface is reduced in neurons of CAG140 Huntington’s disease mice. Acta Neuropathol. Commun. 2014, 2, 179. [Google Scholar] [CrossRef]
- Iuliano, M.; Seeley, C.; Sapp, E.; Jones, E.L.; Martin, C.; Li, X.; DiFiglia, M.; Kegel-Gleason, K.B. Disposition of Proteins and Lipids in Synaptic Membrane Compartments Is Altered in Q175/Q7 Huntington’s Disease Mouse Striatum. Front. Synaptic Neurosci. 2021, 13, 618391. [Google Scholar] [CrossRef]
- Acuna, A.I.; Esparza, M.; Kramm, C.; Beltran, F.A.; Parra, A.V.; Cepeda, C.; Toro, C.A.; Vidal, R.L.; Hetz, C.; Concha, I.I.; et al. A failure in energy metabolism and antioxidant uptake precede symptoms of Huntington’s disease in mice. Nat. Commun. 2013, 4, 2917. [Google Scholar] [CrossRef]
- Ciarmiello, A.; Cannella, M.; Lastoria, S.; Simonelli, M.; Frati, L.; Rubinsztein, D.C.; Squitieri, F. Brain white-matter volume loss and glucose hypometabolism precede the clinical symptoms of Huntington’s disease. J. Nucl. Med. 2006, 47, 215–222. [Google Scholar]
- Gamberino, W.C.; Brennan, W.A., Jr. Glucose transporter isoform expression in Huntington’s disease brain. J. Neurochem. 1994, 63, 1392–1397. [Google Scholar] [CrossRef]
- Li, X.; Valencia, A.; McClory, H.; Sapp, E.; Kegel, K.B.; Difiglia, M. Deficient Rab11 activity underlies glucose hypometabolism in primary neurons of Huntington’s disease mice. Biochem. Biophys. Res. Commun. 2012, 421, 727–730. [Google Scholar] [CrossRef]
- Vittori, A.; Breda, C.; Repici, M.; Orth, M.; Roos, R.A.; Outeiro, T.F.; Giorgini, F.; Hollox, E.J.; Network, R.i.o.t.E.H.s.D. Copy-number variation of the neuronal glucose transporter gene SLC2A3 and age of onset in Huntington’s disease. Hum. Mol. Genet. 2014, 23, 3129–3137. [Google Scholar] [CrossRef]
- Simpson, I.A.; Chundu, K.R.; Davies-Hill, T.; Honer, W.G.; Davies, P. Decreased concentrations of GLUT1 and GLUT3 glucose transporters in the brains of patients with Alzheimer’s disease. Ann. Neurol. 1994, 35, 546–551. [Google Scholar] [CrossRef]
- Liu, Y.; Liu, F.; Iqbal, K.; Grundke-Iqbal, I.; Gong, C.X. Decreased glucose transporters correlate to abnormal hyperphosphorylation of tau in Alzheimer disease. FEBS Lett. 2008, 582, 359–364. [Google Scholar] [CrossRef]
- An, Y.; Varma, V.R.; Varma, S.; Casanova, R.; Dammer, E.; Pletnikova, O.; Chia, C.W.; Egan, J.M.; Ferrucci, L.; Troncoso, J.; et al. Evidence for brain glucose dysregulation in Alzheimer’s disease. Alzheimers Dement. 2018, 14, 318–329. [Google Scholar] [CrossRef]
- Shabani, K.; Hassan, B.A. The brain on time: Links between development and neurodegeneration. Development 2023, 150, dev200397. [Google Scholar] [CrossRef]
- Cepeda, C.; Oikonomou, K.D.; Cummings, D.; Barry, J.; Yazon, V.W.; Chen, D.T.; Asai, J.; Williams, C.K.; Vinters, H.V. Developmental origins of cortical hyperexcitability in Huntington’s disease: Review and new observations. J. Neurosci. Res. 2019, 97, 1624–1635. [Google Scholar] [CrossRef]
- Ratie, L.; Humbert, S. A developmental component to Huntington’s disease. Rev. Neurol. 2024, 180, 357–362. [Google Scholar] [CrossRef]
- Arendt, T.; Stieler, J.; Ueberham, U. Is sporadic Alzheimer’s disease a developmental disorder? J. Neurochem. 2017, 143, 396–408. [Google Scholar] [CrossRef]
- Merker, S.; Reif, A.; Ziegler, G.C.; Weber, H.; Mayer, U.; Ehlis, A.C.; Conzelmann, A.; Johansson, S.; Muller-Reible, C.; Nanda, I.; et al. SLC2A3 single-nucleotide polymorphism and duplication influence cognitive processing and population-specific risk for attention-deficit/hyperactivity disorder. J. Child Psychol. Psychiatry 2017, 58, 798–809. [Google Scholar] [CrossRef]
- Skeide, M.A.; Kirsten, H.; Kraft, I.; Schaadt, G.; Muller, B.; Neef, N.; Brauer, J.; Wilcke, A.; Emmrich, F.; Boltze, J.; et al. Genetic dyslexia risk variant is related to neural connectivity patterns underlying phonological awareness in children. Neuroimage 2015, 118, 414–421. [Google Scholar] [CrossRef]
- Schmidt, S.; Richter, M.; Montag, D.; Sartorius, T.; Gawlik, V.; Hennige, A.M.; Scherneck, S.; Himmelbauer, H.; Lutz, S.Z.; Augustin, R.; et al. Neuronal functions, feeding behavior, and energy balance in Slc2a3+/− mice. Am. J. Physiol. Endocrinol. Metab. 2008, 295, E1084–E1094. [Google Scholar] [CrossRef]
- Stuart, C.A.; Ross, I.R.; Howell, M.E.; McCurry, M.P.; Wood, T.G.; Ceci, J.D.; Kennel, S.J.; Wall, J. Brain glucose transporter (Glut3) haploinsufficiency does not impair mouse brain glucose uptake. Brain Res. 2011, 1384, 15–22. [Google Scholar] [CrossRef]
- Chu, S.Y.; Bachman, D.J.; Callaghan, W.M.; Whitlock, E.P.; Dietz, P.M.; Berg, C.J.; O’Keeffe-Rosetti, M.; Bruce, F.C.; Hornbrook, M.C. Association between obesity during pregnancy and increased use of health care. N. Engl. J. Med. 2008, 358, 1444–1453. [Google Scholar] [CrossRef]
- Yu, Y.; Ma, Q.; Groth, S.W. Prepregnancy dieting and obstetrical and neonatal outcomes: Findings from a national surveillance project in the United States. Midwifery 2024, 132, 103972. [Google Scholar] [CrossRef]
- Bilbo, S.D.; Tsang, V. Enduring consequences of maternal obesity for brain inflammation and behavior of offspring. FASEB J. 2010, 24, 2104–2115. [Google Scholar] [CrossRef]
- Boney, C.M.; Verma, A.; Tucker, R.; Vohr, B.R. Metabolic syndrome in childhood: Association with birth weight, maternal obesity, and gestational diabetes mellitus. Pediatrics 2005, 115, e290–e296. [Google Scholar] [CrossRef]
- Pinney, S.E.; Simmons, R.A. Metabolic programming, epigenetics, and gestational diabetes mellitus. Curr. Diabetes Rep. 2012, 12, 67–74. [Google Scholar] [CrossRef]
- Urbonaite, G.; Knyzeliene, A.; Bunn, F.S.; Smalskys, A.; Neniskyte, U. The impact of maternal high-fat diet on offspring neurodevelopment. Front. Neurosci. 2022, 16, 909762. [Google Scholar] [CrossRef]
- Sathyapalan, T.; Mellor, D.; Atkin, S.L. Obesity and gestational diabetes. Semin. Fetal Neonatal Med. 2010, 15, 89–93. [Google Scholar] [CrossRef]
- Sweeting, A.; Wong, J.; Murphy, H.R.; Ross, G.P. A Clinical Update on Gestational Diabetes Mellitus. Endocr. Rev. 2022, 43, 763–793. [Google Scholar] [CrossRef]
- Grayson, B.E.; Levasseur, P.R.; Williams, S.M.; Smith, M.S.; Marks, D.L.; Grove, K.L. Changes in melanocortin expression and inflammatory pathways in fetal offspring of nonhuman primates fed a high-fat diet. Endocrinology 2010, 151, 1622–1632. [Google Scholar] [CrossRef]
- Srinivasan, M.; Katewa, S.D.; Palaniyappan, A.; Pandya, J.D.; Patel, M.S. Maternal high-fat diet consumption results in fetal malprogramming predisposing to the onset of metabolic syndrome-like phenotype in adulthood. Am. J. Physiol. Endocrinol. Metab. 2006, 291, E792–E799. [Google Scholar] [CrossRef]
- Ganguly, A.; Devaskar, S.U. High-fat diet affects pregestational adiposity and glucose tolerance perturbing gestational placental macronutrient transporters culminating in an obese offspring in wild-type and glucose transporter isoform 3 heterozygous null mice. J. Nutr. Biochem. 2018, 62, 192–201. [Google Scholar] [CrossRef]
- Jones, H.N.; Woollett, L.A.; Barbour, N.; Prasad, P.D.; Powell, T.L.; Jansson, T. High-fat diet before and during pregnancy causes marked up-regulation of placental nutrient transport and fetal overgrowth in C57/BL6 mice. FASEB J. 2009, 23, 271–278. [Google Scholar] [CrossRef]
- Rosario, F.J.; Kanai, Y.; Powell, T.L.; Jansson, T. Increased placental nutrient transport in a novel mouse model of maternal obesity with fetal overgrowth. Obesity 2015, 23, 1663–1670. [Google Scholar] [CrossRef] [PubMed]
- Ganguly, A.; Touma, M.; Thamotharan, S.; De Vivo, D.C.; Devaskar, S.U. Maternal Calorie Restriction Causing Uteroplacental Insufficiency Differentially Affects Mammalian Placental Glucose and Leucine Transport Molecular Mechanisms. Endocrinology 2016, 157, 4041–4054. [Google Scholar] [CrossRef] [PubMed]
- Chizhikov, D.; Buddington, R.K.; Iskusnykh, I.Y. Effects of Phosphatidylserine Source of Docosahexaenoic Acid on Cerebellar Development in Preterm Pigs. Brain Sci. 2020, 10, 475. [Google Scholar] [CrossRef]
- Colombo, J.; Carlson, S.E.; Cheatham, C.L.; Shaddy, D.J.; Kerling, E.H.; Thodosoff, J.M.; Gustafson, K.M.; Brez, C. Long-term effects of LCPUFA supplementation on childhood cognitive outcomes. Am. J. Clin. Nutr. 2013, 98, 403–412. [Google Scholar] [CrossRef]
- Neuringer, M.; Connor, W.E. n-3 fatty acids in the brain and retina: Evidence for their essentiality. Nutr. Rev. 1986, 44, 285–294. [Google Scholar] [CrossRef]
- Uauy, R.; Birch, E.; Birch, D.; Peirano, P. Visual and brain function measurements in studies of n-3 fatty acid requirements of infants. J. Pediatr. 1992, 120, S168–S180. [Google Scholar] [CrossRef]
- Shin, B.C.; Ghosh, S.; Dai, Y.; Byun, S.Y.; Calkins, K.L.; Devaskar, S.U. Early life high-fat diet exposure maintains glucose tolerance and insulin sensitivity with a fatty liver and small brain size in the adult offspring. Nutr. Res. 2019, 69, 67–81. [Google Scholar] [CrossRef] [PubMed]
- Stachowiak, E.K.; Srinivasan, M.; Stachowiak, M.K.; Patel, M.S. Maternal obesity induced by a high fat diet causes altered cellular development in fetal brains suggestive of a predisposition of offspring to neurological disorders in later life. Metab. Brain Dis. 2013, 28, 721–725. [Google Scholar] [CrossRef]
- Boison, D. New insights into the mechanisms of the ketogenic diet. Curr. Opin. Neurol. 2017, 30, 187–192. [Google Scholar] [CrossRef]
- Dynka, D.; Kowalcze, K.; Paziewska, A. The Role of Ketogenic Diet in the Treatment of Neurological Diseases. Nutrients 2022, 14, 5003. [Google Scholar] [CrossRef] [PubMed]
- Wheless, J.W. History of the ketogenic diet. Epilepsia 2008, 49 (Suppl. S8), 3–5. [Google Scholar] [CrossRef] [PubMed]
- Klepper, J.; Akman, C.; Armeno, M.; Auvin, S.; Cervenka, M.; Cross, H.J.; De Giorgis, V.; Della Marina, A.; Engelstad, K.; Heussinger, N.; et al. Glut1 Deficiency Syndrome (Glut1DS): State of the art in 2020 and recommendations of the international Glut1DS study group. Epilepsia Open 2020, 5, 354–365. [Google Scholar] [CrossRef] [PubMed]
- Seidner, G.; Alvarez, M.G.; Yeh, J.I.; O’Driscoll, K.R.; Klepper, J.; Stump, T.S.; Wang, D.; Spinner, N.B.; Birnbaum, M.J.; De Vivo, D.C. GLUT-1 deficiency syndrome caused by haploinsufficiency of the blood-brain barrier hexose carrier. Nat. Genet. 1998, 18, 188–191. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.H.; Shaw, A.; Blackford, R.; Lowman, W.; Laux, L.C.; Millichap, J.J.; Nordli, D.R., Jr. The ketogenic diet in children 3 years of age or younger: A 10-year single-center experience. Sci. Rep. 2019, 9, 8736. [Google Scholar] [CrossRef] [PubMed]
- Lyons, L.; Schoeler, N.E.; Langan, D.; Cross, J.H. Use of ketogenic diet therapy in infants with epilepsy: A systematic review and meta-analysis. Epilepsia 2020, 61, 1261–1281. [Google Scholar] [CrossRef] [PubMed]
- Martin-McGill, K.J.; Bresnahan, R.; Levy, R.G.; Cooper, P.N. Ketogenic diets for drug-resistant epilepsy. Cochrane Database Syst. Rev. 2020, 6, CD001903. [Google Scholar] [CrossRef] [PubMed]
- Bertoli, S.; Trentani, C.; Ferraris, C.; De Giorgis, V.; Veggiotti, P.; Tagliabue, A. Long-term effects of a ketogenic diet on body composition and bone mineralization in GLUT-1 deficiency syndrome: A case series. Nutrition 2014, 30, 726–728. [Google Scholar] [CrossRef] [PubMed]
- Kang, H.C.; Chung, D.E.; Kim, D.W.; Kim, H.D. Early- and late-onset complications of the ketogenic diet for intractable epilepsy. Epilepsia 2004, 45, 1116–1123. [Google Scholar] [CrossRef]
- Paoli, A.; Rubini, A.; Volek, J.S.; Grimaldi, K.A. Beyond weight loss: A review of the therapeutic uses of very-low-carbohydrate (ketogenic) diets. Eur. J. Clin. Nutr. 2013, 67, 789–796. [Google Scholar] [CrossRef]
- Evangeliou, A.; Vlachonikolis, I.; Mihailidou, H.; Spilioti, M.; Skarpalezou, A.; Makaronas, N.; Prokopiou, A.; Christodoulou, P.; Liapi-Adamidou, G.; Helidonis, E.; et al. Application of a ketogenic diet in children with autistic behavior: Pilot study. J. Child. Neurol. 2003, 18, 113–118. [Google Scholar] [CrossRef]
- Connealy, B.D.; Northrup, H.; Au, K.S. Genetic variations in the GLUT3 gene associated with myelomeningocele. Am. J. Obstet. Gynecol. 2014, 211, 305.e1–305.e8. [Google Scholar] [CrossRef]
- Cheng, C.M.; Kelley, B.; Wang, J.; Strauss, D.; Eagles, D.A.; Bondy, C.A. A ketogenic diet increases brain insulin-like growth factor receptor and glucose transporter gene expression. Endocrinology 2003, 144, 2676–2682. [Google Scholar] [CrossRef]
- Dai, Y.; Zhao, Y.; Tomi, M.; Shin, B.C.; Thamotharan, S.; Mazarati, A.; Sankar, R.; Wang, E.A.; Cepeda, C.; Levine, M.S.; et al. Sex-Specific Life Course Changes in the Neuro-Metabolic Phenotype of Glut3 Null Heterozygous Mice: Ketogenic Diet Ameliorates Electroencephalographic Seizures and Improves Sociability. Endocrinology 2017, 158, 936–949. [Google Scholar] [CrossRef]
- Herrera, E.; Gomez-Coronado, D.; Lasuncion, M.A. Lipid metabolism in pregnancy. Biol. Neonate 1987, 51, 70–77. [Google Scholar] [CrossRef]
- Sussman, D.; Ellegood, J.; Henkelman, M. A gestational ketogenic diet alters maternal metabolic status as well as offspring physiological growth and brain structure in the neonatal mouse. BMC Pregnancy Childbirth 2013, 13, 198. [Google Scholar] [CrossRef]
- Sussman, D.; Germann, J.; Henkelman, M. Gestational ketogenic diet programs brain structure and susceptibility to depression & anxiety in the adult mouse offspring. Brain Behav. 2015, 5, e00300. [Google Scholar] [CrossRef]
- Brodsky, D.; Christou, H. Current concepts in intrauterine growth restriction. J. Intensive Care Med. 2004, 19, 307–319. [Google Scholar] [CrossRef]
- Mandruzzato, G.; Antsaklis, A.; Botet, F.; Chervenak, F.A.; Figueras, F.; Grunebaum, A.; Puerto, B.; Skupski, D.; Stanojevic, M. Intrauterine restriction (IUGR). J. Perinat. Med. 2008, 36, 277–281. [Google Scholar] [CrossRef]
- Barker, D.J.; Godfrey, K.M.; Fall, C.; Osmond, C.; Winter, P.D.; Shaheen, S.O. Relation of birth weight and childhood respiratory infection to adult lung function and death from chronic obstructive airways disease. BMJ 1991, 303, 671–675. [Google Scholar] [CrossRef]
- Baschat, A.A. Neurodevelopment following fetal growth restriction and its relationship with antepartum parameters of placental dysfunction. Ultrasound Obstet. Gynecol. 2011, 37, 501–514. [Google Scholar] [CrossRef] [PubMed]
- Jacobsson, B.; Ahlin, K.; Francis, A.; Hagberg, G.; Hagberg, H.; Gardosi, J. Cerebral palsy and restricted growth status at birth: Population-based case-control study. BJOG 2008, 115, 1250–1255. [Google Scholar] [CrossRef] [PubMed]
- Jarvis, S.; Glinianaia, S.V.; Torrioli, M.G.; Platt, M.J.; Miceli, M.; Jouk, P.S.; Johnson, A.; Hutton, J.; Hemming, K.; Hagberg, G.; et al. Cerebral palsy and intrauterine growth in single births: European collaborative study. Lancet 2003, 362, 1106–1111. [Google Scholar] [CrossRef] [PubMed]
- McCowan, L.; Horgan, R.P. Risk factors for small for gestational age infants. Best Pract. Res. Clin. Obstet. Gynaecol. 2009, 23, 779–793. [Google Scholar] [CrossRef] [PubMed]
- Miller, S.L.; Huppi, P.S.; Mallard, C. The consequences of fetal growth restriction on brain structure and neurodevelopmental outcome. J. Physiol. 2016, 594, 807–823. [Google Scholar] [CrossRef] [PubMed]
- Pryor, J.; Silva, P.A.; Brooke, M. Growth, development and behaviour in adolescents born small-for-gestational-age. J. Paediatr. Child Health 1995, 31, 403–407. [Google Scholar] [CrossRef] [PubMed]
- Vollmer, B.; Edmonds, C.J. School Age Neurological and Cognitive Outcomes of Fetal Growth Retardation or Small for Gestational Age Birth Weight. Front. Endocrinol. 2019, 10, 186. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Fu, W.; Liu, J. Neurodevelopment in children with intrauterine growth restriction: Adverse effects and interventions. J. Matern. Fetal Neonatal Med. 2016, 29, 660–668. [Google Scholar] [CrossRef] [PubMed]
- Sharma, D.; Sharma, P.; Shastri, S. Genetic, metabolic and endocrine aspect of intrauterine growth restriction: An update. J. Matern. Fetal Neonatal Med. 2017, 30, 2263–2275. [Google Scholar] [CrossRef]
- Gazzolo, D.; Marinoni, E.; Di Iorio, R.; Lituania, M.; Marras, M.; Bruschettini, M.; Bruschettini, P.; Frulio, R.; Michetti, F.; Petraglia, F.; et al. High maternal blood S100B concentrations in pregnancies complicated by intrauterine growth restriction and intraventricular hemorrhage. Clin. Chem. 2006, 52, 819–826. [Google Scholar] [CrossRef]
- Gazzolo, D.; Marinoni, E.; di Iorio, R.; Lituania, M.; Bruschettini, P.L.; Michetti, F. Circulating S100beta protein is increased in intrauterine growth-retarded fetuses. Pediatr. Res. 2002, 51, 215–219. [Google Scholar] [CrossRef] [PubMed]
- Florio, P.; Marinoni, E.; Di Iorio, R.; Bashir, M.; Ciotti, S.; Sacchi, R.; Bruschettini, M.; Lituania, M.; Serra, G.; Michetti, F.; et al. Urinary S100B protein concentrations are increased in intrauterine growth-retarded newborns. Pediatrics 2006, 118, e747–e754. [Google Scholar] [CrossRef] [PubMed]
- Iskusnykh, I.Y.; Fattakhov, N.; Buddington, R.K.; Chizhikov, V.V. Intrauterine growth restriction compromises cerebellar development by affecting radial migration of granule cells via the JamC/Pard3a molecular pathway. Exp. Neurol. 2021, 336, 113537. [Google Scholar] [CrossRef] [PubMed]
- Janzen, C.; Lei, M.Y.; Cho, J.; Sullivan, P.; Shin, B.C.; Devaskar, S.U. Placental glucose transporter 3 (GLUT3) is up-regulated in human pregnancies complicated by late-onset intrauterine growth restriction. Placenta 2013, 34, 1072–1078. [Google Scholar] [CrossRef] [PubMed]
- Kainulainen, H.; Jarvinen, T.; Heinonen, P.K. Placental glucose transporters in fetal intrauterine growth retardation and macrosomia. Gynecol. Obstet. Investig. 1997, 44, 89–92. [Google Scholar] [CrossRef] [PubMed]
- Ma, Y.; Zhu, M.J.; Uthlaut, A.B.; Nijland, M.J.; Nathanielsz, P.W.; Hess, B.W.; Ford, S.P. Upregulation of growth signaling and nutrient transporters in cotyledons of early to mid-gestational nutrient restricted ewes. Placenta 2011, 32, 255–263. [Google Scholar] [CrossRef] [PubMed]
- Das, U.G.; He, J.; Ehrhardt, R.A.; Hay, W.W., Jr.; Devaskar, S.U. Time-dependent physiological regulation of ovine placental GLUT-3 glucose transporter protein. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2000, 279, R2252–R2261. [Google Scholar] [CrossRef] [PubMed]
- Coan, P.M.; Vaughan, O.R.; Sekita, Y.; Finn, S.L.; Burton, G.J.; Constancia, M.; Fowden, A.L. Adaptations in placental phenotype support fetal growth during undernutrition of pregnant mice. J. Physiol. 2010, 588, 527–538. [Google Scholar] [CrossRef] [PubMed]
- Ganguly, A.; Collis, L.; Devaskar, S.U. Placental glucose and amino acid transport in calorie-restricted wild-type and Glut3 null heterozygous mice. Endocrinology 2012, 153, 3995–4007. [Google Scholar] [CrossRef]
- Lesage, J.; Hahn, D.; Leonhardt, M.; Blondeau, B.; Breant, B.; Dupouy, J.P. Maternal undernutrition during late gestation-induced intrauterine growth restriction in the rat is associated with impaired placental GLUT3 expression, but does not correlate with endogenous corticosterone levels. J. Endocrinol. 2002, 174, 37–43. [Google Scholar] [CrossRef]
- Reinisch, J.M.; Simon, N.G.; Karow, W.G.; Gandelman, R. Prenatal exposure to prednisone in humans and animals retards intrauterine growth. Science 1978, 202, 436–438. [Google Scholar] [CrossRef] [PubMed]
- Motta, K.; Gomes, P.R.L.; Sulis, P.M.; Bordin, S.; Rafacho, A. Dexamethasone Administration During Late Gestation Has No Major Impact on Lipid Metabolism, but Reduces Newborn Survival Rate in Wistar Rats. Front. Physiol. 2018, 9, 783. [Google Scholar] [CrossRef] [PubMed]
- Crowther, C.A.; Ashwood, P.; Andersen, C.C.; Middleton, P.F.; Tran, T.; Doyle, L.W.; Robinson, J.S.; Harding, J.E.; Group, A.S. Maternal intramuscular dexamethasone versus betamethasone before preterm birth (ASTEROID): A multicentre, double-blind, randomised controlled trial. Lancet Child Adolesc. Health 2019, 3, 769–780. [Google Scholar] [CrossRef] [PubMed]
- Alawadhi, M.; Mouihate, A.; Kilarkaje, N.; Al-Bader, M. Progesterone partially recovers placental glucose transporters in dexamethasone-induced intrauterine growth restriction. Reprod. Biomed. Online 2022, 44, 595–607. [Google Scholar] [CrossRef] [PubMed]
- Sadiq, H.F.; Das, U.G.; Tracy, T.F.; Devaskar, S.U. Intra-uterine growth restriction differentially regulates perinatal brain and skeletal muscle glucose transporters. Brain Res. 1999, 823, 96–103. [Google Scholar] [CrossRef]
- Ye, X.; Shin, B.C.; Baldauf, C.; Ganguly, A.; Ghosh, S.; Devaskar, S.U. Developing Brain Glucose Transporters, Serotonin, Serotonin Transporter, and Oxytocin Receptor Expression in Response to Early-Life Hypocaloric and Hypercaloric Dietary, and Air Pollutant Exposures. Dev. Neurosci. 2021, 43, 27–42. [Google Scholar] [CrossRef] [PubMed]
- Baldauf, C.; Sondhi, M.; Shin, B.C.; Ko, Y.E.; Ye, X.; Lee, K.W.; Devaskar, S.U. Murine maternal dietary restriction affects neural Humanin expression and cellular profile. J. Neurosci. Res. 2020, 98, 902–920. [Google Scholar] [CrossRef]
- Garg, M.; Thamotharan, M.; Dai, Y.; Lagishetty, V.; Matveyenko, A.V.; Lee, W.N.; Devaskar, S.U. Glucose intolerance and lipid metabolic adaptations in response to intrauterine and postnatal calorie restriction in male adult rats. Endocrinology 2013, 154, 102–113. [Google Scholar] [CrossRef]
- Thamotharan, S.; Raychaudhuri, N.; Tomi, M.; Shin, B.C.; Devaskar, S.U. Hypoxic adaptation engages the CBP/CREST-induced coactivator complex of Creb-HIF-1alpha in transactivating murine neuroblastic glucose transporter. Am. J. Physiol. Endocrinol. Metab. 2013, 304, E583–E598. [Google Scholar] [CrossRef]
- Maggiotto, L.V.; Sondhi, M.; Shin, B.C.; Garg, M.; Devaskar, S.U. Circulating blood cellular glucose transporters—Surrogate biomarkers for neonatal hypoxic-ischemic encephalopathy assessed by novel scoring systems. Mol. Genet. Metab. 2019, 127, 166–173. [Google Scholar] [CrossRef]
| Brain GLUTs Expression and Neurodevelopment | Placenta GLUTs Expression | ||
|---|---|---|---|
| High-fat diet | Maternal | - Reduced GLUT3 expression (rat) [129]. - n-3 fatty acids improve vision development (human) [126,127]. - No significant change in the first 2 years of age, but significant positive effects in cognitive tests 3–6 years of age (human) [125,126,127]. | - Increased GLUT1, no change GLUT3(mouse) [120,121]. - High-fat + high-sucrose diet increased GLUT1 and GLUT3 (mouse) [122]. - GLUT3+/− mice increased GLUT1 and GLUT3 [120]. |
| Postnatal | - Exposure to postnatal n-3 fatty acid-enriched high-fat diet after maternal n-6 fatty acid-enriched high-fat diet reduced GLUT3, with no change in GLUT1 (mouse) [128]. | - | |
| Ketogenic diet | Maternal | - Brain structural change (decreased the volume of cortex, hippocampus, corpus callosum) (mouse) [146]. - Reduced susceptibility to anxiety and depression, elevated hyperactivity (mouse) [147]. | - No data regarding GLUTs. |
| Maternal and Postnatal | - GLUT3+/− mice decreased ASD-like behaviors and decreased sociability in wild-type mice [144]. | - | |
| Energy restriction | Maternal | - Maternal uterine artery ligation in late gestation decreased GLUT1 (rat) [175]. - 50% dietary restriction during mid-to-late gestation decreased fetal brain GLUT3 with reduction in brain 5-HT and SERT concentrations with anxiety and reduced cortical thickness (mouse) [176,177]. | Variable results depending on the model and severity of the IUGR state [164,165,166,167,168,169,170,174]. - Late-gestation mild-form IUGR showed increased GLUT3, with no change in. GLUT1 (human) [164]. - Moderately severe-form IUGR showed no change in GLUT3 and GLUT4 (human) [165]. - 50% nutrient restriction during the first half of gestation increased GLUT1 and GLUT3 during mid-gestation (ewe) [166]. - Chronic hypoglycemia by insulin decreased GLUT1, with no GLUT3 change (sheep) [167]. - 80% nutrient restriction throughout pregnancy increased GLUT1 (mouse) [168]. - Mid-to-late gestation 25% dietary restriction increased GLUT3, and 50% restriction decreased GLUT3 (mouse) [169,170]. - Late gestation dexamethasone-induced IUGR decreased GLUT1 and GLUT3 (mouse) [174]. |
| Postnatal | - Offspring of 50% dietary-restricted rats displayed an adaptive increase in GLUT1, GLUT3, and BDNF with heightened anxiety and cognitive impairment (rat) [9,178]. | - |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Daida, T.; Shin, B.-C.; Cepeda, C.; Devaskar, S.U. Neurodevelopment Is Dependent on Maternal Diet: Placenta and Brain Glucose Transporters GLUT1 and GLUT3. Nutrients 2024, 16, 2363. https://doi.org/10.3390/nu16142363
Daida T, Shin B-C, Cepeda C, Devaskar SU. Neurodevelopment Is Dependent on Maternal Diet: Placenta and Brain Glucose Transporters GLUT1 and GLUT3. Nutrients. 2024; 16(14):2363. https://doi.org/10.3390/nu16142363
Chicago/Turabian StyleDaida, Tomoko, Bo-Chul Shin, Carlos Cepeda, and Sherin U. Devaskar. 2024. "Neurodevelopment Is Dependent on Maternal Diet: Placenta and Brain Glucose Transporters GLUT1 and GLUT3" Nutrients 16, no. 14: 2363. https://doi.org/10.3390/nu16142363