
Role of Gut Microbial Metabolites in the Pathogenesis of Primary Liver Cancers
 , , , and
, , , and
Abstract
:1. Introduction
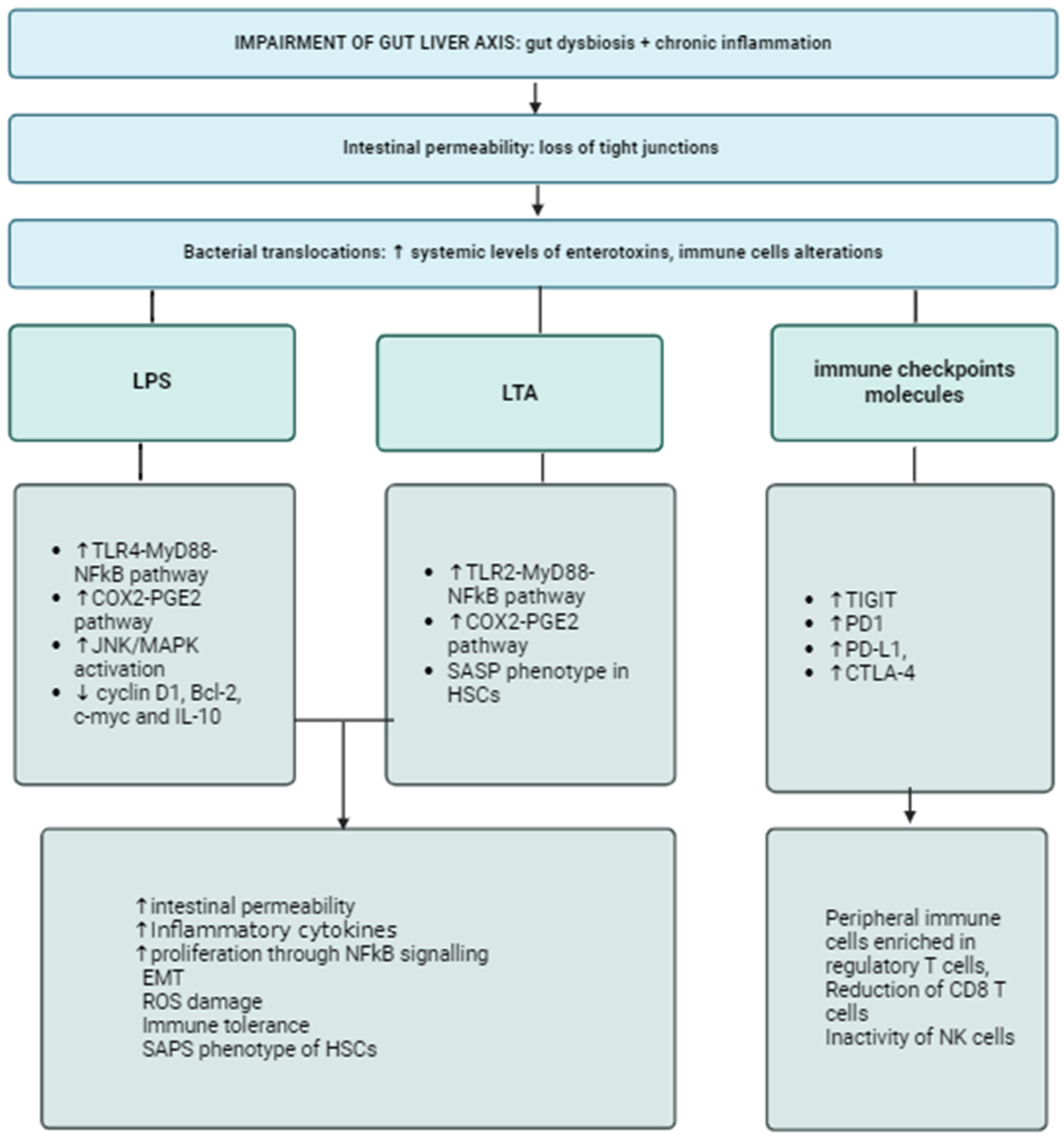
1.1. The Gut-Biliary-Liver Axis: Between Inflammation and Immunosuppression
1.2. Gut Dysbiosis and Hepatobiliary Carcinogenesis
2. Gut Metabolome
2.1. Bile Acids
2.2. Choline and TMAO
2.3. Indoles
2.4. Short-Chain Fatty Acids
2.5. Ethanol
2.6. Branched-Chain Amino Acids
3. Discussion and Future Perspectives
4. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
References
- Sung, H.; Ferlay, J.; Siegel, R.L.; Laversanne, M.; Soerjomataram, I.; Jemal, A.; Bray, F. Global Cancer Statistics 2020: GLOBOCAN Estimates of Incidence and Mortality Worldwide for 36 Cancers in 185 Countries. CA Cancer J. Clin. 2021, 71, 209–249. [Google Scholar] [CrossRef]
- Rich, N.E. Changing Epidemiology of Hepatocellular Carcinoma Within the United States and Worldwide. Surg. Oncol. Clin. N. Am. 2024, 33, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Pascale, A.; Rosmorduc, O.; Duclos-Vallée, J.-C. New epidemiologic trends in cholangiocarcinoma. Clin. Res. Hepatol. Gastroenterol. 2023, 47, 102223. [Google Scholar] [CrossRef] [PubMed]
- Jandhyala, S.M.; Talukdar, R.; Subramanyam, C.; Vuyyuru, H.; Sasikala, M.; Nageshwar Reddy, D. Role of the normal gut microbiota. World J. Gastroenterol. 2015, 21, 8787–8803. [Google Scholar] [CrossRef] [PubMed]
- Yu, Z.; Morrison, M. Improved extraction of PCR-quality community DNA from digesta and fecal samples. Biotechniques 2004, 36, 808–812. [Google Scholar] [CrossRef] [PubMed]
- Luo, W.; Guo, S.; Zhou, Y.; Zhao, J.; Wang, M.; Sang, L.; Chang, B.; Wang, B. Hepatocellular Carcinoma: How the Gut Microbiota Contributes to Pathogenesis, Diagnosis, and Therapy. Front. Microbiol. 2022, 13, 873160. [Google Scholar] [CrossRef] [PubMed]
- Arumugam, M.; Raes, J.; Pelletier, E.; Le Paslier, D.; Yamada, T.; Mende, D.R.; Fernandes, G.R.; Tap, J.; Bruls, T.; Batto, J.M.; et al. Enterotypes of the human gut microbiome. Nature 2011, 473, 174–180. [Google Scholar] [CrossRef] [PubMed]
- Nardone, G.; Compare, D. The human gastric microbiota: Is it time to rethink the pathogenesis of stomach diseases? United Eur. Gastroenterol. J. 2015, 3, 255–260. [Google Scholar] [CrossRef]
- Osman, M.-A.; Neoh, H.-M.; Ab Mutalib, N.-S.; Chin, S.-F.; Jamal, R. 16S rRNA Gene Sequencing for Deciphering the Colorectal Cancer Gut Microbiome: Current Protocols and Workflows. Front. Microbiol. 2018, 9, 767. [Google Scholar] [CrossRef]
- Wei, L.Q.; Cheong, I.H.; Yang, G.H.; Li, X.G.; Kozlakidis, Z.; Ding, L.; Liu, N.N.; Wang, H. The Application of High-Throughput Technologies for the Study of Microbiome and Cancer. Front. Genet. 2021, 12, 699793. [Google Scholar] [CrossRef]
- Zhou, C.; Bisseling, T.M.; van der Post, R.S.; Boleij, A. The influence of Helicobacter pylori, proton pump inhibitor, and obesity on the gastric microbiome in relation to gastric cancer development. Comput. Struct. Biotechnol. J. 2023, 23, 186–198. [Google Scholar] [CrossRef] [PubMed]
- Meng, Y.; Duan, Q.; Jiao, K.; Xue, J. A screened predictive model for esophageal squamous cell carcinoma based on salivary flora data. Math. Biosci. Eng. MBE 2023, 20, 18368–18385. [Google Scholar] [CrossRef] [PubMed]
- Pleguezuelos-Manzano, C.; Puschhof, J.; Rosendahl Huber, A.; Van Hoeck, A.; Wood, H.M.; Nomburg, J.; Gurjao, C.; Manders, F.; Dalmasso, G.; Stege, P.B.; et al. Mutational signature in colorectal cancer caused by genotoxic pks+ E. coli. Nature 2020, 580, 269–273. [Google Scholar] [CrossRef] [PubMed]
- de Almeida, C.V.; Taddei, A.; Amedei, A. The controversial role of Enterococcus faecalis in colorectal cancer. Ther. Adv. Gastroenterol. 2018, 11, 1756284818783606. [Google Scholar] [CrossRef] [PubMed]
- Wong, C.C.; Yu, J. Gut microbiota in colorectal cancer development and therapy. Nat. Rev. Clin. Oncol. 2023, 20, 429–452. [Google Scholar] [CrossRef] [PubMed]
- Ayuningtyas, N.F.; Mahdani, F.Y.; Pasaribu, T.A.S.; Chalim, M.; Ayna, V.K.P.; Santosh, A.B.R.; Santacroce, L.; Surboyo, M.D.C. Role of Candida albicans in Oral Carcinogenesis. Pathophysiol. Off. J. Int. Soc. Pathophysiol. 2022, 29, 650–662. [Google Scholar] [CrossRef]
- Liu, J.; Tan, Y.; Cheng, H.; Zhang, D.; Feng, W.; Peng, C. Functions of Gut Microbiota Metabolites, Current Status and Future Perspectives. Aging Dis. 2022, 13, 1106–1126. [Google Scholar] [CrossRef] [PubMed]
- Albillos, A.; de Gottardi, A.; Rescigno, M. The gut-liver axis in liver disease: Pathophysiological basis for therapy. J. Hepatol. 2020, 72, 558–577. [Google Scholar] [CrossRef]
- Juneja, P.; Tripathi, D.M.; Kaur, S. Revisiting the gut-liver axis: Gut lymphatic system in liver cirrhosis and portal hypertension. Am. J. Physiol. Liver Physiol. 2022, 322, G473–G479. [Google Scholar] [CrossRef]
- Brescia, P.; Rescigno, M. The gut vascular barrier: A new player in the gut–liver–brain axis. Trends Mol. Med. 2021, 27, 844–855. [Google Scholar] [CrossRef]
- Milosevic, I.; Vujovic, A.; Barac, A.; Djelic, M.; Korac, M.; Radovanovic Spurnic, A.; Gmizic, I.; Stevanovic, O.; Djordjevic, V.; Lekic, N.; et al. Gut-Liver Axis, Gut Microbiota, and Its Modulation in the Management of Liver Diseases: A Review of the Literature. Int. J. Mol. Sci. 2019, 20, 395. [Google Scholar] [CrossRef] [PubMed]
- Song, Q.; Zhang, X. The Role of Gut–Liver Axis in Gut Microbiome Dysbiosis Associated NAFLD and NAFLD-HCC. Biomedicines 2022, 10, 524. [Google Scholar] [CrossRef] [PubMed]
- Ram, A.K.; Pottakat, B.; Vairappan, B. Increased systemic zonula occludens 1 associated with inflammation and independent biomarker in patients with hepatocellular carcinoma. BMC Cancer 2018, 18, 572. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Trivedi, Y.; Bolgarina, Z.; Desai, H.N.; Senaratne, M.; Swami, S.S.; Aye, S.L.; Mohammed, L. The Role of Gut Microbiome in Hepatocellular Carcinoma: A Systematic Review. Cureus 2023, 15, e43862. [Google Scholar] [CrossRef] [PubMed]
- Schwabe, R.F.; Greten, T.F. Gut microbiome in HCC—Mechanisms, diagnosis and therapy. J. Hepatol. 2020, 72, 230–238. [Google Scholar] [CrossRef] [PubMed]
- De Muynck, K.; Vanderborght, B.; Van Vlierberghe, H.; Devisscher, L. The Gut–Liver Axis in Chronic Liver Disease: A Macrophage Perspective. Cells 2021, 10, 2959. [Google Scholar] [CrossRef]
- Guo, J.; Friedman, S.L. Toll-like receptor 4 signaling in liver injury and hepatic fibrogenesis. Fibrogenesis Tissue Repair 2010, 3, 21. [Google Scholar] [CrossRef] [PubMed]
- Manilla, V.; Di Tommaso, N.; Santopaolo, F.; Gasbarrini, A.; Ponziani, F.R. Endotoxemia and Gastrointestinal Cancers: Insight into the Mechanisms Underlying a Dangerous Relationship. Microorganisms 2023, 11, 267. [Google Scholar] [CrossRef] [PubMed]
- Khan, H.U.; Aamir, K.; Jusuf, P.R.; Sethi, G.; Sisinthy, S.P.; Ghildyal, R.; Arya, A. Lauric acid ameliorates lipopolysaccharide (LPS)-induced liver inflammation by mediating TLR4/MyD88 pathway in Sprague Dawley (SD) rats. Life Sci. 2021, 265, 118750. [Google Scholar] [CrossRef]
- Getachew, A.; Hussain, M.; Huang, X.; Li, Y. Toll-like receptor 2 signaling in liver pathophysiology. Life Sci. 2021, 284, 119941. [Google Scholar] [CrossRef]
- Ramírez-Pérez, O.; Cruz-Ramón, V.; Chinchilla-López, P.; Méndez-Sánchez, N. The Role of the Gut Microbiota in Bile Acid Metabolism. Ann. Hepatol. 2017, 16 Suppl. 1: s3-105, s15–s20. [Google Scholar] [CrossRef] [PubMed]
- Yoshimoto, S.; Loo, T.M.; Atarashi, K.; Kanda, H.; Sato, S.; Oyadomari, S.; Iwakura, Y.; Oshima, K.; Morita, H.; Hattori, M.; et al. Obesity-induced gut microbial metabolite promotes liver cancer through senescence secretome. Nature 2013, 499, 97–101. [Google Scholar] [CrossRef]
- Larabi, A.B.; Masson, H.L.P.; Bäumler, A.J. Bile acids as modulators of gut microbiota composition and function. Gut Microbes 2023, 15, 2172671. [Google Scholar] [CrossRef]
- Litmanovich, A.; Khazim, K.; Cohen, I. The Role of Interleukin-1 in the Pathogenesis of Cancer and its Potential as a Therapeutic Target in Clinical Practice. Oncol. Ther. 2018, 6, 109–127. [Google Scholar] [CrossRef]
- Garlanda, C.; Mantovani, A. Interleukin-1 in tumor progression, therapy, and prevention. Cancer Cell 2021, 39, 1023–1027. [Google Scholar] [CrossRef]
- Loo, T.M.; Kamachi, F.; Watanabe, Y.; Yoshimoto, S.; Kanda, H.; Arai, Y.; Nakajima-Takagi, Y.; Iwama, A.; Koga, T.; Sugimoto, Y.; et al. Gut Microbiota Promotes Obesity-Associated Liver Cancer through PGE2-Mediated Suppression of Antitumor Immunity. Cancer Discov. 2017, 7, 522–538. [Google Scholar] [CrossRef]
- Dapito, D.H.; Mencin, A.; Gwak, G.-Y.; Pradere, J.-P.; Jang, M.-K.; Mederacke, I.; Caviglia, J.M.; Khiabanian, H.; Adeyemi, A.; Bataller, R.; et al. Promotion of Hepatocellular Carcinoma by the Intestinal Microbiota and TLR4. Cancer Cell 2012, 21, 504–516. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Li, Y.; Liu, D.; Liu, J. LPS promotes epithelial–mesenchymal transition and activation of TLR4/JNK signaling. Tumour Biol. J. Int. Soc. Oncodevelopmental Biol. Med. 2014, 35, 10429–10435. [Google Scholar] [CrossRef]
- Rooks, M.G.; Garrett, W.S. Gut microbiota, metabolites and host immunity. Nat. Rev. Immunol. 2016, 16, 341–352. [Google Scholar] [CrossRef]
- Kiriyama, Y.; Nochi, H. The Role of Gut Microbiota-Derived Lithocholic Acid, Deoxycholic Acid and Their Derivatives on the Function and Differentiation of Immune Cells. Microorganisms 2023, 11, 2730. [Google Scholar] [CrossRef]
- Schroeder, B.O.; Bäckhed, F. Signals from the gut microbiota to distant organs in physiology and disease. Nat. Med. 2016, 22, 1079–1089. [Google Scholar] [CrossRef]
- Gur, C.; Ibrahim, Y.; Isaacson, B.; Yamin, R.; Abed, J.; Gamliel, M.; Enk, J.; Bar-On, Y.; Stanietsky-Kaynan, N.; Coppenhagen-Glazer, S.; et al. Binding of the Fap2 Protein of Fusobacterium nucleatum to Human Inhibitory Receptor TIGIT Protects Tumors from Immune Cell Attack. Immunity 2015, 42, 344–355. [Google Scholar] [CrossRef]
- Geissmann, F.; Cameron, T.O.; Sidobre, S.; Manlongat, N.; Kronenberg, M.; Briskin, M.J.; Dustin, M.L.; Littman, D.R. Intravascular Immune Surveillance by CXCR6+ NKT Cells Patrolling Liver Sinusoids. PLoS Biol. 2005, 3, e113. [Google Scholar] [CrossRef]
- Albillos, A.; Lario, M.; Álvarez-Mon, M. Cirrhosis-associated immune dysfunction: Distinctive features and clinical relevance. J. Hepatol. 2014, 61, 1385–1396. [Google Scholar] [CrossRef]
- Zheng, Y.; Wang, T.; Tu, X.; Huang, Y.; Zhang, H.; Tan, D.; Jiang, W.; Cai, S.; Zhao, P.; Song, R.; et al. Gut microbiome affects the response to anti-PD-1 immunotherapy in patients with hepatocellular carcinoma. J. Immunother. Cancer 2019, 7, 193. [Google Scholar] [CrossRef]
- Behary, J.; Amorim, N.; Jiang, X.-T.; Raposo, A.; Gong, L.; McGovern, E.; Ibrahim, R.; Chu, F.; Stephens, C.; Jebeili, H.; et al. Gut microbiota impact on the peripheral immune response in non-alcoholic fatty liver disease related hepatocellular carcinoma. Nat. Commun. 2021, 12, 187. [Google Scholar] [CrossRef] [PubMed]
- Pfister, D.; Núñez, N.G.; Pinyol, R.; Govaere, O.; Pinter, M.; Szydlowska, M.; Gupta, R.; Qiu, M.; Deczkowska, A.; Weiner, A.; et al. NASH limits anti-tumour surveillance in immunotherapy-treated HCC. Nature 2021, 592, 450–456. [Google Scholar] [CrossRef] [PubMed]
- Nomura, M.; Nagatomo, R.; Doi, K.; Shimizu, J.; Baba, K.; Saito, T.; Matsumoto, S.; Inoue, K.; Muto, M. Association of Short-Chain Fatty Acids in the Gut Microbiome With Clinical Response to Treatment With Nivolumab or Pembrolizumab in Patients With Solid Cancer Tumors. JAMA Netw. Open 2020, 3, e202895. [Google Scholar] [CrossRef]
- Li, M.; van Esch, B.C.A.M.; Henricks, P.A.J.; Folkerts, G.; Garssen, J. The Anti-inflammatory Effects of Short Chain Fatty Acids on Lipopolysaccharide- or Tumor Necrosis Factor α-Stimulated Endothelial Cells via Activation of GPR41/43 and Inhibition of HDACs. Front. Pharmacol. 2018, 9, 533. [Google Scholar] [CrossRef]
- Woods, D.M.; Sodré, A.L.; Villagra, A.; Sarnaik, A.A.; Sotomayor, E.M.; Weber, J. HDAC Inhibition Upregulates PD-1 Ligands in Melanoma and Augments Immunotherapy with PD-1 Blockade. Cancer Immunol. Res. 2015, 3, 1375–1385. [Google Scholar] [CrossRef]
- Coutzac, C.; Jouniaux, J.-M.; Paci, A.; Schmidt, J.; Mallardo, D.; Seck, A.; Asvatourian, V.; Cassard, L.; Saulnier, P.; Lacroix, L.; et al. Systemic short chain fatty acids limit antitumor effect of CTLA-4 blockade in hosts with cancer. Nat. Commun. 2020, 11, 2168. [Google Scholar] [CrossRef] [PubMed]
- Ma, C.; Han, M.; Heinrich, B.; Fu, Q.; Zhang, Q.; Sandhu, M.; Agdashian, D.; Terabe, M.; Berzofsky, J.A.; Fako, V.; et al. Gut microbiome–mediated bile acid metabolism regulates liver cancer via NKT cells. Science 2018, 360, eaan5931. [Google Scholar] [CrossRef] [PubMed]
- Elvevi, A.; Laffusa, A.; Gallo, C.; Invernizzi, P.; Massironi, S. Any Role for Microbiota in Cholangiocarcinoma? A Comprehensive Review. Cells 2023, 12, 370. [Google Scholar] [CrossRef] [PubMed]
- Wheatley, R.C.; Kilgour, E.; Jacobs, T.; Lamarca, A.; Hubner, R.A.; Valle, J.W.; McNamara, M.G. Potential influence of the microbiome environment in patients with biliary tract cancer and implications for therapy. Br. J. Cancer 2022, 126, 693–705. [Google Scholar] [CrossRef] [PubMed]
- Chagani, S.; Kwong, L.N. Cholangiocarcinoma Risk Factors Open the Floodgates for Gut Microbes and Immunosuppressive Myeloid Cells. Cancer Discov. 2021, 11, 1014–1015. [Google Scholar] [CrossRef] [PubMed]
- Jusakul, A.; Cutcutache, I.; Yong, C.H.; Lim, J.Q.; Huang, M.N.; Padmanabhan, N.; Nellore, V.; Kongpetch, S.; Ng, A.W.T.; Ng, L.M.; et al. Whole-Genome and Epigenomic Landscapes of Etiologically Distinct Subtypes of Cholangiocarcinoma. Cancer Discov. 2017, 7, 1116–1135. [Google Scholar] [CrossRef] [PubMed]
- Chaisaingmongkol, J.; Budhu, A.; Dang, H.; Rabibhadana, S.; Pupacdi, B.; Kwon, S.M.; Forgues, M.; Pomyen, Y.; Bhudhisawasdi, V.; Lertprasertsuke, N.; et al. Common Molecular Subtypes Among Asian Hepatocellular Carcinoma and Cholangiocarcinoma. Cancer Cell 2017, 32, 57–70.e3. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.; Yang, L.; Chu, H. Targeting Gut Microbiota for the Treatment of Primary Biliary Cholangitis: From Bench to Bedside. J. Clin. Transl. Hepatol. 2023, 11, 958–966. [Google Scholar] [CrossRef]
- Bogdanos, D.-P.; Baum, H.; Grasso, A.; Okamoto, M.; Butler, P.; Ma, Y.; Rigopoulou, E.; Montalto, P.; Davies, E.T.; Burroughs, A.K.; et al. Microbial mimics are major targets of crossreactivity with human pyruvate dehydrogenase in primary biliary cirrhosis. J. Hepatol. 2004, 40, 31–39. [Google Scholar] [CrossRef]
- Bogdanos, D.; Choudhuri, K.; Vergani, D. Molecular mimicry and autoimmune liver disease: Virtuous intentions, malign consequences. Liver Int. 2001, 21, 225–232. [Google Scholar] [CrossRef]
- Huang, L.; Yu, Q.; Peng, H.; Zhen, Z. Alterations of gut microbiome and effects of probiotic therapy in patients with liver cirrhosis: A systematic review and meta-analysis. Medicine 2022, 101, e32335. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Ren, Z.; Li, A.; Jiang, J.; Zhou, L.; Yu, Z.; Lu, H.; Xie, H.; Chen, X.; Shao, L.; Zhang, R.; et al. Gut microbiome analysis as a tool towards targeted non-invasive biomarkers for early hepatocellular carcinoma. Gut 2019, 68, 1014–1023. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Ponziani, F.R.; Bhoori, S.; Castelli, C.; Putignani, L.; Rivoltini, L.; Del Chierico, F.; Sanguinetti, M.; Morelli, D.; Sterbini, F.P.; Petito, V.; et al. Hepatocellular Carcinoma Is Associated With Gut Microbiota Profile and Inflammation in Nonalcoholic Fatty Liver Disease. Hepatology 2019, 69, 107–120. [Google Scholar] [CrossRef]
- Bendixen, S.M.; Jakobsgaard, P.R.; Hansen, D.; Hejn, K.H.; Terkelsen, M.K.; Bjerre, F.A.; Thulesen, A.P.; Eriksen, N.G.; Hallenborg, P.; Geng, Y.; et al. Single cell-resolved study of advanced murine MASH reveals a homeostatic pericyte signaling module. J. Hepatol. 2024, 80, 467–481. [Google Scholar] [CrossRef]
- Wiest, R.; Garcia-Tsao, G. Bacterial translocation (BT) in cirrhosis. Hepatology 2005, 41, 422–433. [Google Scholar] [CrossRef]
- Grąt, M.; Wronka, K.; Krasnodębski, M.; Masior, Ł.; Lewandowski, Z.; Kosińska, I.; Grąt, K.; Stypułkowski, J.; Rejowski, S.; Wasilewicz, M.; et al. Profile of Gut Microbiota Associated With the Presence of Hepatocellular Cancer in Patients With Liver Cirrhosis. Transplant. Proc. 2016, 48, 1687–1691. [Google Scholar] [CrossRef]
- Liu, Q.; Li, F.; Zhuang, Y.; Xu, J.; Wang, J.; Mao, X.; Zhang, Y.; Liu, X. Alteration in gut microbiota associated with hepatitis B and non-hepatitis virus related hepatocellular carcinoma. Gut Pathog. 2019, 11, 1. [Google Scholar] [CrossRef]
- Zhang, X.; Coker, O.O.; Chu, E.S.; Fu, K.; Lau, H.C.H.; Wang, Y.-X.; Chan, A.W.H.; Wei, H.; Yang, X.; Sung, J.J.Y.; et al. Dietary cholesterol drives fatty liver-associated liver cancer by modulating gut microbiota and metabolites. Gut 2020, 70, 761–774. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Xie, G.; Wang, X.; Liu, P.; Wei, R.; Chen, W.; Rajani, C.; Hernandez, B.Y.; Alegado, R.; Dong, B.; Li, D.; et al. Distinctly altered gut microbiota in the progression of liver disease. Oncotarget 2016, 7, 19355–19366. [Google Scholar] [CrossRef]
- Long, C.; Zhou, X.; Xia, F.; Zhou, B. Intestinal Barrier Dysfunction and Gut Microbiota in Non-Alcoholic Fatty Liver Disease: Assessment, Mechanisms, and Therapeutic Considerations. Biology 2024, 13, 243. [Google Scholar] [CrossRef]
- Nejman, D.; Livyatan, I.; Fuks, G.; Gavert, N.; Zwang, Y.; Geller, L.T.; Rotter-Maskowitz, A.; Weiser, R.; Mallel, G.; Gigi, E.; et al. The human tumor microbiome is composed of tumor type–specific intracellular bacteria. Science 2020, 368, 973–980. [Google Scholar] [CrossRef]
- Poore, G.D.; Kopylova, E.; Zhu, Q.; Carpenter, C.; Fraraccio, S.; Wandro, S.; Kosciolek, T.; Janssen, S.; Metcalf, J.; Song, S.J.; et al. Microbiome analyses of blood and tissues suggest cancer diagnostic approach. Nature 2020, 579, 567–574. [Google Scholar] [CrossRef]
- Li, S.; Xia, H.; Wang, Z.; Zhang, X.; Song, T.; Li, J.; Xu, L.; Zhang, N.; Fan, S.; Li, Q.; et al. Intratumoral microbial heterogeneity affected tumor immune microenvironment and determined clinical outcome of HBV-related HCC. Hepatology 2023, 78, 1079–1091. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Effenberger, M.; Waschina, S.; Bronowski, C.; Sturm, G.; Tassiello, O.; Sommer, F.; Zollner, A.; Watschinger, C.; Grabherr, F.; Gstir, R.; et al. A gut bacterial signature in blood and liver tissue characterizes cirrhosis and hepatocellular carcinoma. Hepatol. Commun. 2023, 7, e00182. [Google Scholar] [CrossRef]
- Purohit, H.J. Gut-Bioreactor and Human Health in Future. Indian J. Microbiol. 2018, 58, 3–7. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Blanchet, M.; Brunel, J.M. Bile Acid Derivatives: From Old Molecules to a New Potent Therapeutic Use: An Overview. Curr. Med. Chem. 2018, 25, 3613–3636. [Google Scholar] [CrossRef]
- Winston, J.A.; Theriot, C.M. Diversification of host bile acids by members of the gut microbiota. Gut Microbes 2020, 11, 158–171. [Google Scholar] [CrossRef]
- Jia, W.; Xie, G.; Jia, W. Bile acid–microbiota crosstalk in gastrointestinal inflammation and carcinogenesis. Nat. Rev. Gastroenterol. Hepatol. 2018, 15, 111–128. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Thomas, C.; Pellicciari, R.; Pruzanski, M.; Auwerx, J.; Schoonjans, K. Targeting bile-acid signalling for metabolic diseases. Nat. Rev. Drug Discov. 2008, 7, 678–693. [Google Scholar] [CrossRef]
- Wei, S.; Ma, X.; Zhao, Y. Mechanism of Hydrophobic Bile Acid-Induced Hepatocyte Injury and Drug Discovery. Front. Pharmacol. 2020, 11, 1084. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Meng, X.; Chang, Z.; Che, N.; Wu, J.; Dang, T.; Chai, J. Acid/bile exposure triggers TRAIL-mediated apoptosis in esophageal cancer cells by suppressing the decoy receptors and c-FLIPR. Int. J. Biochem. Cell Biol. 2020, 122, 105736. [Google Scholar] [CrossRef]
- Higuchi, H.; Bronk, S.F.; Taniai, M.; Canbay, A.; Gores, G.J. Cholestasis Increases Tumor Necrosis Factor-Related Apoptotis-Inducing Ligand (TRAIL)-R2/DR5 Expression and Sensitizes the Liver to TRAIL-Mediated Cytotoxicity. J. Pharmacol. Exp. Ther. 2002, 303, 461–467. [Google Scholar] [CrossRef]
- Maresca, M.; Yahi, N.; Younès-Sakr, L.; Boyron, M.; Caporiccio, B.; Fantini, J. Both direct and indirect effects account for the pro-inflammatory activity of enteropathogenic mycotoxins on the human intestinal epithelium: Stimulation of interleukin-8 secretion, potentiation of interleukin-1β effect and increase in the transepithelial passage of commensal bacteria. Toxicol. Appl. Pharmacol. 2008, 228, 84–92. [Google Scholar] [CrossRef]
- Yan, S.-F.; Fujita, T.; Lu, J.; Okada, K.; Zou, Y.S.; Mackman, N.; Pinsky, D.J.; Stern, D.M. Egr-1, a master switch coordinating upregulation of divergent gene families underlying ischemic stress. Nat. Med. 2000, 6, 1355–1361. [Google Scholar] [CrossRef]
- Anwer, M.S. Intracellular Signaling by Bile Acids. J. Bio-Sci. 2012, 20, 1–23. [Google Scholar] [CrossRef]
- Huang, X.-F.; Zhao, W.-Y.; Huang, W.-D. FXR and liver carcinogenesis. Acta Pharmacol. Sin. 2015, 36, 37–43. [Google Scholar] [CrossRef]
- Fiorucci, S.; Baldelli, F. Farnesoid X receptor agonists in biliary tract disease. Curr. Opin. Gastroenterol. 2009, 25, 252–259. [Google Scholar] [CrossRef]
- Zhao, Q.; Liu, F.; Cheng, Y.; Xiao, X.-R.; Hu, D.-D.; Tang, Y.-M.; Bao, W.-M.; Yang, J.-H.; Jiang, T.; Hu, J.-P.; et al. Celastrol Protects from Cholestatic Liver Injury Through Modulation of SIRT1-FXR Signaling. Mol. Cell. Proteom. MCP 2019, 18, 520–533. [Google Scholar] [CrossRef]
- Wolfe, A.; Thomas, A.; Edwards, G.; Jaseja, R.; Guo, G.L.; Apte, U. Increased activation of the Wnt/β-catenin pathway in spontaneous hepatocellular carcinoma observed in farnesoid X receptor knockout mice. J. Pharmacol. Exp. Ther. 2011, 338, 12–21. [Google Scholar] [CrossRef]
- Said, I.; Ahad, H.; Said, A. Gut microbiome in non-alcoholic fatty liver disease associated hepatocellular carcinoma: Current knowledge and potential for therapeutics. World J. Gastrointest. Oncol. 2022, 14, 947–958. [Google Scholar] [CrossRef]
- Fu, T.; Coulter, S.; Yoshihara, E.; Oh, T.G.; Fang, S.; Cayabyab, F.; Zhu, Q.; Zhang, T.; Leblanc, M.; Liu, S.; et al. FXR Regulates Intestinal Cancer Stem Cell Proliferation. Cell 2019, 176, 1098–1112.e18. [Google Scholar] [CrossRef]
- Sun, L.; Cai, J.; Gonzalez, F.J. The role of farnesoid X receptor in metabolic diseases, and gastrointestinal and liver cancer. Nat. Rev. Gastroenterol. Hepatol. 2021, 18, 335–347. [Google Scholar] [CrossRef] [PubMed]
- Yang, M.; Khoukaz, L.; Qi, X.; Kimchi, E.T.; Staveley-O’carroll, K.F.; Li, G. Diet and Gut Microbiota Interaction-Derived Metabolites and Intrahepatic Immune Response in NAFLD Development and Treatment. Biomedicines 2021, 9, 1893. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Herraez, E.; Romero, M.R.; Macias, R.I.R.; Monte, M.J.; Marin, J.J.G. Clinical relevance of the relationship between changes in gut microbiota and bile acid metabolism in patients with intrahepatic cholangiocarcinoma. Hepatobiliary Surg. Nutr. 2020, 9, 211–214. [Google Scholar] [CrossRef]
- Zhen, J.; Zhou, Z.; He, M.; Han, H.-X.; Lv, E.-H.; Wen, P.-B.; Liu, X.; Wang, Y.-T.; Cai, X.-C.; Tian, J.-Q.; et al. The gut microbial metabolite trimethylamine N-oxide and cardiovascular diseases. Front. Endocrinol. 2023, 14, 1085041. [Google Scholar] [CrossRef] [PubMed]
- Canyelles, M.; Borràs, C.; Rotllan, N.; Tondo, M.; Escolà-Gil, J.C.; Blanco-Vaca, F. Gut Microbiota-Derived TMAO: A Causal Factor Promoting Atherosclerotic Cardiovascular Disease? Int. J. Mol. Sci. 2023, 24, 1940. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Zheng, Y.; He, J.-Q. Pathogenic Mechanisms of Trimethylamine N-Oxide-induced Atherosclerosis and Cardiomyopathy. Curr. Vasc. Pharmacol. 2022, 20, 29–36. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Seldin, M.M.; Meng, Y.; Qi, H.; Zhu, W.; Wang, Z.; Hazen, S.L.; Lusis, A.J.; Shih, D.M. Trimethylamine N-Oxide Promotes Vascular Inflammation Through Signaling of Mitogen-Activated Protein Kinase and Nuclear Factor-κB. J. Am. Heart Assoc. 2016, 5, e002767. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Matthews, D.R.; Li, H.; Zhou, J.; Li, Q.; Glaser, S.; Francis, H.; Alpini, G.; Wu, C. Methionine- and Choline-Deficient Diet–Induced Nonalcoholic Steatohepatitis Is Associated with Increased Intestinal Inflammation. Am. J. Pathol. 2021, 191, 1743–1753. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Shanmugham, M.; Bellanger, S.; Leo, C.H. Gut-Derived Metabolite, Trimethylamine-N-oxide (TMAO) in Cardio-Metabolic Diseases: Detection, Mechanism, and Potential Therapeutics. Pharmaceuticals 2023, 16, 504. [Google Scholar] [CrossRef]
- Seyyedsalehi, M.S.; Rossi, M.; Hadji, M.; Rashidian, H.; Marzban, M.; Parpinel, M.; Fiori, F.; Naghibzadeh-Tahami, A.; Hannun, Y.A.; Luberto, C.; et al. Dietary Choline and Betaine Intake and Risk of Colorectal Cancer in an Iranian Population. Cancers 2023, 15, 2557. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Richman, E.L.; Kenfield, S.A.; Stampfer, M.J.; Giovannucci, E.L.; Zeisel, S.H.; Willett, W.C.; Chan, J.M. Choline intake and risk of lethal prostate cancer: Incidence and survival. Am. J. Clin. Nutr. 2012, 96, 855–863. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Sun, S.; Li, X.; Ren, A.; Du, M.; Du, H.; Shu, Y.; Zhu, L.; Wang, W. Choline and betaine consumption lowers cancer risk: A meta-analysis of epidemiologic studies. Sci. Rep. 2016, 6, 35547. [Google Scholar] [CrossRef]
- Chai, C.; Chen, L.; Deng, M.-G.; Liang, Y.; Liu, F.; Nie, J.-Q. Dietary choline intake and non-alcoholic fatty liver disease (NAFLD) in U.S. adults: National Health and Nutrition Examination Survey (NHANES) 2017–2018. Eur. J. Clin. Nutr. 2023, 77, 1160–1166. [Google Scholar] [CrossRef]
- Caballero, F.; Fernández, A.; Matías, N.; Martínez, L.; Fucho, R.; Elena, M.; Caballeria, J.; Morales, A.; Fernández-Checa, J.C.; García-Ruiz, C. Specific contribution of methionine and choline in nutritional nonalcoholic steatohepatitis: Impact on mitochondrial S-adenosyl-L-methionine and glutathione. J. Biol. Chem. 2010, 285, 18528–18536. [Google Scholar] [CrossRef] [PubMed]
- Ikawa-Yoshida, A.; Matsuo, S.; Kato, A.; Ohmori, Y.; Higashida, A.; Kaneko, E.; Matsumoto, M. Hepatocellular carcinoma in a mouse model fed a choline-deficient, L-amino acid-defined, high-fat diet. Int. J. Exp. Pathol. 2017, 98, 221–233. [Google Scholar] [CrossRef]
- Liu, Z.-Y.; Tan, X.-Y.; Li, Q.-J.; Liao, G.-C.; Fang, A.-P.; Zhang, D.-M.; Chen, P.-Y.; Wang, X.-Y.; Luo, Y.; Long, J.-A.; et al. Trimethylamine N-oxide, a gut microbiota-dependent metabolite of choline, is positively associated with the risk of primary liver cancer: A case-control study. Nutr. Metab. 2018, 15, 81. [Google Scholar] [CrossRef] [PubMed]
- Wu, Y.; Rong, X.; Pan, M.; Wang, T.; Yang, H.; Chen, X.; Xiao, Z.; Zhao, C. Integrated Analysis Reveals the Gut Microbial Metabolite TMAO Promotes Inflammatory Hepatocellular Carcinoma by Upregulating POSTN. Front. Cell Dev. Biol. 2022, 10, 840171. [Google Scholar] [CrossRef]
- González-González, L.; Alonso, J. Periostin: A Matricellular Protein With Multiple Functions in Cancer Development and Progression. Front. Oncol. 2018, 8, 225. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Chen, L.; Tian, X.; Gong, W.; Sun, B.; Li, G.; Liu, D.; Guo, P.; He, Y.; Chen, Z.; Xia, Y.; et al. Periostin mediates epithelial-mesenchymal transition through the MAPK/ERK pathway in hepatoblastoma. Cancer Biol. Med. 2019, 16, 89–100. [Google Scholar] [CrossRef]
- Cho, C.E.; Taesuwan, S.; Malysheva, O.V.; Bender, E.; Tulchinsky, N.F.; Yan, J.; Sutter, J.L.; Caudill, M.A. Trimethylamine-N-oxide (TMAO) response to animal source foods varies among healthy young men and is influenced by their gut microbiota composition: A randomized controlled trial. Mol. Nutr. Food Res. 2017, 61, 1600324. [Google Scholar] [CrossRef]
- Tan, X.; Liu, Y.; Long, J.; Chen, S.; Liao, G.; Wu, S.; Li, C.; Wang, L.; Ling, W.; Zhu, H. Trimethylamine N-Oxide Aggravates Liver Steatosis through Modulation of Bile Acid Metabolism and Inhibition of Farnesoid X Receptor Signaling in Nonalcoholic Fatty Liver Disease. Mol. Nutr. Food Res. 2019, 63, e1900257. [Google Scholar] [CrossRef]
- Lombardi, B.; Shinozuka, H. Enhancement of 2-acetylaminofluorene liver carcinogenesis in rats fed a choline-devoid diet. Int. J. Cancer 1979, 23, 565–570. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Cao, L.; Chen, R.; Zhou, X.; Fan, X.; Liang, Y.; Jia, R.; Wang, H.; Liu, G.; Guo, Y.; et al. Osteopontin Promotes Hepatic Progenitor Cell Expansion and Tumorigenicity via Activation of β-Catenin in Mice. Stem Cells 2015, 33, 3569–3580. [Google Scholar] [CrossRef]
- Li, X.; Zhang, B.; Hu, Y.; Zhao, Y. New Insights Into Gut-Bacteria-Derived Indole and Its Derivatives in Intestinal and Liver Diseases. Front. Pharmacol. 2021, 12, 769501. [Google Scholar] [CrossRef]
- Gillam, E.M.J.; Notley, L.M.; Cai, H.; De Voss, J.J.; Guengerich, F.P. Oxidation of Indole by Cytochrome P450 Enzymes. Biochemistry 2000, 39, 13817–13824. [Google Scholar] [CrossRef]
- Liu, Y.; Hou, Y.; Wang, G.; Zheng, X.; Hao, H. Gut Microbial Metabolites of Aromatic Amino Acids as Signals in Host–Microbe Interplay. Trends Endocrinol. Metab. TEM 2020, 31, 818–834. [Google Scholar] [CrossRef] [PubMed]
- Ding, Y.; Yanagi, K.; Yang, F.; Callaway, E.; Cheng, C.; Hensel, M.E.; Menon, R.; Alaniz, R.C.; Lee, K.; Jayaraman, A. Oral supplementation of gut microbial metabolite indole-3-acetate alleviates diet-induced steatosis and inflammation in mice. eLife 2024, 12, RP87458. [Google Scholar] [CrossRef]
- Nasta, T.Z.; Tabandeh, M.R.; Amini, K.; Abbasi, A.; Dayer, D.; Jalili, C. The influence of indole propionic acid on molecular markers of steroidogenesis, ER stress, and apoptosis in rat granulosa cells exposed to high glucose conditions. J. Steroid Biochem. Mol. Biol. 2024, 240, 106509. [Google Scholar] [CrossRef] [PubMed]
- Den Besten, G.; van Eunen, K.; Groen, A.K.; Venema, K.; Reijngoud, D.-J.; Bakker, B.M. The role of short-chain fatty acids in the interplay between diet, gut microbiota, and host energy metabolism. J. Lipid Res. 2013, 54, 2325–2340. [Google Scholar] [CrossRef]
- Visekruna, A.; Luu, M. The Role of Short-Chain Fatty Acids and Bile Acids in Intestinal and Liver Function, Inflammation, and Carcinogenesis. Front. Cell Dev. Biol. 2021, 9, 703218. [Google Scholar] [CrossRef] [PubMed]
- Pérez-Reytor, D.; Puebla, C.; Karahanian, E.; García, K. Use of Short-Chain Fatty Acids for the Recovery of the Intestinal Epithelial Barrier Affected by Bacterial Toxins. Front. Physiol. 2021, 12, 650313. [Google Scholar] [CrossRef] [PubMed]
- Willemsen, L.E.M.; Koetsier, M.A.; van Deventer, S.J.H.; van Tol, E.A.F. Short chain fatty acids stimulate epithelial mucin 2 expression through differential effects on prostaglandin E(1) and E(2) production by intestinal myofibroblasts. Gut 2003, 52, 1442–1447. [Google Scholar] [CrossRef] [PubMed]
- Singh, N.; Gurav, A.; Sivaprakasam, S.; Brady, E.; Padia, R.; Shi, H.; Thangaraju, M.; Prasad, P.D.; Manicassamy, S.; Munn, D.H.; et al. Activation of Gpr109a, receptor for niacin and the commensal metabolite butyrate, suppresses colonic inflammation and carcinogenesis. Immunity 2014, 40, 128–139. [Google Scholar] [CrossRef] [PubMed]
- Zhang, D.; Jian, Y.-P.; Zhang, Y.-N.; Li, Y.; Gu, L.-T.; Sun, H.-H.; Liu, M.-D.; Zhou, H.-L.; Wang, Y.-S.; Xu, Z.-X. Short-chain fatty acids in diseases. Cell Commun. Signal. CCS 2023, 21, 212. [Google Scholar] [CrossRef] [PubMed]
- Brütting, C.; Bisch, M.L.; Brandsch, C.; Hirche, F.; Stangl, G.I. Impact of dietary propionate on fructose-induced changes in lipid metabolism, gut microbiota and short-chain fatty acids in mice. Int. J. Food Sci. Nutr. 2021, 72, 160–173. [Google Scholar] [CrossRef] [PubMed]
- Singh, V.; Lee, G.; Son, H.; Koh, H.; Kim, E.S.; Unno, T.; Shin, J.-H. Butyrate producers, “The Sentinel of Gut”: Their intestinal significance with and beyond butyrate, and prospective use as microbial therapeutics. Front. Microbiol. 2023, 13, 1103836. [Google Scholar] [CrossRef] [PubMed]
- van der Hee, B.; Wells, J.M. Microbial Regulation of Host Physiology by Short-chain Fatty Acids. Trends Microbiol. 2021, 29, 700–712. [Google Scholar] [CrossRef]
- Kim, M.H.; Kang, S.G.; Park, J.H.; Yanagisawa, M.; Kim, C.H. Short-chain fatty acids activate GPR41 and GPR43 on intestinal epithelial cells to promote inflammatory responses in mice. Gastroenterology 2013, 145, 396–406.e10. [Google Scholar] [CrossRef]
- Lapidot, Y.; Amir, A.; Nosenko, R.; Uzan-Yulzari, A.; Veitsman, E.; Cohen-Ezra, O.; Davidov, Y.; Weiss, P.; Bradichevski, T.; Segev, S.; et al. Alterations in the Gut Microbiome in the Progression of Cirrhosis to Hepatocellular Carcinoma. mSystems 2020, 5, e00153-20. [Google Scholar] [CrossRef]
- Singh, V.; San Yeoh, B.; Chassaing, B.; Xiao, X.; Saha, P.; Olvera, R.A.; Lapek, J.D., Jr.; Zhang, L.; Wang, W.B.; Hao, S.; et al. Dysregulated Microbial Fermentation of Soluble Fiber Induces Cholestatic Liver Cancer. Cell 2018, 175, 679–694.e22. [Google Scholar] [CrossRef] [PubMed]
- Zhou, D.; Pan, Q.; Xin, F.-Z.; Zhang, R.-N.; He, C.-X.; Chen, G.-Y.; Liu, C.; Chen, Y.-W.; Fan, J.-G. Sodium butyrate attenuates high-fat diet-induced steatohepatitis in mice by improving gut microbiota and gastrointestinal barrier. World J. Gastroenterol. 2017, 23, 60–75. [Google Scholar] [CrossRef] [PubMed]
- Mollica, M.P.; Mattace Raso, G.; Cavaliere, G.; Trinchese, G.; De Filippo, C.; Aceto, S.; Prisco, M.; Pirozzi, C.; Di Guida, F.; Lama, A.; et al. Butyrate Regulates Liver Mitochondrial Function, Efficiency, and Dynamics in Insulin-Resistant Obese Mice. Diabetes 2017, 66, 1405–1418. [Google Scholar] [CrossRef] [PubMed]
- Rajapakse, J.; Khatiwada, S.; Akon, A.C.; Yu, K.L.; Shen, S.; Zekry, A. Unveiling the complex relationship between gut microbiota and liver cancer: Opportunities for novel therapeutic interventions. Gut Microbes 2023, 15, 2240031. [Google Scholar] [CrossRef] [PubMed]
- Lasitschka, F.; Giese, T.; Paparella, M.; Kurzhals, S.R.; Wabnitz, G.; Jacob, K.; Gras, J.; Bode, K.A.; Heninger, A.; Sziskzai, T.; et al. Human monocytes downregulate innate response receptors following exposure to the microbial metabolite n-butyrate. Immunity, Inflamm. Dis. 2017, 5, 480–492. [Google Scholar] [CrossRef] [PubMed]
- Chang, P.V.; Hao, L.; Offermanns, S.; Medzhitov, R. The microbial metabolite butyrate regulates intestinal macrophage function via histone deacetylase inhibition. Proc. Natl. Acad. Sci. USA 2014, 111, 2247–2252. [Google Scholar] [CrossRef] [PubMed]
- McBrearty, N.; Arzumanyan, A.; Bichenkov, E.; Merali, S.; Merali, C.; Feitelson, M. Short chain fatty acids delay the development of hepatocellular carcinoma in HBx transgenic mice. Neoplasia 2021, 23, 529–538. [Google Scholar] [CrossRef] [PubMed]
- Son, M.-Y.; Cho, H.-S. Anticancer Effects of Gut Microbiota-Derived Short-Chain Fatty Acids in Cancers. J. Microbiol. Biotechnol. 2023, 33, 849–856. [Google Scholar] [CrossRef]
- Ropero, S.; Esteller, M. The role of histone deacetylases (HDACs) in human cancer. Mol. Oncol. 2007, 1, 19–25. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Hu, C.; Xu, B.; Wang, X.; Wan, W.; Lu, J.; Kong, D.; Jin, Y.; You, W.; Sun, H.; Mu, X.; et al. Gut microbiota–derived short-chain fatty acids regulate group 3 innate lymphoid cells in HCC. Hepatology 2023, 77, 48–64. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Ma, H.-Y.; Yamamoto, G.; Xu, J.; Liu, X.; Karin, D.; Kim, J.Y.; Alexandrov, L.B.; Koyama, Y.; Nishio, T.; Benner, C.; et al. IL-17 signaling in steatotic hepatocytes and macrophages promotes hepatocellular carcinoma in alcohol-related liver disease. J. Hepatol. 2020, 72, 946–959. [Google Scholar] [CrossRef]
- Vivier, E.; Artis, D.; Colonna, M.; Diefenbach, A.; Di Santo, J.P.; Eberl, G.; Koyasu, S.; Locksley, R.M.; McKenzie, A.N.J.; Mebius, R.E.; et al. Innate Lymphoid Cells: 10 Years On. Cell 2018, 174, 1054–1066. [Google Scholar] [CrossRef] [PubMed]
- Song, Q.; Zhang, X.; Liu, W.; Wei, H.; Liang, W.; Zhou, Y.; Ding, Y.; Ji, F.; Cheung, A.H.-K.; Wong, N.; et al. Bifidobacterium pseudolongum-generated acetate suppresses non-alcoholic fatty liver disease-associated hepatocellular carcinoma. J. Hepatol. 2023, 79, 1352–1365. [Google Scholar] [CrossRef]
- Zhu, X.; Li, K.; Liu, G.; Wu, R.; Zhang, Y.; Wang, S.; Xu, M.; Lu, L.; Li, P. Microbial metabolite butyrate promotes anti-PD-1 antitumor efficacy by modulating T cell receptor signaling of cytotoxic CD8 T cell. Gut Microbes 2023, 15, 2249143. [Google Scholar] [CrossRef]
- Che, Y.; Chen, G.; Guo, Q.; Duan, Y.; Feng, H.; Xia, Q. Gut microbial metabolite butyrate improves anticancer therapy by regulating intracellular calcium homeostasis. Hepatology 2023, 78, 88–102. [Google Scholar] [CrossRef] [PubMed]
- Pant, K.; Richard, S.; Gradilone, S.A. Short-Chain Fatty Acid Butyrate Induces Cilia Formation and Potentiates the Effects of HDAC6 Inhibitors in Cholangiocarcinoma Cells. Front. Cell Dev. Biol. 2022, 9, 809382. [Google Scholar] [CrossRef] [PubMed]
- Wang, G.; Santerre, J.; Labow, R. High-performance liquid chromatographic separation and tandem mass spectrometric identification of breakdown products associated with the biological hydrolysis of a biomedical polyurethane. J. Chromatogr. B Biomed. Sci. Appl. 1997, 698, 69–80. [Google Scholar] [CrossRef]
- Xue, G.; Feng, J.; Zhang, R.; Du, B.; Sun, Y.; Liu, S.; Yan, C.; Liu, X.; Du, S.; Feng, Y.; et al. Three Klebsiella species as potential pathobionts generating endogenous ethanol in a clinical cohort of patients with auto-brewery syndrome: A case control study. EBioMedicine 2023, 91, 104560. [Google Scholar] [CrossRef]
- Chen, X.; Zhang, Z.; Li, H.; Zhao, J.; Wei, X.; Lin, W.; Zhao, X.; Jiang, A.; Yuan, J. Endogenous ethanol produced by intestinal bacteria induces mitochondrial dysfunction in non-alcoholic fatty liver disease. J. Gastroenterol. Hepatol. 2020, 35, 2009–2019. [Google Scholar] [CrossRef]
- Chen, J.; Vitetta, L. Gut Microbiota Metabolites in NAFLD Pathogenesis and Therapeutic Implications. Int. J. Mol. Sci. 2020, 21, 5214. [Google Scholar] [CrossRef]
- Yuan, J.; Chen, C.; Cui, J.; Lu, J.; Yan, C.; Wei, X.; Zhao, X.; Li, N.; Li, S.; Xue, G.; et al. Fatty Liver Disease Caused by High-Alcohol-Producing Klebsiella pneumoniae. Cell Metab. 2019, 30, 675–688.e7. [Google Scholar] [CrossRef] [PubMed]
- Bayoumy, A.B.; Mulder, C.J.J.; Mol, J.J.; Tushuizen, M.E. Gut fermentation syndrome: A systematic review of case reports. United Eur. Gastroenterol. J. 2021, 9, 332–342. [Google Scholar] [CrossRef] [PubMed]
- Zakhari, S. Overview: How is alcohol metabolized by the body? Alcohol Res. Health J. Natl. Inst. Alcohol Abus. Alcohol. 2006, 29, 245–254. [Google Scholar]
- Seitz, H.K.; Stickel, F. Molecular mechanisms of alcohol-mediated carcinogenesis. Nat. Rev. Cancer 2007, 7, 599–612. [Google Scholar] [CrossRef] [PubMed]
- Seitz, H.K.; Stickel, F. Acetaldehyde as an underestimated risk factor for cancer development: Role of genetics in ethanol metabolism. Genes Nutr. 2010, 5, 121–128. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Helander, A.; Lindahl-Kiessling, K. Increased frequency of acetaldehyde-induced sister-chromatid exchanges in human lymphocytes treated with an aldehyde dehydrogenase inhibitor. Mutat. Res. Lett. 1991, 264, 103–107. [Google Scholar] [CrossRef] [PubMed]
- Dellarco, V.L. A mutagenicity assessment of acetaldehyde. Mutat. Res. Genet. Toxicol. 1988, 195, 1–20. [Google Scholar] [CrossRef]
- Roos, W.P.; Thomas, A.D.; Kaina, B. DNA damage and the balance between survival and death in cancer biology. Nat. Rev. Cancer 2016, 16, 20–33. [Google Scholar] [CrossRef]
- Matsuo, K.; Hamajima, N.; Shinoda, M.; Hatooka, S.; Inoue, M.; Takezaki, T.; Tajima, K. Gene-environment interaction between an aldehyde dehydrogenase-2 (ALDH2) polymorphism and alcohol consumption for the risk of esophageal cancer. Carcinog. 2001, 22, 913–916. [Google Scholar] [CrossRef]
- Mbaye, B.; Wasfy, R.M.; Alou, M.T.; Borentain, P.; Gerolami, R.; Dufour, J.-C.; Million, M. A catalog of ethanol-producing microbes in humans. Futur. Microbiol. 2024, 19, 697–714. [Google Scholar] [CrossRef]
- Kocaelli, H.; Apaydin, A.; Aydil, B.; Ayhan, M.; Karadeniz, A.; Ozel, S.; Yılmaz, E.; Akgün, B.; Eren, B. Evaluation of potential salivary acetaldehyde production from ethanol in oral cancer patients and healthy subjects. Hippokratia 2014, 18, 269–274. [Google Scholar]
- Seitz, H.K.; Simanowski, U.A.; Garzon, F.T.; Rideout, J.M.; Peters, T.J.; Koch, A.; Berger, M.R.; Einecke, H.; Maiwald, M. Possible role of acetaldehyde in ethanol-related rectal cocarcinogenesis in the rat. Gastroenterology 1990, 98, 406–413. [Google Scholar] [CrossRef] [PubMed]
- Cao, L.; Shao, N.; Du, J.; Zhu, H.; Gao, J.; Li, Q.; Sun, Y.; Hu, J.; Yin, G.; Xu, G. Involvement of reactive oxygen species (ROS) in the hepatopancreatic cytotoxicity, oxidative stress, and apoptosis induced by microcystin-LR in Eriocheir sinensis. Comp. Biochem. Physiol. Part C Toxicol. Pharmacol. 2024, 276, 109801. [Google Scholar] [CrossRef] [PubMed]
- Blomstrand, R. Observations on the formation of ethanol in the intestinal tract in man. Life Sci. Pt. 2 Biochem. Gen. Mol. Biol. 1971, 10, 575–582. [Google Scholar] [CrossRef]
- Zhu, L.; Baker, S.S.; Gill, C.; Liu, W.; Alkhouri, R.; Baker, R.D.; Gill, S.R. Characterization of gut microbiomes in nonalcoholic steatohepatitis (NASH) patients: A connection between endogenous alcohol and NASH. Hepatology 2013, 57, 601–609. [Google Scholar] [CrossRef] [PubMed]
- Engstler, A.J.; Aumiller, T.; Degen, C.; Dürr, M.; Weiss, E.; Maier, I.B.; Schattenberg, J.M.; Jin, C.J.; Sellmann, C.; Bergheim, I. Insulin resistance alters hepatic ethanol metabolism: Studies in mice and children with non-alcoholic fatty liver disease. Gut 2016, 65, 1564–1571. [Google Scholar] [CrossRef]
- Biswas, D.; Duffley, L.; Pulinilkunnil, T. Role of branched-chain amino acid–catabolizing enzymes in intertissue signaling, metabolic remodeling, and energy homeostasis. FASEB J. Off. Publ. Fed. Am. Soc. Exp. Biol. 2019, 33, 8711–8731. [Google Scholar] [CrossRef] [PubMed]
- Zhang, S.; Zeng, X.; Ren, M.; Mao, X.; Qiao, S. Novel metabolic and physiological functions of branched chain amino acids: A review. J. Anim. Sci. Biotechnol. 2017, 8, 10. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- She, P.; Reid, T.M.; Bronson, S.K.; Vary, T.C.; Hajnal, A.; Lynch, C.J.; Hutson, S.M. Disruption of BCATm in mice leads to increased energy expenditure associated with the activation of a futile protein turnover cycle. Cell Metab. 2007, 6, 181–194. [Google Scholar] [CrossRef]
- Sun, E.J.; Wankell, M.; Palamuthusingam, P.; McFarlane, C.; Hebbard, L. Targeting the PI3K/Akt/mTOR Pathway in Hepatocellular Carcinoma. Biomedicines 2021, 9, 1639. [Google Scholar] [CrossRef]
- Moberg, M.; Apró, W.; Ekblom, B.; van Hall, G.; Holmberg, H.-C.; Blomstrand, E. Activation of mTORC1 by leucine is potentiated by branched-chain amino acids and even more so by essential amino acids following resistance exercise. Am. J. Physiol. Physiol. 2016, 310, C874–C884. [Google Scholar] [CrossRef] [PubMed]
- Lo, E.K.K.; Felicianna; Xu, J.-H.; Zhan, Q.; Zeng, Z.; El-Nezami, H. The Emerging Role of Branched-Chain Amino Acids in Liver Diseases. Biomedicines 2022, 10, 1444. [Google Scholar] [CrossRef]
- Kitagawa, A.; Osawa, T.; Noda, M.; Kobayashi, Y.; Aki, S.; Nakano, Y.; Saito, T.; Shimizu, D.; Komatsu, H.; Sugaya, M.; et al. Convergent genomic diversity and novel BCAA metabolism in intrahepatic cholangiocarcinoma. Br. J. Cancer 2023, 128, 2206–2217. [Google Scholar] [CrossRef]
- Bi, C.; Xiao, G.; Liu, C.; Yan, J.; Chen, J.; Si, W.; Zhang, J.; Liu, Z. Molecular Immune Mechanism of Intestinal Microbiota and Their Metabolites in the Occurrence and Development of Liver Cancer. Front. Cell Dev. Biol. 2021, 9, 702414. [Google Scholar] [CrossRef] [PubMed]
- Chen, F.; Dai, X.; Zhou, C.-C.; Li, K.-X.; Zhang, Y.-J.; Lou, X.-Y.; Zhu, Y.-M.; Sun, Y.-L.; Peng, B.-X.; Cui, W. Integrated analysis of the faecal metagenome and serum metabolome reveals the role of gut microbiome-associated metabolites in the detection of colorectal cancer and adenoma. Gut 2022, 71, 1315–1325. [Google Scholar] [CrossRef]
- Wang, W.; Zhen, S.; Ping, Y.; Wang, L.; Zhang, Y. Metabolomic biomarkers in liquid biopsy: Accurate cancer diagnosis and prognosis monitoring. Front. Oncol. 2024, 14, 1331215. [Google Scholar] [CrossRef]
- Xin, H.-Y.; Zou, J.-X.; Sun, R.-Q.; Hu, Z.-Q.; Chen, Z.; Luo, C.-B.; Zhou, Z.-J.; Wang, P.-C.; Li, J.; Yu, S.-Y.; et al. Characterization of tumor microbiome and associations with prognosis in intrahepatic cholangiocarcinoma. J. Gastroenterol. 2024, 59, 411–423. [Google Scholar] [CrossRef]
- Ponziani, F.R.; Nicoletti, A.; Gasbarrini, A.; Pompili, M. Diagnostic and therapeutic potential of the gut microbiota in patients with early hepatocellular carcinoma. Ther. Adv. Med. Oncol. 2019, 11, 1758835919848184. [Google Scholar] [CrossRef]
- Yavuz, B.G.; Datar, S.; Chamseddine, S.; Mohamed, Y.I.; LaPelusa, M.; Lee, S.S.; Hu, Z.I.; Koay, E.J.; Cao, H.S.T.; Jalal, P.K.; et al. The Gut Microbiome as a Biomarker and Therapeutic Target in Hepatocellular Carcinoma. Cancers 2023, 15, 4875. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Lee, P.-C.; Wu, C.-J.; Hung, Y.-W.; Lee, C.J.; Chi, C.-T.; Lee, I.-C.; Yu-Lun, K.; Chou, S.-H.; Luo, J.-C.; Hou, M.-C.; et al. Gut microbiota and metabolites associate with outcomes of immune checkpoint inhibitor–treated unresectable hepatocellular carcinoma. J. Immunother. Cancer 2022, 10, e004779. [Google Scholar] [CrossRef]
- Gopalakrishnan, V.; Spencer, C.N.; Nezi, L.; Reuben, A.; Andrews, M.C.; Karpinets, T.V.; Prieto, P.A.; Vicente, D.; Hoffman, K.; Wei, S.C.; et al. Gut microbiome modulates response to anti–PD-1 immunotherapy in melanoma patients. Science 2018, 359, 97–103. [Google Scholar] [CrossRef] [PubMed]
- Matson, V.; Fessler, J.; Bao, R.; Chongsuwat, T.; Zha, Y.; Alegre, M.-L.; Luke, J.J.; Gajewski, T.F. The commensal microbiome is associated with anti–PD-1 efficacy in metastatic melanoma patients. Science 2018, 359, 104–108. [Google Scholar] [CrossRef] [PubMed]
- Routy, B.; le Chatelier, E.; DeRosa, L.; Duong, C.P.M.; Alou, M.T.; Daillère, R.; Fluckiger, A.; Messaoudene, M.; Rauber, C.; Roberti, M.P.; et al. Gut microbiome influences efficacy of PD-1–based immunotherapy against epithelial tumors. Science 2018, 359, 91–97. [Google Scholar] [CrossRef] [PubMed]
- Pallozzi, M.; Di Tommaso, N.; Maccauro, V.; Santopaolo, F.; Gasbarrini, A.; Ponziani, F.R.; Pompili, M. Non-Invasive Biomarkers for Immunotherapy in Patients with Hepatocellular Carcinoma: Current Knowledge and Future Perspectives. Cancers 2022, 14, 4631. [Google Scholar] [CrossRef] [PubMed]
- Derosa, L.; Hellmann, M.D.; Spaziano, M.; Halpenny, D.; Fidelle, M.; Rizvi, H.; Long, N.; Plodkowski, A.J.; Arbour, K.C.; Chaft, J.E.; et al. Negative association of antibiotics on clinical activity of immune checkpoint inhibitors in patients with advanced renal cell and non-small-cell lung cancer. Ann. Oncol. Off. J. Eur. Soc. Med. Oncol. 2018, 29, 1437–1444. [Google Scholar] [CrossRef] [PubMed]
- Tinsley, N.; Zhou, C.; Tan, G.; Rack, S.; Lorigan, P.; Blackhall, F.; Krebs, M.; Carter, L.; Thistlethwaite, F.; Graham, D.; et al. Cumulative Antibiotic Use Significantly Decreases Efficacy of Checkpoint Inhibitors in Patients with Advanced Cancer. Oncologist 2020, 25, 55–63. [Google Scholar] [CrossRef] [PubMed]
- Binda, C.; Gibiino, G.; Coluccio, C.; Sbrancia, M.; Dajti, E.; Sinagra, E.; Capurso, G.; Sambri, V.; Cucchetti, A.; Ercolani, G.; et al. Biliary Diseases from the Microbiome Perspective: How Microorganisms Could Change the Approach to Benign and Malignant Diseases. Microorganisms 2022, 10, 312. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Allegretti, J.R.; Kassam, Z.; Mullish, B.H.; Chiang, A.; Carrellas, M.; Hurtado, J.; Marchesi, J.R.; McDonald, J.A.K.; Pechlivanis, A.; Barker, G.F.; et al. Effects of Fecal Microbiota Transplantation with Oral Capsules in Obese Patients. Clin. Gastroenterol. Hepatol. Off. Clin. Pract. J. Am. Gastroenterol. Assoc. 2020, 18, 855–863.e2. [Google Scholar] [CrossRef] [PubMed]
- Dao, M.C.; Everard, A.; Aron-Wisnewsky, J.; Sokolovska, N.; Prifti, E.; Verger, E.O.; Kayser, B.D.; Levenez, F.; Chilloux, J.; Hoyles, L.; et al. Akkermansia muciniphila and improved metabolic health during a dietary intervention in obesity: Relationship with gut microbiome richness and ecology. Gut 2016, 65, 426–436. [Google Scholar] [CrossRef]
- Study Details|FMT in IT-refractory HCC—FAB-HCC Pilot Study|ClinicalTrials.gov. Available online: https://clinicaltrials.gov/study/NCT05750030 (accessed on 9 July 2024).
- Allegretti, J.R.; Kassam, Z.; Carrellas, M.; Mullish, B.H.; Marchesi, J.R.; Pechlivanis, A.; Smith, M.; Gerardin, Y.; Timberlake, S.; Pratt, D.S.; et al. Fecal Microbiota Transplantation in Patients with Primary Sclerosing Cholangitis: A Pilot Clinical Trial. Am. J. Gastroenterol. 2019, 114, 1071–1079. [Google Scholar] [CrossRef]
- Agus, A.; Clément, K.; Sokol, H. Gut microbiota-derived metabolites as central regulators in metabolic disorders. Gut 2021, 70, 1174–1182. [Google Scholar] [CrossRef] [PubMed]
- Meng, X.; Li, S.; Li, Y.; Gan, R.-Y.; Li, H.-B. Gut Microbiota’s Relationship with Liver Disease and Role in Hepatoprotection by Dietary Natural Products and Probiotics. Nutrients 2018, 10, 1457. [Google Scholar] [CrossRef] [PubMed]
- Ichikawa, M.; Nakamoto, N.; Kredo-Russo, S.; Weinstock, E.; Weiner, I.N.; Khabra, E.; Ben-Ishai, N.; Inbar, D.; Kowalsman, N.; Mordoch, R.; et al. Bacteriophage therapy against pathological Klebsiella pneumoniae ameliorates the course of primary sclerosing cholangitis. Nat. Commun. 2023, 14, 3261. [Google Scholar] [CrossRef] [PubMed]


{kind=link}
{kind=link}
| Study Model | Tumor | Microbiota Composition | Other Features |
|---|---|---|---|
| Observational, fecal sample (healthy vs. cirrhosis vs. early HCC) [61] | early HCC | ↑ Actinobacteria, Klebsiella and Haemophilus (producing LPS) ↓ Ruminococcus, Oscillibacter, Faecalibacterium, Clostridium IV, Coprococcus (butyrate-producing bacteria families) | Fecal microbial diversity was decreased from healthy controls to cirrhosis, but it was increased from cirrhosis to early HCC with cirrhosis |
| Observational, HBV-related HCC (B-HCC) vs. non-HBV and non-HCV-related HCC (NBNC-HCC) [67] | HCC | ↑Escherichia, Shigella, Enterococcus ↓ Faecalibacterium, Ruminococcus, Ruminoclostridium | Higher species richness of fecal microbiota of B-HCC vs. others |
| Observational NAFLD-related cirrhosis and HCC vs. NAFLD-related cirrhosis without HCC vs. healthy controls [63] | HCC | ↑ Bacteroides and Ruminococcaceae ↓ Bifidobacterium, Akkermansia | Akkermansia and Bifidobacterium were inversely correlated with calprotectin concentration, which, in turn, was associated with humoral and cellular inflammatory markers |
| Case–control HBV-related HCC tissues vs. chronic hepatitis [69] | HCC | ↑ E. coli S. dysenteriae | ↓ Intratumoral microbial heterogeneity of HCC tissues decreased compared with that of nontumor tissues |
| Observational CCA vs. HCC vs. liver cirrhosis vs. healthy [71] | CCA | ↑ Lactobacillus, Actinomyces, Peptostreptococcaceae, and Alloscardovia | ↑ α-diversities and β-diversities compared to other groups |
| Observational, in vitro tumor tissue vs. paracancerous tumor [72] | CCA | ↑ Burkholderiales, Pseudomonadales, Xanthomonadales, Bacillales and Clostridiales | P. fungorum higher in the paracancerous tissues and negatively correlated with CA19.9 |
| Gut Metabolite | Mechanism of Action | Effects | Reference |
|---|---|---|---|
Bile Acids | DCA induces SASP phenotype in HSCs DCA and G-CDCA induce endoplasmic reticulum (ER) stress with Ca2+ release and promotion of ROS CA, GCA, LCA and CDCA interact with TRAIL and Fas > PKC/MAPK/NFkB and JAk-STAT3 and the PI3-K CDCA and DCA act on EGFR, on Erg-1/MAPK signaling [80] and PKC/MAPK/NFkB BAs reduce FXR activity through SIRT1 in hepatocytes via the Wnt/β-catenin pathway | fibrogenesis chronic inflammation chronic inflammation, fibrogenesis cell proliferation cell proliferation | [32] [6,25,83] [81,82] [79,80,85] [90] |
Choline and TMAo | ↑ ROS ↓ intrahepatic triglycerides, resulting in a higher risk of metabolic-associated disease, including HCC [101,102,103] activation of mTOR signaling upregulation POSTN gene ↑ f FXR-antagonists ↑ Wnt/β-catenin pathway | DNA damage cell necrosis cell proliferation | [98,99,100,101] [105,106,107] [109,110,111] [90,114] |
SCFAs | GPR41, GPR43 or GPR109/PKC/ERK, PKA/ERK activation DNA epigenetic modifications and HDAC inhibition Butyrate ↑ regulatory T cells ↑ IL-10 by microbiota antigen-specific Th 1 cells ↓macrophages in the lamina propria | cell proliferation DNA damage immune suppression | [125,129] [137] [139] [134] |
Ethanol | ↑ ADH, NADH, CYP2E1 | DNA synthesis and repair mucosal injury and cellular DNA instability cell necrosis | [153,154,162,164] |
BCAA | ↑ IRS1/PI3K/AKT/mTORC1 ↑ catabolic enzymes of BCAAs in tumor cells, ↑ accumulation of branched-chain ketoacids | cell proliferation, angiogenesis and apoptosis | [167,168,169,170,171,172] |
Indoles | ↓ indole-3-acetate intestinal levels in HFD ↓ fatty acid oxidation | disruption of the intestinal barrier, overexpression of inflammatory cytokines and inhibition of immune cells | [117,118,119] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Pallozzi, M.; De Gaetano, V.; Di Tommaso, N.; Cerrito, L.; Santopaolo, F.; Stella, L.; Gasbarrini, A.; Ponziani, F.R. Role of Gut Microbial Metabolites in the Pathogenesis of Primary Liver Cancers. Nutrients 2024, 16, 2372. https://doi.org/10.3390/nu16142372
Pallozzi M, De Gaetano V, Di Tommaso N, Cerrito L, Santopaolo F, Stella L, Gasbarrini A, Ponziani FR. Role of Gut Microbial Metabolites in the Pathogenesis of Primary Liver Cancers. Nutrients. 2024; 16(14):2372. https://doi.org/10.3390/nu16142372
Chicago/Turabian StylePallozzi, Maria, Valeria De Gaetano, Natalia Di Tommaso, Lucia Cerrito, Francesco Santopaolo, Leonardo Stella, Antonio Gasbarrini, and Francesca Romana Ponziani. 2024. "Role of Gut Microbial Metabolites in the Pathogenesis of Primary Liver Cancers" Nutrients 16, no. 14: 2372. https://doi.org/10.3390/nu16142372