
High-Fat Diet Delays Liver Fibrosis Recovery and Promotes Hepatocarcinogenesis in Rat Liver Cirrhosis Model
 , , ,
, , ,
Abstract
:1. Introduction
2. Materials and Methods
2.1. Animals, Diets, and Chemicals
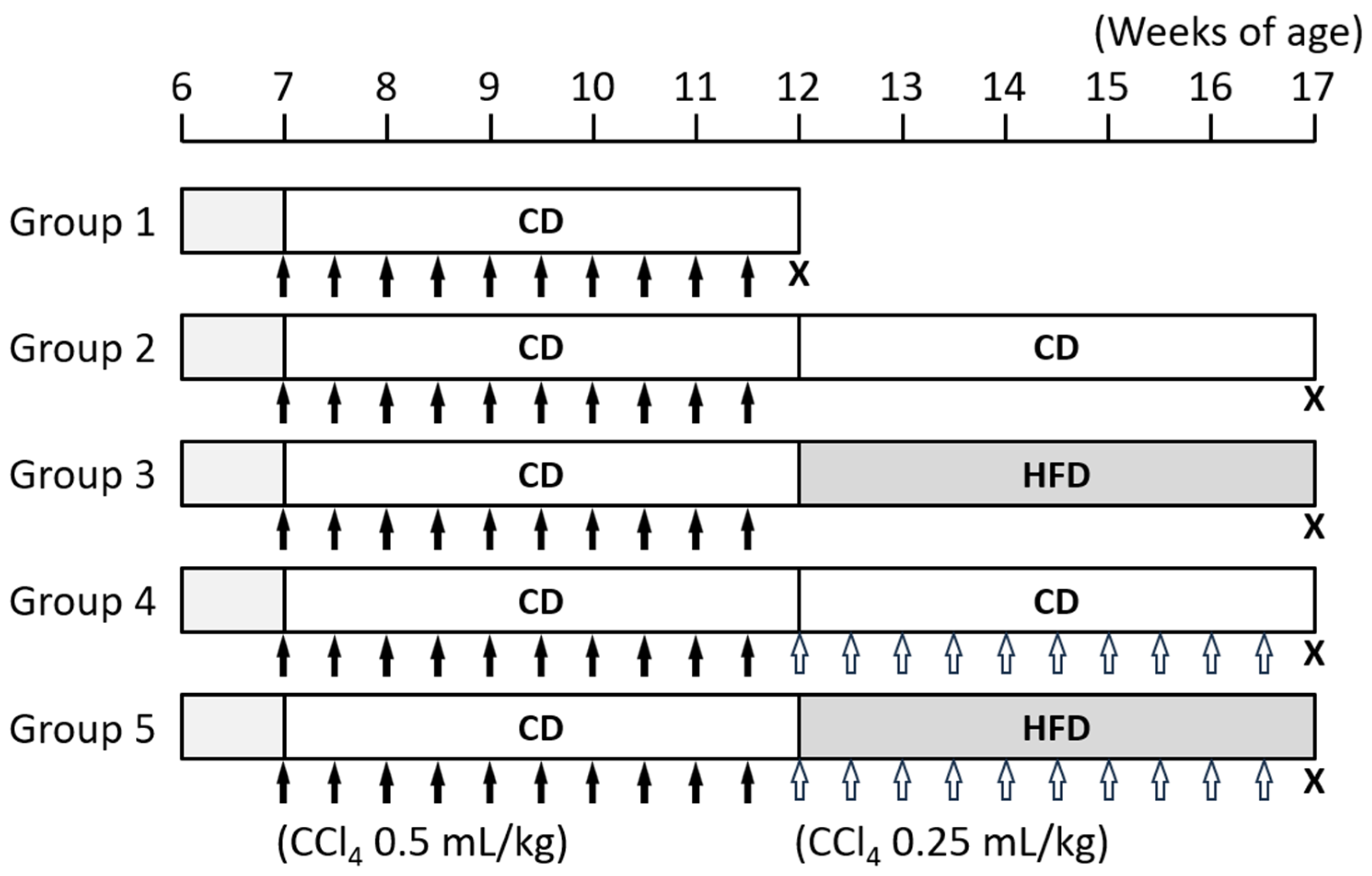
2.2. Procedure for Animal Experiments
2.3. Histology and Immunohistochemistry
2.4. Quantitative Real-Time Reverse Transcription-PCR
2.5. Measurement of Serum Parameters
2.6. Statistical Analysis
3. Results
3.1. General Observations
3.2. HFD Exacerbated CCl4-Induced Liver Fibrosis and Delayed the Reversal of Fibrosis
3.3. HFD Increased Lipogenesis and Induced Steatosis in the Liver
3.4. HFD Promoted Hepatic Inflammation
3.5. Effects of HFD on Insulin Resistance and Oxidative Stress
3.6. HFD Promoted the Development of Hepatic Precancerous Lesions
4. Discussion
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Takehara, T.; Sakamoto, N.; Nishiguchi, S.; Ikeda, F.; Tatsumi, T.; Ueno, Y.; Yatsuhashi, H.; Takikawa, Y.; Kanda, T.; Sakamoto, M.; et al. Efficacy and safety of sofosbuvir-velpatasvir with or without ribavirin in HCV-infected Japanese patients with decompensated cirrhosis: An open-label phase 3 trial. J. Gastroenterol. 2019, 54, 87–95. [Google Scholar] [CrossRef] [PubMed]
- Enomoto, H.; Ueno, Y.; Hiasa, Y.; Nishikawa, H.; Hige, S.; Takikawa, Y.; Taniai, M.; Ishikawa, T.; Yasui, K.; Takaki, A.; et al. Transition in the etiology of liver cirrhosis in Japan: A nationwide survey. J. Gastroenterol. 2020, 55, 353–362. [Google Scholar] [CrossRef] [PubMed]
- Sayiner, M.; Golabi, P.; Younossi, Z.M. Disease Burden of Hepatocellular Carcinoma: A Global Perspective. Dig. Dis. Sci. 2019, 64, 910–917. [Google Scholar] [CrossRef] [PubMed]
- Sung, H.; Ferlay, J.; Siegel, R.L.; Laversanne, M.; Soerjomataram, I.; Jemal, A.; Bray, F. Global Cancer Statistics 2020: GLOBOCAN Estimates of Incidence and Mortality Worldwide for 36 Cancers in 185 Countries. CA Cancer J. Clin. 2021, 71, 209–249. [Google Scholar] [CrossRef] [PubMed]
- Hoshida, Y.; Fuchs, B.C.; Bardeesy, N.; Baumert, T.F.; Chung, R.T. Pathogenesis and prevention of hepatitis C virus-induced hepatocellular carcinoma. J. Hepatol. 2014, 61, S79–S90. [Google Scholar] [CrossRef] [PubMed]
- Aydin, M.M.; Akcali, K.C. Liver fibrosis. Turk. J. Gastroenterol. 2018, 29, 14–21. [Google Scholar] [CrossRef] [PubMed]
- Delgado, M.E.; Cardenas, B.I.; Farran, N.; Fernandez, M. Metabolic Reprogramming of Liver Fibrosis. Cells 2021, 10, 3604. [Google Scholar] [CrossRef]
- Dokmak, A.; Lizaola-Mayo, B.; Trivedi, H.D. The Impact of Nonalcoholic Fatty Liver Disease in Primary Care: A Population Health Perspective. Am. J. Med. 2021, 134, 23–29. [Google Scholar] [CrossRef] [PubMed]
- Sohn, W.; Lee, H.W.; Lee, S.; Lim, J.H.; Lee, M.W.; Park, C.H.; Yoon, S.K. Obesity and the risk of primary liver cancer: A systematic review and meta-analysis. Clin. Mol. Hepatol. 2021, 27, 157–174. [Google Scholar] [CrossRef]
- Zhang, C.Y.; Liu, S.; Yang, M. Treatment of liver fibrosis: Past, current, and future. World J. Hepatol. 2023, 15, 755–774. [Google Scholar] [CrossRef]
- Muto, Y.; Sato, S.; Watanabe, A.; Moriwaki, H.; Suzuki, K.; Kato, A.; Kato, M.; Nakamura, T.; Higuchi, K.; Nishiguchi, S.; et al. Effects of oral branched-chain amino acid granules on event-free survival in patients with liver cirrhosis. Clin. Gastroenterol. Hepatol. 2005, 3, 705–713. [Google Scholar] [CrossRef] [PubMed]
- Iwasa, M.; Kobayashi, Y.; Mifuji-Moroka, R.; Hara, N.; Miyachi, H.; Sugimoto, R.; Tanaka, H.; Fujita, N.; Gabazza, E.C.; Takei, Y. Branched-chain amino acid supplementation reduces oxidative stress and prolongs survival in rats with advanced liver cirrhosis. PLoS ONE 2013, 8, e70309. [Google Scholar] [CrossRef] [PubMed]
- Gluud, L.L.; Dam, G.; Les, I.; Marchesini, G.; Borre, M.; Aagaard, N.K.; Vilstrup, H. Branched-chain amino acids for people with hepatic encephalopathy. Cochrane Database Syst. Rev. 2017, 5, CD001939. [Google Scholar] [CrossRef] [PubMed]
- Yoshiji, H.; Nagoshi, S.; Akahane, T.; Asaoka, Y.; Ueno, Y.; Ogawa, K.; Kawaguchi, T.; Kurosaki, M.; Sakaida, I.; Shimizu, M.; et al. Evidence-based clinical practice guidelines for Liver Cirrhosis 2020. J. Gastroenterol. 2021, 56, 593–619. [Google Scholar] [CrossRef] [PubMed]
- Lai, J.C.; Tandon, P.; Bernal, W.; Tapper, E.B.; Ekong, U.; Dasarathy, S.; Carey, E.J. Malnutrition, Frailty, and Sarcopenia in Patients with Cirrhosis: 2021 Practice Guidance by the American Association for the Study of Liver Diseases. Hepatology 2021, 74, 1611–1644. [Google Scholar] [CrossRef] [PubMed]
- Xiao, J.; Ching, Y.P.; Liong, E.C.; Nanji, A.A.; Fung, M.L.; Tipoe, G.L. Garlic-derived S-allylmercaptocysteine is a hepato-protective agent in non-alcoholic fatty liver disease in vivo animal model. Eur. J. Nutr. 2013, 52, 179–191. [Google Scholar] [CrossRef] [PubMed]
- Sumi, T.; Shirakami, Y.; Shimizu, M.; Kochi, T.; Ohno, T.; Kubota, M.; Shiraki, M.; Tsurumi, H.; Tanaka, T.; Moriwaki, H. (−)-Epigallocatechin-3-gallate suppresses hepatic preneoplastic lesions developed in a novel rat model of non-alcoholic steatohepatitis. Springerplus 2013, 2, 690. [Google Scholar] [CrossRef] [PubMed]
- Shirakami, Y.; Kochi, T.; Kubota, M.; Sakai, H.; Ibuka, T.; Yoshimi, K.; Kuramoto, T.; Tanaka, T.; Shimizu, M.; Seishima, M. Inhibitory effects of pentoxifylline on inflammation-related tumorigenesis in rat colon. Oncotarget 2018, 9, 33972–33981. [Google Scholar] [CrossRef] [PubMed]
- Miao, D.; Zhang, L. Leptin modulates the expression of catabolic genes in rat nucleus pulposus cells through the mitogen-activated protein kinase and Janus kinase 2/signal transducer and activator of transcription 3 pathways. Mol. Med. Rep. 2015, 12, 1761–1768. [Google Scholar] [CrossRef]
- Dong, D.; Yin, L.; Qi, Y.; Xu, L.; Peng, J. Protective Effect of the Total Saponins from Rosa laevigata Michx Fruit against Carbon Tetrachloride-Induced Liver Fibrosis in Rats. Nutrients 2015, 7, 4829–4850. [Google Scholar] [CrossRef]
- Kochi, T.; Shimizu, M.; Terakura, D.; Baba, A.; Ohno, T.; Kubota, M.; Shirakami, Y.; Tsurumi, H.; Tanaka, T.; Moriwaki, H. Non-alcoholic steatohepatitis and preneoplastic lesions develop in the liver of obese and hypertensive rats: Suppressing effects of EGCG on the development of liver lesions. Cancer Lett. 2014, 342, 60–69. [Google Scholar] [CrossRef] [PubMed]
- Imai, K.; Takai, K.; Nishigaki, Y.; Shimizu, S.; Naiki, T.; Hayashi, H.; Uematsu, T.; Sugihara, J.; Tomita, E.; Shimizu, M.; et al. Insulin resistance raises the risk for recurrence of stage I hepatocellular carcinoma after curative radiofrequency ablation in hepatitis C virus-positive patients: A prospective, case series study. Hepatol. Res. 2010, 40, 376–382. [Google Scholar] [CrossRef] [PubMed]
- Phoolchund, A.G.S.; Khakoo, S.I. MASLD and the Development of HCC: Pathogenesis and Therapeutic Challenges. Cancers 2024, 16, 259. [Google Scholar] [CrossRef] [PubMed]
- Asgharpour, A.; Cazanave, S.C.; Pacana, T.; Seneshaw, M.; Vincent, R.; Banini, B.A.; Kumar, D.P.; Daita, K.; Min, H.K.; Mirshahi, F.; et al. A diet-induced animal model of non-alcoholic fatty liver disease and hepatocellular cancer. J. Hepatol. 2016, 65, 579–588. [Google Scholar] [CrossRef] [PubMed]
- Wei, G.; An, P.; Vaid, K.A.; Nasser, I.; Huang, P.; Tan, L.; Zhao, S.; Schuppan, D.; Popov, Y.V. Comparison of murine steatohepatitis models identifies a dietary intervention with robust fibrosis, ductular reaction, and rapid progression to cirrhosis and cancer. Am. J. Physiol. Gastrointest. Liver Physiol. 2020, 318, G174–G188. [Google Scholar] [CrossRef] [PubMed]
- Setyaningsih, W.A.W.; Sari, D.C.R.; Romi, M.M.; Arfian, N. Liver fibrosis associated with adipose tissue and liver inflammation in an obesity model. Med. J. Malays. 2021, 76, 304–310. [Google Scholar]
- Farooq, M.; Hameed, H.; Dimanche-Boitrel, M.T.; Piquet-Pellorce, C.; Samson, M.; Le Seyec, J. Switching to Regular Diet Partially Resolves Liver Fibrosis Induced by High-Fat, High-Cholesterol Diet in Mice. Nutrients 2022, 14, 386. [Google Scholar] [CrossRef] [PubMed]
- Hui, J.M.; Sud, A.; Farrell, G.C.; Bandara, P.; Byth, K.; Kench, J.G.; McCaughan, G.W.; George, J. Insulin resistance is associated with chronic hepatitis C virus infection and fibrosis progression [corrected]. Gastroenterology 2003, 125, 1695–1704. [Google Scholar] [CrossRef] [PubMed]
- Chen, X.; Yamamoto, M.; Fujii, K.; Nagahama, Y.; Ooshio, T.; Xin, B.; Okada, Y.; Furukawa, H.; Nishikawa, Y. Differential reactivation of fetal/neonatal genes in mouse liver tumors induced in cirrhotic and non-cirrhotic conditions. Cancer Sci. 2015, 106, 972–981. [Google Scholar] [CrossRef]
- Eguchi, A.; Mizukami, S.; Nakamura, M.; Masuda, S.; Murayama, H.; Kawashima, M.; Inohana, M.; Nagahara, R.; Kobayashi, M.; Yamashita, R.; et al. Metronidazole enhances steatosis-related early-stage hepatocarcinogenesis in high fat diet-fed rats through DNA double-strand breaks and modulation of autophagy. Environ. Sci. Pollut. Res. Int. 2022, 29, 779–789. [Google Scholar] [CrossRef]
- Masuda, S.; Mizukami, S.; Eguchi, A.; Ichikawa, R.; Nakamura, M.; Nakamura, K.; Okada, R.; Tanaka, T.; Shibutani, M.; Yoshida, T. Immunohistochemical expression of autophagosome markers LC3 and p62 in preneoplastic liver foci in high fat diet-fed rats. J. Toxicol. Sci. 2019, 44, 565–574. [Google Scholar] [CrossRef] [PubMed]
- Park, E.J.; Lee, J.H.; Yu, G.Y.; He, G.; Ali, S.R.; Holzer, R.G.; Osterreicher, C.H.; Takahashi, H.; Karin, M. Dietary and genetic obesity promote liver inflammation and tumorigenesis by enhancing IL-6 and TNF expression. Cell 2010, 140, 197–208. [Google Scholar] [CrossRef] [PubMed]
- Siegel, A.B.; Zhu, A.X. Metabolic syndrome and hepatocellular carcinoma: Two growing epidemics with a potential link. Cancer 2009, 115, 5651–5661. [Google Scholar] [CrossRef] [PubMed]
- Deberardinis, R.J.; Sayed, N.; Ditsworth, D.; Thompson, C.B. Brick by brick: Metabolism and tumor cell growth. Curr. Opin. Genet. Dev. 2008, 18, 54–61. [Google Scholar] [CrossRef] [PubMed]
- Drury, J.; Geisen, M.E.; Tessmann, J.W.; Rychahou, P.G.; Kelson, C.O.; He, D.; Wang, C.; Evers, B.M.; Zaytseva, Y.Y. Overexpression of Fatty Acid Synthase Upregulates Glutamine-Fructose-6-Phosphate Transaminase 1 and O-Linked N-Acetylglucosamine Transferase to Increase O-GlcNAc Protein Glycosylation and Promote Colorectal Cancer Growth. Int. J. Mol. Sci. 2024, 25, 4883. [Google Scholar] [CrossRef] [PubMed]
- He, Y.; Qi, S.; Chen, L.; Zhu, J.; Liang, L.; Chen, X.; Zhang, H.; Zhuo, L.; Zhao, S.; Liu, S.; et al. The roles and mechanisms of SREBP1 in cancer development and drug response. Genes Dis. 2024, 11, 100987. [Google Scholar] [CrossRef] [PubMed]
- Shirakami, Y.; Shimizu, M.; Kubota, M.; Ohno, T.; Kochi, T.; Nakamura, N.; Sumi, T.; Tanaka, T.; Moriwaki, H.; Seishima, M. Pentoxifylline prevents nonalcoholic steatohepatitis-related liver pre-neoplasms by inhibiting hepatic inflammation and lipogenesis. Eur. J. Cancer Prev. 2016, 25, 206–215. [Google Scholar] [CrossRef] [PubMed]
- Sakai, H.; Shirakami, Y.; Shimizu, M. Chemoprevention of obesity-related liver carcinogenesis by using pharmaceutical and nutraceutical agents. World J. Gastroenterol. 2016, 22, 394–406. [Google Scholar] [CrossRef] [PubMed]
- Gaggini, M.; Morelli, M.; Buzzigoli, E.; DeFronzo, R.A.; Bugianesi, E.; Gastaldelli, A. Non-alcoholic fatty liver disease (NAFLD) and its connection with insulin resistance, dyslipidemia, atherosclerosis and coronary heart disease. Nutrients 2013, 5, 1544–1560. [Google Scholar] [CrossRef]
- Loft, A.; Alfaro, A.J.; Schmidt, S.F.; Pedersen, F.B.; Terkelsen, M.K.; Puglia, M.; Chow, K.K.; Feuchtinger, A.; Troullinaki, M.; Maida, A.; et al. Liver-fibrosis-activated transcriptional networks govern hepatocyte reprogramming and intra-hepatic communication. Cell Metab. 2021, 33, 1685–1700.e9. [Google Scholar] [CrossRef]
- Szablewski, L. Insulin Resistance: The Increased Risk of Cancers. Curr. Oncol. 2024, 31, 998–1027. [Google Scholar] [CrossRef] [PubMed]
- da Silva, B.S.; Paulino, A.M.B.; Taffarel, M.; Borba, I.G.; Telles, L.O.; Lima, V.V.; Aguiar, D.H.; Dias, M.C.; Nascimento, A.F.; Sinhorin, V.D.G.; et al. High sucrose diet attenuates oxidative stress, inflammation and liver injury in thioacetamide-induced liver cirrhosis. Life Sci. 2021, 267, 118944. [Google Scholar] [CrossRef] [PubMed]
- Nie, G.; Zhu, X.; Zhang, H.; Wang, H.; Yan, J.; Li, X. Identifying the predictive role and the related drugs of oxidative stress genes in the hepatocellular carcinoma. Cancer Rep. 2024, 7, e1978. [Google Scholar] [CrossRef] [PubMed]
- Choudhari, S.K.; Chaudhary, M.; Gadbail, A.R.; Sharma, A.; Tekade, S. Oxidative and antioxidative mechanisms in oral cancer and precancer: A review. Oral Oncol. 2014, 50, 10–18. [Google Scholar] [CrossRef] [PubMed]
- Jimenez-Cortegana, C.; Garcia-Galey, A.; Tami, M.; Del Pino, P.; Carmona, I.; Lopez, S.; Alba, G.; Sanchez-Margalet, V. Role of Leptin in Non-Alcoholic Fatty Liver Disease. Biomedicines 2021, 9, 762. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.; Liu, X.; Zou, Y.; Gong, J.; Ge, Z.; Lin, X.; Zhang, W.; Huang, H.; Zhao, J.; Saw, P.E.; et al. A high-fat diet promotes cancer progression by inducing gut microbiota-mediated leucine production and PMN-MDSC differentiation. Proc. Natl. Acad. Sci. USA 2024, 121, e2306776121. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Zhang, H.; Dai, Z. Cancer Treatment with the Ketogenic Diet: A Systematic Review and Meta-analysis of Animal Studies. Front. Nutr. 2021, 8, 594408. [Google Scholar] [CrossRef] [PubMed]
- Römer, M.; Dörfler, J.; Huebner, J. The use of ketogenic diets in cancer patients: A systematic review. Clin. Exp. Med. 2021, 21, 501–536. [Google Scholar] [CrossRef]
- Goswami, S.; Zhang, Q.; Celik, C.E.; Reich, E.M.; Yilmaz, Ö.H. Dietary fat and lipid metabolism in the tumor microenvironment. Biochim. Biophys. Acta Rev. Cancer 2023, 1878, 188984. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Ingredients (g/kg) | Control Chow | HFD |
|---|---|---|
| Casein | 140 | 256 |
| Corn starch | 465.7 | 160 |
| Sucrose | 100 | 55 |
| Dextrose | 155 | 60 |
| Cellulose | 50 | 66.1 |
| Soybean oil | 40 | 20 |
| Lard | 0 | 330 |
| Vitamin mixture | 35 | 35 |
| Mineral mixture | 10 | 10 |
| Calcium carbonate | 0 | 1.8 |
| L-cysteine | 1.8 | 3.6 |
| Choline bitartrate | 2.5 | 2.5 |
| Energy (kcal/g) | 3.85 | 5.06 |
| Gene | Forward | Reverse |
|---|---|---|
| Acta2 | CTGGAGAAGAGCTACGAACTGC | CTGATCCACATCTGCTGGAAGG |
| Ccl2 | CGTGCTGTCTCAGCCAGAT | GGATCATCTTGCCAGTGAATG |
| Fasn | TCGTCTGCCTCCAGATCC | GGCAATTTCCCGGACATAC |
| Gapdh | TGGGAAGCTGGTCATCAAC | GCATCACCCCATTTGATGTT |
| Il1b | AGGCTTCCTTGTGCAAGTGT | TCCTGGGGAAGGCATTAGGA |
| Il6 | CCCTTCAGGAACAGCTATGAA | ACAACATCAGTCCCAAGAAGG |
| Serpin1 | CCCCGGCACTGGTAAATCTT | TCTGTCTATCTGCTGCCCCT |
| Srebf1 | GGAGCCATGGATTGCACATT | GCTTCCAGAGAGGAGCCCAG |
| Tgfb1 | CTTCAGCTCCACAGAGAAGAACTGC | CACGATCATGTTGGACAACTGCTCC |
| Timp1 | ACAGCTTTCTGCAACTCGGA | AGTTTGCAAGGGATGGCTGA |
| Tnf | AGTTGGGGAGGGAGACCTT | CATCCACCCAAGGATGTTTAG |
| Group | Treatment | Number of Rats | Body Weight (g) | Relative Organ Weight (g/100 g Body Weight) | |
|---|---|---|---|---|---|
| Liver | White Adipose Tissue | ||||
| G2 | CCl4 (−)/CD | 5 | 367.2 ± 12.0 a | 2.8 ± 0.2 | 0.9 ± 0.3 |
| G3 | CCl4 (−)/HFD | 5 | 438.4 ± 37.6 b | 2.8 ± 0.2 | 1.8 ± 0.4 b |
| G4 | CCl4 (+)/CD | 5 | 372.2 ± 19.8 | 3.0 ± 1.5 | 0.8 ± 0.2 |
| G5 | CCl4 (+)/HFD | 5 | 367.0 ± 34.4 | 3.0 ± 0.2 | 1.1 ± 0.3 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Taguchi, D.; Shirakami, Y.; Sakai, H.; Maeda, T.; Miwa, T.; Kubota, M.; Imai, K.; Ibuka, T.; Shimizu, M. High-Fat Diet Delays Liver Fibrosis Recovery and Promotes Hepatocarcinogenesis in Rat Liver Cirrhosis Model. Nutrients 2024, 16, 2506. https://doi.org/10.3390/nu16152506
Taguchi D, Shirakami Y, Sakai H, Maeda T, Miwa T, Kubota M, Imai K, Ibuka T, Shimizu M. High-Fat Diet Delays Liver Fibrosis Recovery and Promotes Hepatocarcinogenesis in Rat Liver Cirrhosis Model. Nutrients. 2024; 16(15):2506. https://doi.org/10.3390/nu16152506
Chicago/Turabian StyleTaguchi, Daisuke, Yohei Shirakami, Hiroyasu Sakai, Toshihide Maeda, Takao Miwa, Masaya Kubota, Kenji Imai, Takashi Ibuka, and Masahito Shimizu. 2024. "High-Fat Diet Delays Liver Fibrosis Recovery and Promotes Hepatocarcinogenesis in Rat Liver Cirrhosis Model" Nutrients 16, no. 15: 2506. https://doi.org/10.3390/nu16152506