
Congenital Diarrhoeas and Enteropathies
Abstract
1. Introduction
2. Materials and Methods
2.1. Defects in Epithelial Ionic Transport or Digestion and Absorption of Nutrients
- (a)
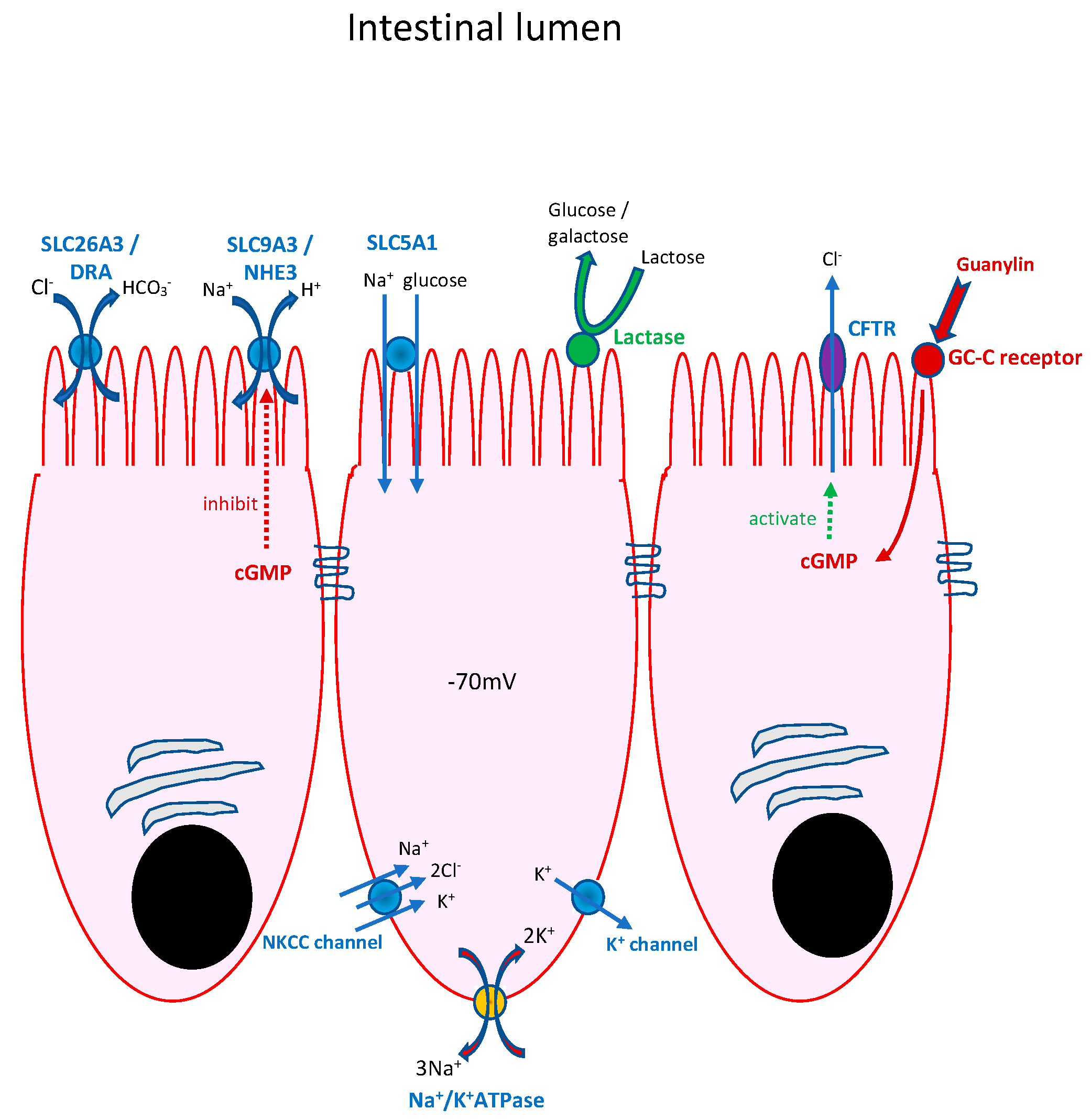
- Congenital chloride-losing diarrhoea (CCD)
- (b)
- Congenital sodium diarrhoea (CSD)
- (i)
- “Classical” CSD presents antenatally, similar to CCD, but is much rarer and is genetically heterogeneous, with syndromic and non-syndromic forms [9]. CSD arises as a result of inadequate functioning of the apical enterocyte membrane sodium proton antiporter 3 (NHE3), a Na+/H+ exchanger. Biallelic mutations in the gene SLC9A3 (seen in 40% of infants with CSD) can result in the absence or dysfunction of NHE3, with diarrhoea of antenatal onset presenting as polyhydramnios, dilated loops of bowel and premature delivery. As with CCD, there is failure to pass meconium, rapid postnatal dehydration and, in this instance, hyponatraemic acidosis with high stool sodium.
- (ii)
- “Syndromic” CSD comprises a group of infants with choanal atresia and recurrent punctate keratitis. Less commonly, bowel and anal atresias, polydactyly and cleft palate are seen. In contrast to “classical” CSD, small bowel biopsy demonstrates villus atrophy and epithelial tufting (“tufting enteropathy”). Diarrhoea arises as a result of the misregulation of the heterotrimeric epithelial sodium channel, which is critical for sodium reabsorption in the distal colon but is expressed throughout the gastrointestinal tract. It arises as a consequence of a mutation in the gene SPINT2, which encodes a serine peptidase inhibitor. The activity of ENaC depends upon its proteolytic activation by the matriptase–prostasin system. SPINT2 targets the serine proteases matriptase and prostasin. The loss of SPINT2 from the intestinal epithelium of the small and large intestines results in a decrease in the cell surface expression of matriptase and, as a result, a decrease in epithelial ENaC expression.
- (c)
- Congenital Glucose–Galactose malabsorption (GGM)
- (d)
- Congenital lactase deficiency
- (e)
- Sucrase-isomaltase deficiency
- (f)
- DGAT1 deficiency
- (g)
- Other defects in intestinal epithelial lipid re-esterification and export
2.2. Defects in Epithelial Structure
- (a)
- Microvillus inclusion disease
- (b)
- Tufting enteropathy
- (c)
- Trichohepatoenteric syndrome (Phenotypic or Syndromic Diarrhoea)
- (d)
- Syndromic protein-losing enteropathies (PLEs) with early-onset diarrhoea
- (i)
- Plasmalemma Vesicle-Associated Protein mutations
- (ii)
- Hennekam lymphangiectasia–lymphoedema Syndrome
- (iii)
- Congenital disorder of glycosylation type 1b (MPI-CDG)
- (iv)
- Hereditary PLE in association with CD55 deficiency (CHAPLE syndrome)
- (v)
- Infantile Systemic Hyalinosis
2.3. Enteroendocrine Cell Defects
- (a)
- Enteric anendocrinosis
- (b)
- Mitchell–Riley Syndrome
- (c)
- ARX deficiency
- (d)
- Proprotein convertase (PPC) 1/3 deficiency—enteric dysendocrinosis
- (e)
- ICR-related gastrointestinal endocrinopathy
2.4. Defects in Intestinal Immune Homeostasis
- (a)
- Epithelial barrier and response defects
- (i)
- TTC7A deficiency
- (b)
- Neutrophil granule dysfunction
- (c)
- Autoinflammatory and hyperinflammatory disorders
- (i)
- Mevalonate kinase deficiency (MKD)
- (ii)
- XLP2/XIAP
- (iii)
- Familial haemophagocytic lymphohistiocytosis type 5 (FHL-5)
- (d)
- Defects in T- and B-cell function
- (i)
- RIPK1 deficiency
- (e)
- Defects in regulatory T cells and IL-10 signalling
- (i)
- X-linked immune dysregulation, polyendocrinology enteropathy syndrome (IPEX)
- (ii)
- IL-2 receptor a chain defects (IL2RA encoding CD25)
- (iii)
- LRBA and CTLA4 deficiency
- (iv)
- IL-10 and IL10R deficiency
3. Conclusions
Author Contributions
Funding
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Avery, G.B.; Villavicencio, O.; Lilly, J.R.; Randolph, J.G. Intractable diarrhea in early infancy. Pediatrics 1968, 41, 712–722. [Google Scholar] [CrossRef] [PubMed]
- Canani, R.B.; Castaldo, G.; Bacchetta, R.; Martín, M.G.; Goulet, O. Congenital diarrhoeal disorders: Advances in this evolving web of inherited enteropathies. Nat. Rev. Gastroenterol. Hepatol. 2015, 12, 293–302. [Google Scholar] [CrossRef] [PubMed]
- Thiagarajah, J.R.; Kamin, D.S.; Acra, S.; Goldsmith, J.D.; Roland, J.T.; Lencer, W.I.; Muise, A.M.; Goldenring, J.R.; Avitzur, Y.; Martin, M.G. Advances in Evaluation of Chronic Diarrhea in Infants. Gastroenterology 2018, 154, 2045–2046. [Google Scholar] [CrossRef]
- Gupta, A.; Sanville, J.; Menz, T.; Warner, N.; Muise, A.M.; Fiedler, K.; Martin, M.G.; Padbury, J.; Phornphutkul, C.; Sanchez-Esteban, J.; et al. Application of Whole Exome Sequencing in Congenital Secretory Diarrhea Diagnosis. J. Pediatr. Gastroenterol. Nutr. 2019, 68, e106–e108. [Google Scholar] [CrossRef] [PubMed]
- Oz-Levi, D.; Olender, T.; Bar-Joseph, I.; Zhu, Y.; Marek-Yagel, D.; Barozzi, I.; Osterwalder, M.; Alkelai, A.; Ruozzo, E.K.; Han, Y.; et al. Noncoding deletions reveal a gene that is critical for intestinal function. Nature 2019, 571, 107–111. [Google Scholar] [CrossRef]
- Elkadri, A.A. Congenital Diarrheal Syndromes. Clin. Perinatol. 2020, 47, 87–104. [Google Scholar] [CrossRef]
- Uhlig, H.H.; Schwerd, T.; Koletzko, S.; Shah, N.; Kammermeier, J.; Elkadri, A.; Quahed, J.; Wilson, D.C.; Travis, S.P.; Turner, D.; et al. The diagnostic approach to monogenic very early onset inflammatory bowel disease. Gastroenterology 2014, 147, 990–993. [Google Scholar] [CrossRef]
- Overeem, A.W.; Posovszky, C.; Rings, E.H.M.M.; Giepmans, B.N.G.; van Ijzendoorn, S.C.D. The role of enterocyte defects in the pathogenesis of congenital diarrheal disorders. Dis. Model. Mech. 2016, 9, 1–12. [Google Scholar] [CrossRef]
- Wedenoja, S.; Höglund, P.; Holmberg, C. Review article: The clinical management of congenital chloride diarrhoea. Aliment. Pharmacol. Ther. 2010, 31, 477–485. [Google Scholar] [CrossRef]
- Janecke, A.R.; Heinz-Erian, P.; Müller, T. Congenital Sodium Diarrhea: A Form of Intractable Diarrhea, with a Link to Inflammatory Bowel Disease. J. Pediatr. Gastroenterol. Nutr. 2016, 63, 170–176. [Google Scholar] [CrossRef]
- Wright, E.M.; Turk, E.; Martín, M.G. Molecular basis for glucose-galactose malabsorption. Cell Biochem. Biophys. 2002, 36, 115–121. [Google Scholar] [CrossRef] [PubMed]
- Martín, M.G.; Turk, E.; Lostao, M.P.; Kerner, C.; Wright, E.M. Defects in Na+/glucose cotransporter (SGLT1) trafficking and function cause glucose-galactose malabsorption. Nat. Genet. 1996, 12, 216–220. [Google Scholar] [CrossRef]
- Robayo-Torres, C.C.; Nichols, B.L. Molecular differentiation of congenital lactase deficiency from adult-type hypolactasia. Nutr. Rev. 2007, 65, 95–98. [Google Scholar] [CrossRef]
- Haas, J.T.; Winter, H.S.; Lim, E.; Kirby, A.; Blumenstiel, B.; DeFelice, M.; Gabriel, S.; Jalas, C.; Branski, D.; Grueter, C.A.; et al. DGAT1 mutation is linked to a congenital diarrheal disorder. J. Clin. Investig. 2012, 122, 4680–4684. [Google Scholar] [CrossRef] [PubMed]
- van Rijn, J.M.; Ardy, R.C.; Kuloğlu, Z.; Härter, B.; van Haaften-Visser, D.Y.; van der Doef, H.P.J.; van Hoesel, M.; Kansu, A.; van Vug, A.H.M.; Thian, M.; et al. Intestinal Failure and Aberrant Lipid Metabolism in Patients with DGAT1 Deficiency. Gastroenterology 2018, 155, 130–143.e15. [Google Scholar] [CrossRef] [PubMed]
- Berriot-Varoqueaux, N.; Aggerbeck, L.P.; Samson-Bouma, M.; Wetterau, J.R. The role of the microsomal triglygeride transfer protein in abetalipoproteinemia. Annu. Rev. Nutr. 2000, 20, 663–697. [Google Scholar] [CrossRef] [PubMed]
- Peretti, N.; Sassolas, A.; Roy, C.C.; Deslandres, C.; Charcosset, M.; Castagnetti, J.; Pugnet-Chardon, L.; Moulin, P.; Labarge, S.; Bouthillier, L.; et al. Guidelines for the diagnosis and management of chylomicron retention disease based on a review of the literature and the experience of two centers. Orphanet J. Rare Dis. 2010, 5, 24. [Google Scholar] [CrossRef]
- Müller, T.; Hess, M.W.; Schiefermeier, N.; Pfaller, K.; Ebner, H.L.; Heinz-Erian, P.; Postingl, H.; Partsch, J.; Röllinghoff, B.; Köhler, H.; et al. MYO5B mutations cause microvillus inclusion disease and disrupt epithelial cell polarity. Nat. Genet. 2008, 40, 1163–1165. [Google Scholar] [CrossRef]
- Pathak, S.J.; Mueller, J.L.; Okamoto, K.; Das, B.; Hertecant, J.; Greenhalgh, L.; Cole, T.; Pinsk, V.; Yerushalmi, B.; Gurkan, O.E.; et al. EPCAM mutation update: Variants associated with congenital tufting enteropathy and Lynch syndrome. Hum. Mutat. 2019, 40, 142–161. [Google Scholar] [CrossRef]
- Salomon, J.; Goulet, O.; Canioni, D.; Brousse, N.; Lemale, J.; Tounian, P.; Coulomb, A.; Marinier, E.; Hugot, J.P.; Ruemmele, F.; et al. Genetic characterization of congenital tufting enteropathy: Epcam associated phenotype and involvement of SPINT2 in the syndromic form. Hum. Genet. 2014, 133, 299–310. [Google Scholar] [CrossRef]
- Lemale, J.; Coulomb, A.; Dubern, B.; Boudjemaa, S.; Viola, S.; Josset, P.; Tounian, P.; Girardet, J.P. Intractable diarrhea with tufting enteropathy: A favorable outcome is possible. J. Pediatr. Gastroenterol. Nutr. 2011, 52, 734–739. [Google Scholar] [CrossRef] [PubMed]
- Vély, F.; Barlogis, V.; Marinier, E.; Coste, M.-E.; Dubern, B.; Dugelay, E.; Lemale, J.; Martinez-Vinson, C.; Peretti, N.; Perry, A.; et al. Combined Immunodeficiency in Patients with Trichohepatoenteric Syndrome. Front. Immunol. 2018, 9, 1036. [Google Scholar] [CrossRef]
- Bourgeois, P.; Esteve, C.; Chaix, C.; Béroud, C.; Levy, N.; THES Clinical Consortium; Fabre, A.; Badens, C. Tricho-Hepato-Enteric Syndrome mutation update: Mutations spectrum of TTC37 and SKIV2L, clinical analysis and future prospects. Hum. Mutat. 2018, 39, 774–789. [Google Scholar] [CrossRef]
- Broekaert, I.J.; Becker, K.; Gottschalk, I.; Körber, F.; Dötsch, J.; Thiele, H.; Altmüller, J.; Nürnberg, P.; Hünseler, C.; Cirak, S. Mutations in plasmalemma vesicle-associated protein cause severe syndromic protein-losing enteropathy. J. Med. Genet. 2018, 55, 637–640. [Google Scholar] [CrossRef]
- Kurolap, A.; Eshach Adiv, O.; Gonzaga-Jauregui, C.; Dolnikov, K.; Mory, A.; Paperna, T.; Hershkovitz, T.; Overton, J.D.; Kaplan, M.; Glaser, F.; et al. Establishing the role of PLVAP in protein-losing enteropathy: A homozygous missense variant leads to an attenuated phenotype. J. Med. Genet. 2018, 55, 779–784. [Google Scholar] [CrossRef] [PubMed]
- Hennekam, R.C.; Geerdink, R.A.; Hamel, B.C.; Hennekam, F.A.; Kraus, P.; Rammeloo, J.A.; Tillemans, A.A. Autosomal recessive intestinal lymphangiectasia and lymphedema, with facial anomalies and mental retardation. Am. J. Med. Genet. 1989, 34, 593–600. [Google Scholar] [CrossRef] [PubMed]
- Pujol, F.; Hodgson, T.; Martinez-Corral, I.; Prats, A.-C.; Devenport, D.; Takeichi, M.; Genot, E.; Mäkinen, T.; Francis-West, P.; Garmy-Susini, B.; et al. Dachsous1-Fat4 Signaling Controls Endothelial Cell Polarization During Lymphatic Valve Morphogenesis-Brief Report. Arterioscler. Thromb. Vasc. Biol. 2017, 37, 1732–1735. [Google Scholar] [CrossRef]
- Connell, F.; Kalidas, K.; Ostergaard, P.; Brice, G.; Homfray, T.; Roberts, L.; Bunyan, D.J.; Mitton, S.; Mansour, S.; Mortimer, P.; et al. Linkage and sequence analysis indicate that CCBE1 is mutated in recessively inherited generalised lymphatic dysplasia. Hum. Genet. 2010, 127, 231–241. [Google Scholar] [CrossRef]
- Jaeken, J.; Matthijs, G.; Saudubray, J.M.; Dionisi-Vici, C.; Bertini, E.; de Lonlay, P.; Henri, H.; Carchon, H.; Schollen, E.; Van Schaftingen, E. Phosphomannose isomerase deficiency: A carbohydrate-deficient glycoprotein syndrome with hepatic-intestinal presentation. Am. J. Hum. Genet. 1998, 62, 1535–1539. [Google Scholar] [CrossRef]
- de Lonlay, P.; Seta, N.; Barrot, S.; Chabrol, B.; Drouin, V.; Gabriel, B.M.; Journel, H.; Kretz, M.; Laurent, J.; Le Merrer, M.; et al. A broad spectrum of clinical presentations in congenital disorders of glycosylation I: A series of 26 cases. J. Med. Genet. 2001, 38, 14–19. [Google Scholar] [CrossRef]
- Niehues, R.; Hasilik, M.; Alton, G.; Körner, C.; Schiebe-Sukumar, M.; Koch, H.G.; Zimmer, K.P.; Wu, R.; Harms, E.; Reiter, K.; et al. Carbohydrate-deficient glycoprotein syndrome type Ib. Phosphomannose isomerase deficiency and mannose therapy. J. Clin. Investig. 1998, 101, 1414–1420. [Google Scholar] [CrossRef]
- Ozen, A.; Comrie, W.A.; Ardy, R.C.; Domínguez Conde, C.; Dalgic, B.; Beser, Ö.F.; Morawski, A.R.; Karakoc-Aydiner, E.; Tutar, E.; Baris, S. CD55 Deficiency, Early-Onset Protein-Losing Enteropathy, and Thrombosis. N. Engl. J. Med. 2017, 377, 52–61. [Google Scholar] [CrossRef] [PubMed]
- Kurolap, A.; Eshach Adiv, O.; Hershkovitz, T.; Tabib, A.; Karbian, N.; Paperna, T.; Mory, A.; Vachyan, A.; Slijper, N.; Steinberg, R.; et al. Eculizumab is safe and effective as a long-term treatment for protein-losing enteropathy due to CD55 deficiency. J. Pediatr. Gastroenterol. Nutr. 2019, 68, 325–333. [Google Scholar] [CrossRef]
- Schussler, E.; Linkner, R.V.; Levitt, J.; Mehta, L.; Martignetti, J.A.; Oishi, K. Protein-losing enteropathy and joint contractures caused by a novel homozygous ANTXR2 mutation. Adv. Genom. Genet. 2018, 8, 17–21. [Google Scholar] [CrossRef] [PubMed]
- Cozma, C.; Hovakimyan, M.; Iurașcu, M.-I.; Makhseed, N.; Selim, L.A.; Alhashem, A.M.; Ben-Omran, T.; Mahmoud, I.G.; Al Menabawy, N.M.; Al-Mureikhi, M.; et al. Genetic, clinical and biochemical characterization of a large cohort of patients with hyaline fibromatosis syndrome. Orphanet J. Rare Dis. 2019, 14, 209–210. [Google Scholar] [CrossRef]
- Germán-Díaz, M.; Rodriguez-Gil, Y.; Cruz-Rojo, J.; Charbit-Henrion, F.; Cerf-Bensussan, N.; Manzanares-López Manzanares, J.; Moreno-Villares, J.M. A New Case of Congenital Malabsorptive Diarrhea and Diabetes Secondary to Mutant Neurogenin-3. Pediatrics 2017, 140, e20162210. [Google Scholar] [CrossRef] [PubMed]
- Terry, N.A.; Lee, R.A.; Walp, E.R.; Kaestner, K.H.; Lee May, C. Dysgenesis of enteroendocrine cells in Aristaless-Related Homeobox polyalanine expansion mutations. J. Pediatr. Gastroenterol. Nutr. 2015, 60, 192–199. [Google Scholar] [CrossRef]
- Jackson, R.S.; Creemers, J.W.M.; Farooqi, I.S.; Raffin-Sanson, M.-L.; Varro, A.; Dockray, G.J.; Holst, J.J.; Brubaker, P.L.; Corvol, P.; Polonsky, K.S.; et al. Small-intestinal dysfunction accompanies the complex endocrinopathy of human proprotein convertase 1 deficiency. J. Clin. Investig. 2003, 112, 1550–1560. [Google Scholar] [CrossRef]
- Uhlig, H.H. Monogenic diseases associated with intestinal inflammation: Implications for the understanding of inflammatory bowel disease. Gut 2013, 62, 1795–1805. [Google Scholar] [CrossRef]
- Jardine, S.; Dhingani, N.; Muise, A.M. TTC7A: Steward of Intestinal Health. Cell. Mol. Gastroenterol. Hepatol. 2019, 7, 555–570. [Google Scholar] [CrossRef]
- Jardine, S.; Anderson, S.; Babcock, S.; Leung, G.; Pan, J.; Dhingani, N.; Warner, N.; Guo, C.; Siddiqui, I.; Kotlarz, D.; et al. Drug Screen Identifies Leflunomide for Treatment of Inflammatory Bowel Diseases Caused by TTC7A Deficiency. Gastroenterology 2019, 158, 1000–1015. [Google Scholar] [CrossRef] [PubMed]
- Levy, M.; Arion, A.; Berrebi, D.; Cuisset, L.; Jeanne-Pasquier, C.; Bader-Meunier, B.; Jung, C. Severe early-onset colitis revealing mevalonate kinase deficiency. Pediatrics 2013, 132, e779–e783. [Google Scholar] [CrossRef] [PubMed]
- Kelsen, J.R.; Dawany, N.; Martinez, A.; Martinez, A.; Grochowski, C.M.; Maurer, K.; Rappaport, E.; Piccoli, D.A.; Baldassano, R.; Mamula, P.; et al. A de novo whole gene deletion of XIAP detected by exome sequencing analysis in very early onset inflammatory bowel disease: A case report. BMC Gastroenterol. 2015, 15, 160–165. [Google Scholar] [CrossRef] [PubMed]
- Pagel, J.; Beutel, K.; Lehmberg, K.; Koch, F.; Maul-Pavicic, A.; Rohlfs, A.-K.; Al-Jefri, A.; Beier, R.; Bomme Ousager, L.; Ehlert, K.; et al. Distinct mutations in STXBP2 are associated with variable clinical presentations in patients with familial hemophagocytic lymphohistiocytosis type 5 (FHL5). Blood 2012, 119, 6016–6024. [Google Scholar] [CrossRef]
- Agarwal, S.; Smereka, P.; Harpaz, N.; Cunningham-Rundles, C.; Mayer, L. Characterization of immunologic defects in patients with common variable immunodeficiency (CVID) with intestinal disease. Inflamm. Bowel Dis. 2011, 17, 251–259. [Google Scholar] [CrossRef]
- Barmettler, S.; Otani, I.M.; Minhas, J.; Abraham, R.S.; Chang, Y.; Dorsey, M.J.; Ballas, Z.K.; Bonilla, F.A.; Ochs, H.D.; Walter, J.E. Gastrointestinal Manifestations in X-linked Agammaglobulinemia. J. Clin. Immunol. 2017, 37, 287–294. [Google Scholar] [CrossRef]
- Levy, J.; Espanol-Boren, T.; Thomas, C.; Fischer, A.; Tovo, P.; Bordigoni, P.; Resnick, I.; Fasth, A.; Baer, M.; Gomez, L.; et al. Clinical spectrum of X-linked hyper-IgM syndrome. J. Pediatr. 1997, 131 Pt 1, 47–54. [Google Scholar] [CrossRef]
- Ohya, T.; Yanagimachi, M.; Iwasawa, K.; Umetsu, S.; Sogo, T.; Inui, A.; Fujisawa, T.; Ito, S. Childhood-onset inflammatory bowel diseases associated with mutation of Wiskott-Aldrich syndrome protein gene. World J. Gastroenterol. 2017, 23, 8544–8552. [Google Scholar] [CrossRef]
- Li, Y.; Führer, M.; Bahrami, E.; Socha, P.; Klaudel-Dreszler, M.; Bouzidi, A.; Liu, Y.; Lehle, A.S.; Magg, T.; Hollizeck, S. Human RIPK1 deficiency causes combined immunodeficiency and inflammatory bowel diseases. Proc. Natl. Acad. Sci. USA 2019, 116, 970–975. [Google Scholar] [CrossRef]
- Duclaux-Loras, R.; Charbit-Henrion, F.; Neven, B.; Nowak, J.; Collardeau-Frachon, S.; Malcus, C.; Ray, P.F.; Moshous, D.; Beltrand, J.; Goulet, O.; et al. Clinical Heterogeneity of Immune Dysregulation, Polyendocrinopathy, Enteropathy, X-Linked Syndrome: A French Multicenter Retrospective Study. Clin. Transl. Gastroenterol. 2018, 9, 201. [Google Scholar] [CrossRef]
- Azizi, G.; Yazdani, R.; Rae, W.; Abolhassani, H.; Rojas, M.; Aghamohammadi, A.; Anaya, J.M. Monogenic polyautoimmunity in primary immunodeficiency diseases. Autoimmun. Rev. 2018, 17, 1028–1039. [Google Scholar] [CrossRef] [PubMed]
- Zheng, C.; Huang, Y.; Hu, W.; Shi, J.; Ye, Z.; Qian, X.; Huang, Z.; Xue, A.; Wang, Y.; Lu, J.; et al. Phenotypic Characterization of Very Early-Onset Inflammatory Bowel Disease with Interleukin-10 Signaling Deficiency: Based on a Large Cohort Study. Inflamm. Bowel Dis. 2019, 25, 756–766. [Google Scholar] [CrossRef] [PubMed]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Defects in Digestion and Absorption of Nutrients or of Ionic Transport | Defects in Enterocyte Structure and Function | Defects in Enteroendocrine Cell Differentiation | Defects in Intestinal Immune Homeostasis |
|---|---|---|---|
|
|
|
|
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Köglmeier, J.; Lindley, K.J. Congenital Diarrhoeas and Enteropathies. Nutrients 2024, 16, 2971. https://doi.org/10.3390/nu16172971
Köglmeier J, Lindley KJ. Congenital Diarrhoeas and Enteropathies. Nutrients. 2024; 16(17):2971. https://doi.org/10.3390/nu16172971
Chicago/Turabian StyleKöglmeier, Jutta, and Keith James Lindley. 2024. "Congenital Diarrhoeas and Enteropathies" Nutrients 16, no. 17: 2971. https://doi.org/10.3390/nu16172971
APA StyleKöglmeier, J., & Lindley, K. J. (2024). Congenital Diarrhoeas and Enteropathies. Nutrients, 16(17), 2971. https://doi.org/10.3390/nu16172971