

Severity Ranking of Missense and Frameshift Genetic Variants in SCD1 by In Silico and In Vitro Functional Analysis



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Chemicals and Materials
2.2. Web-Based Tools for In Silico Analysis
2.3. Expression Plasmid Constructs and Mutagenesis
2.4. Cell Culture and Transfection
2.5. Cell Treatments
2.6. Preparation of Cell Lysates
2.7. GC-MS Analysis of Fatty Acid Profiles
2.8. Immunoblot Analysis
2.9. RNA Isolation, cDNA Synthesis
2.10. qPCR Analysis
2.11. Aggregation Analysis
2.12. Trypsin Sensitivity Assay
2.13. Statistical Analysis
3. Results
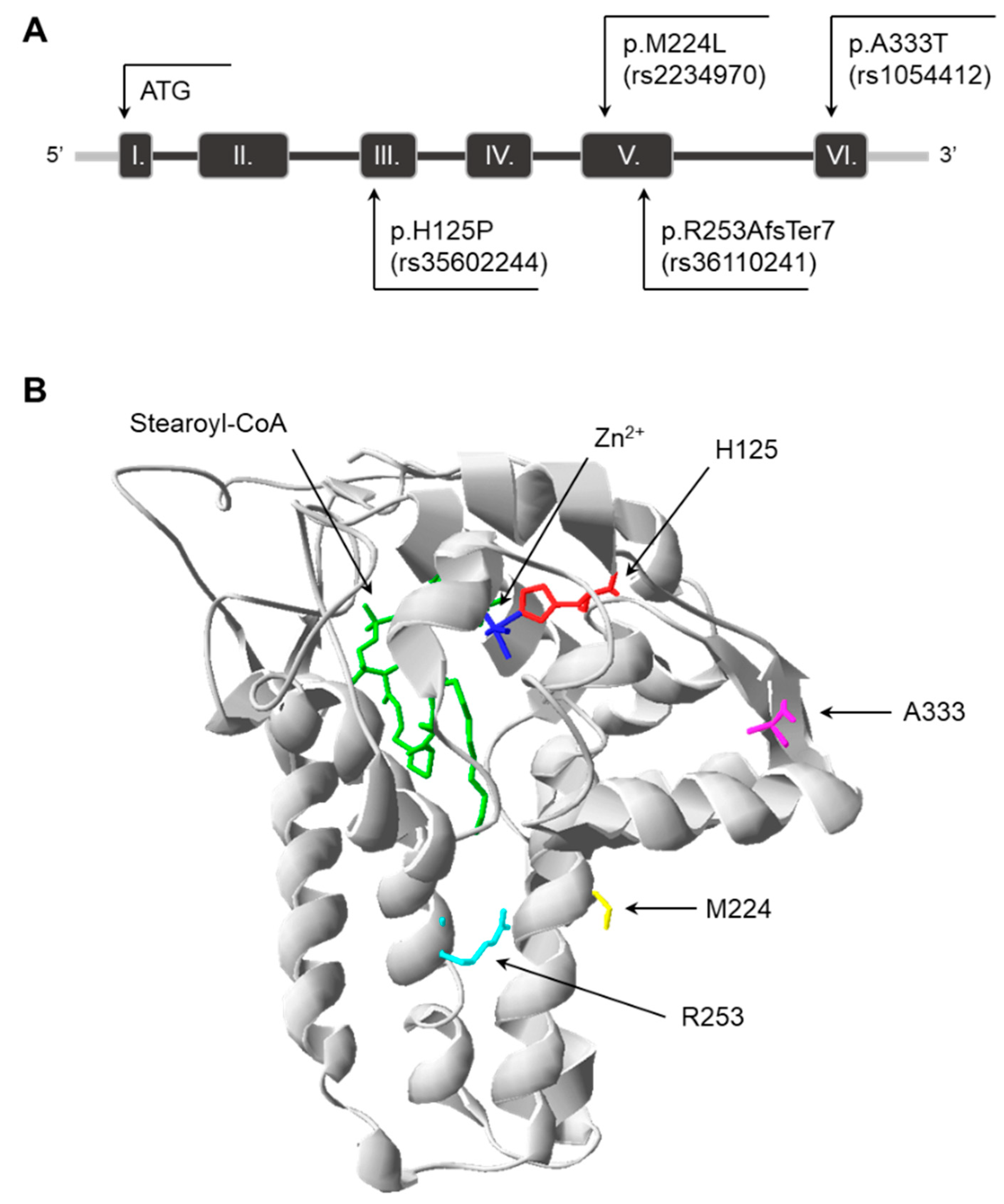
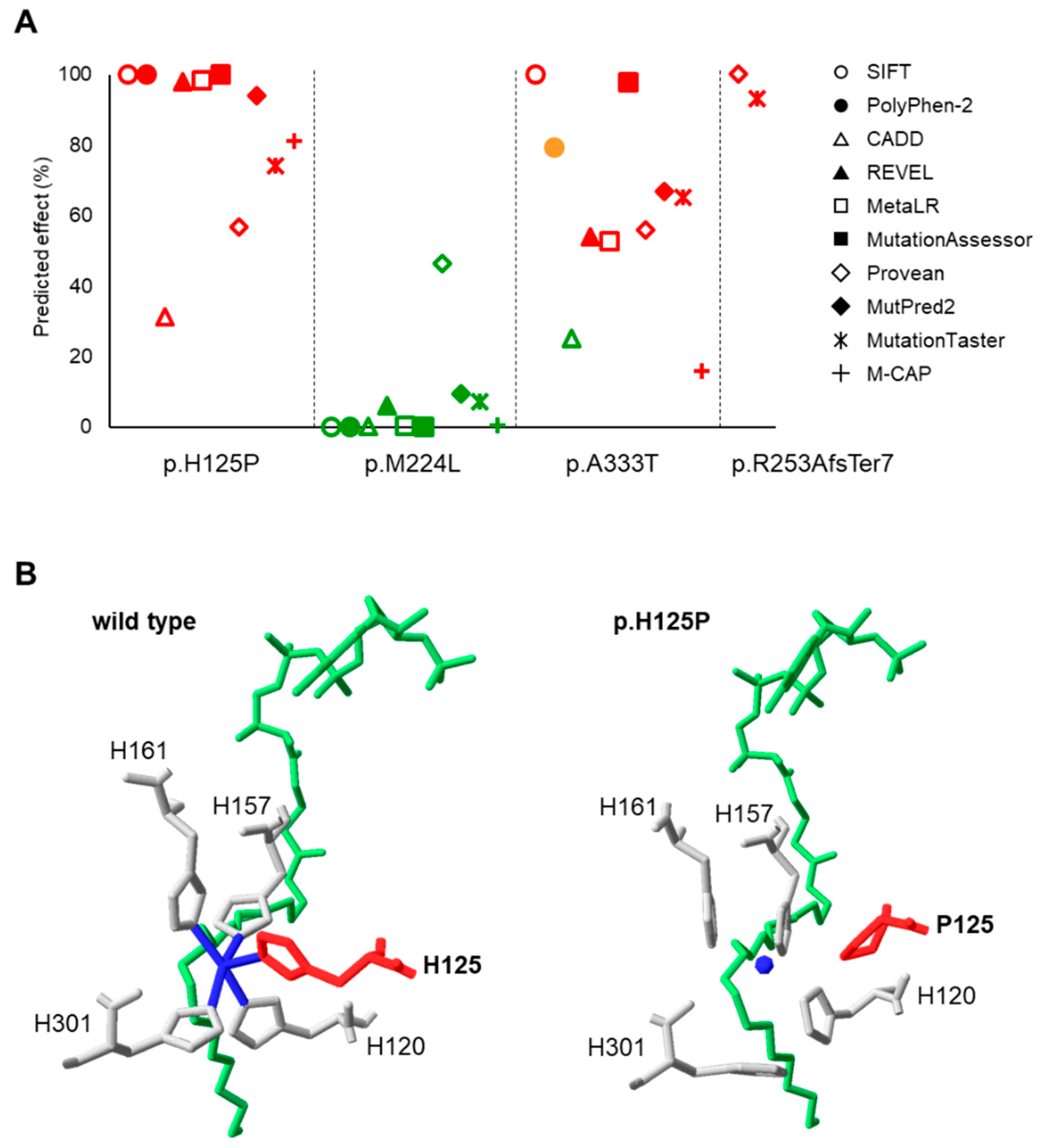
3.1. Predicted Impact of Natural Genetic Variants on SCD1
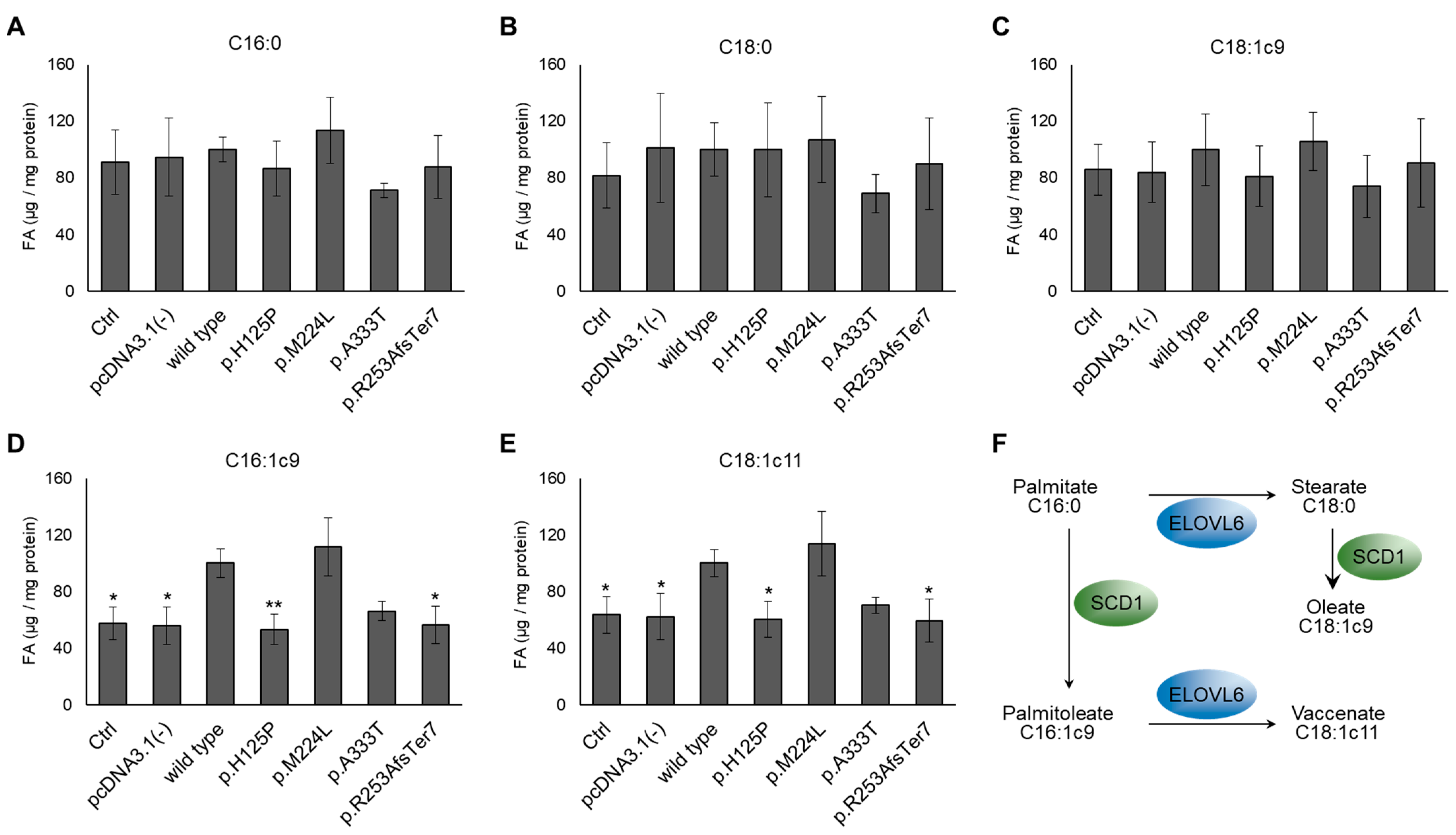
3.2. Altered Cellular Fatty Acid Profile of SCD1 Missense and Frameshift Variants
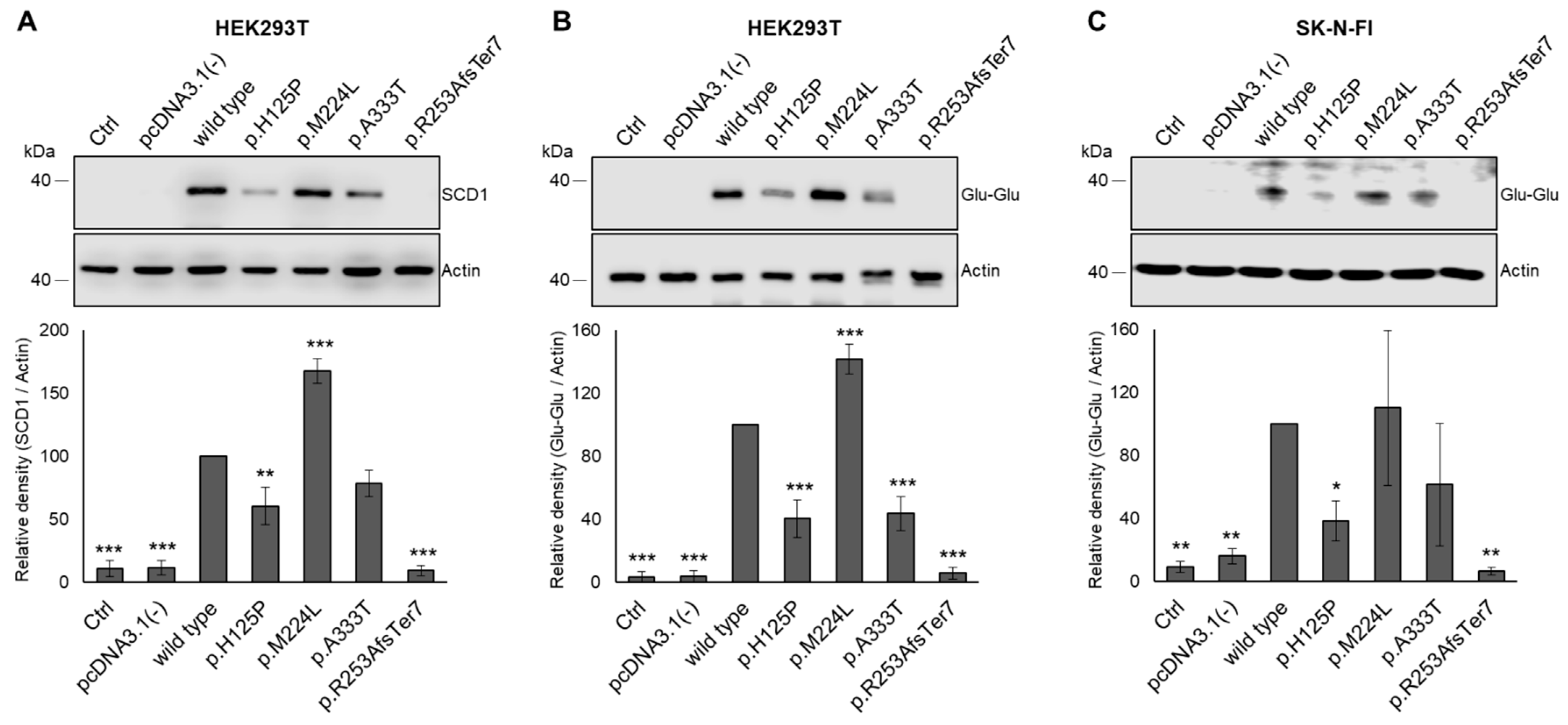
3.3. Modulating Effect of SCD1 Mutations on Intracellular Protein Levels
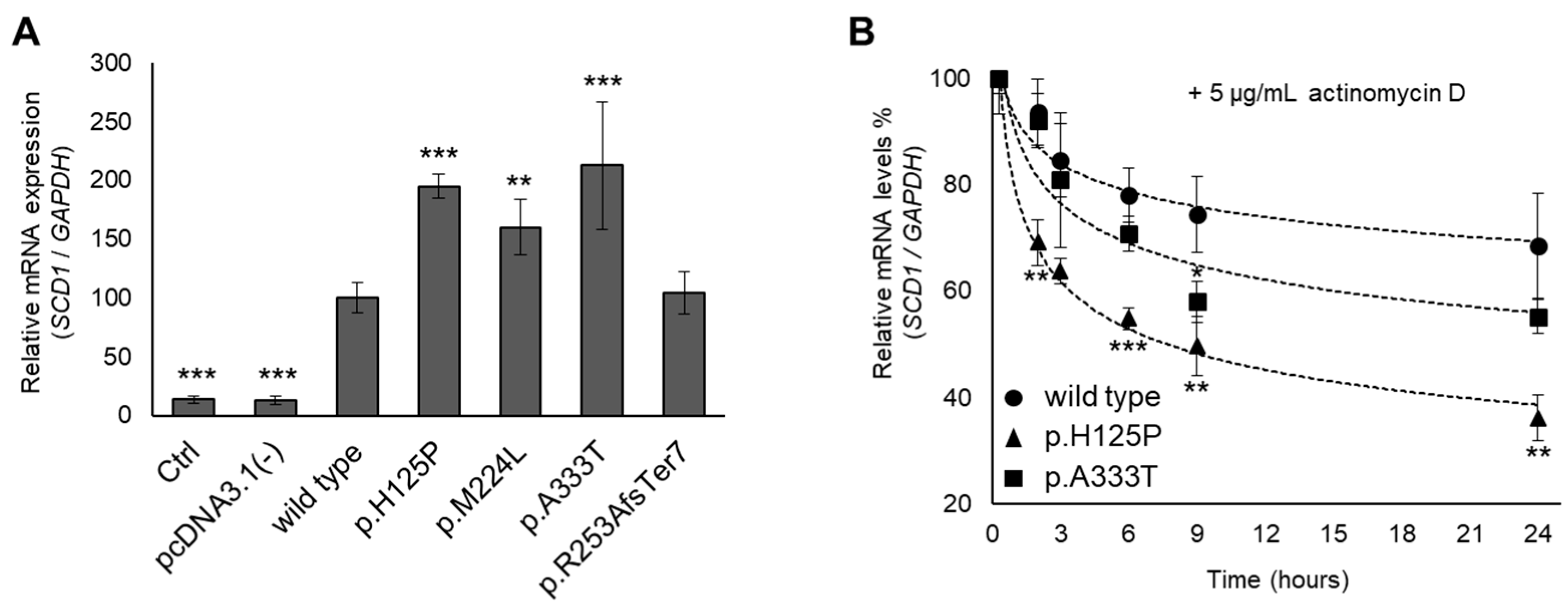
3.4. Altered mRNA Levels and Stability of p.H125P and p.A333T SCD1 Variants
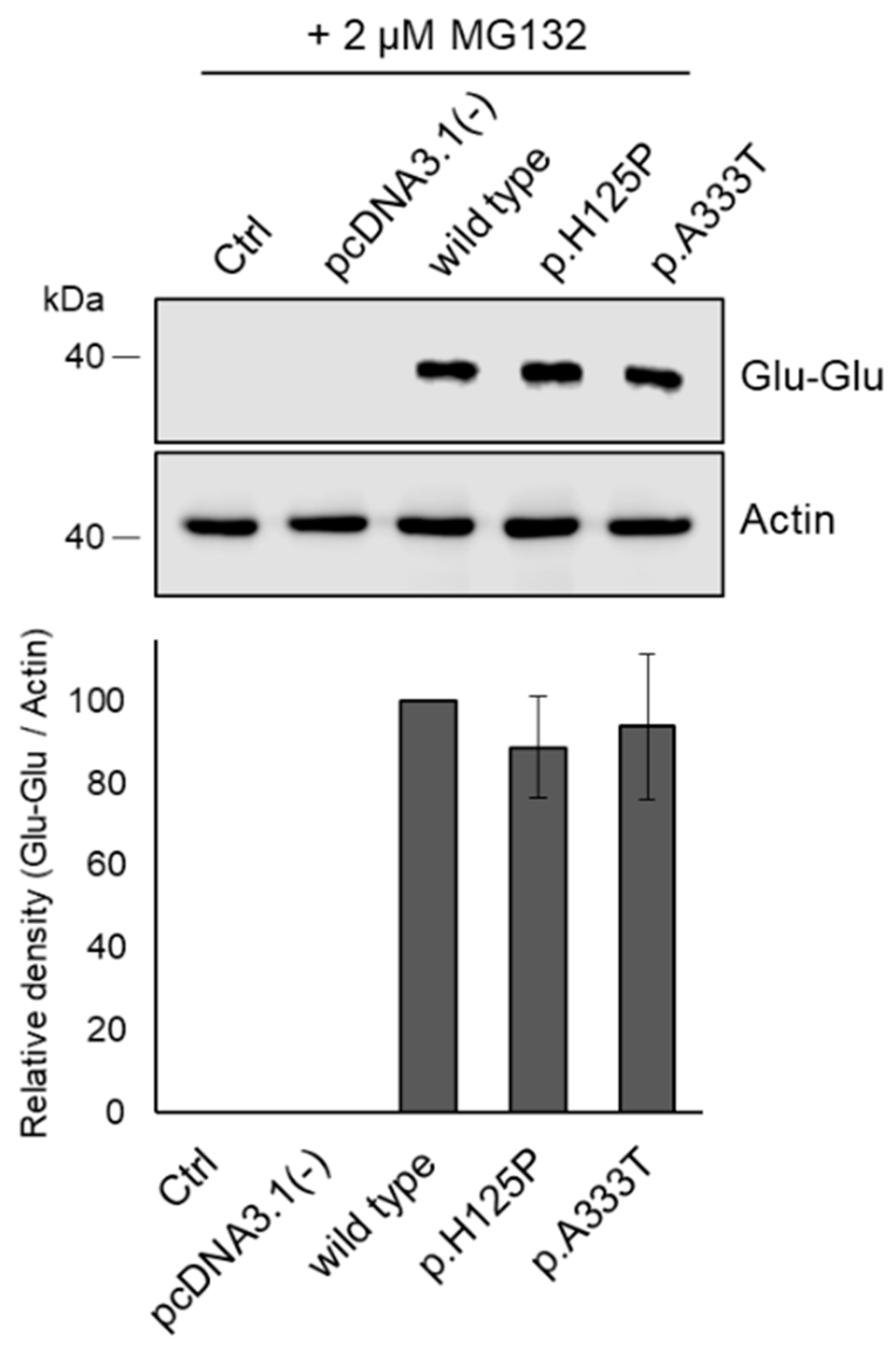
3.5. Proteasome Inhibitor Prevents Intracellular Degradation of p.H125P and p.A333T SCD1 Variants
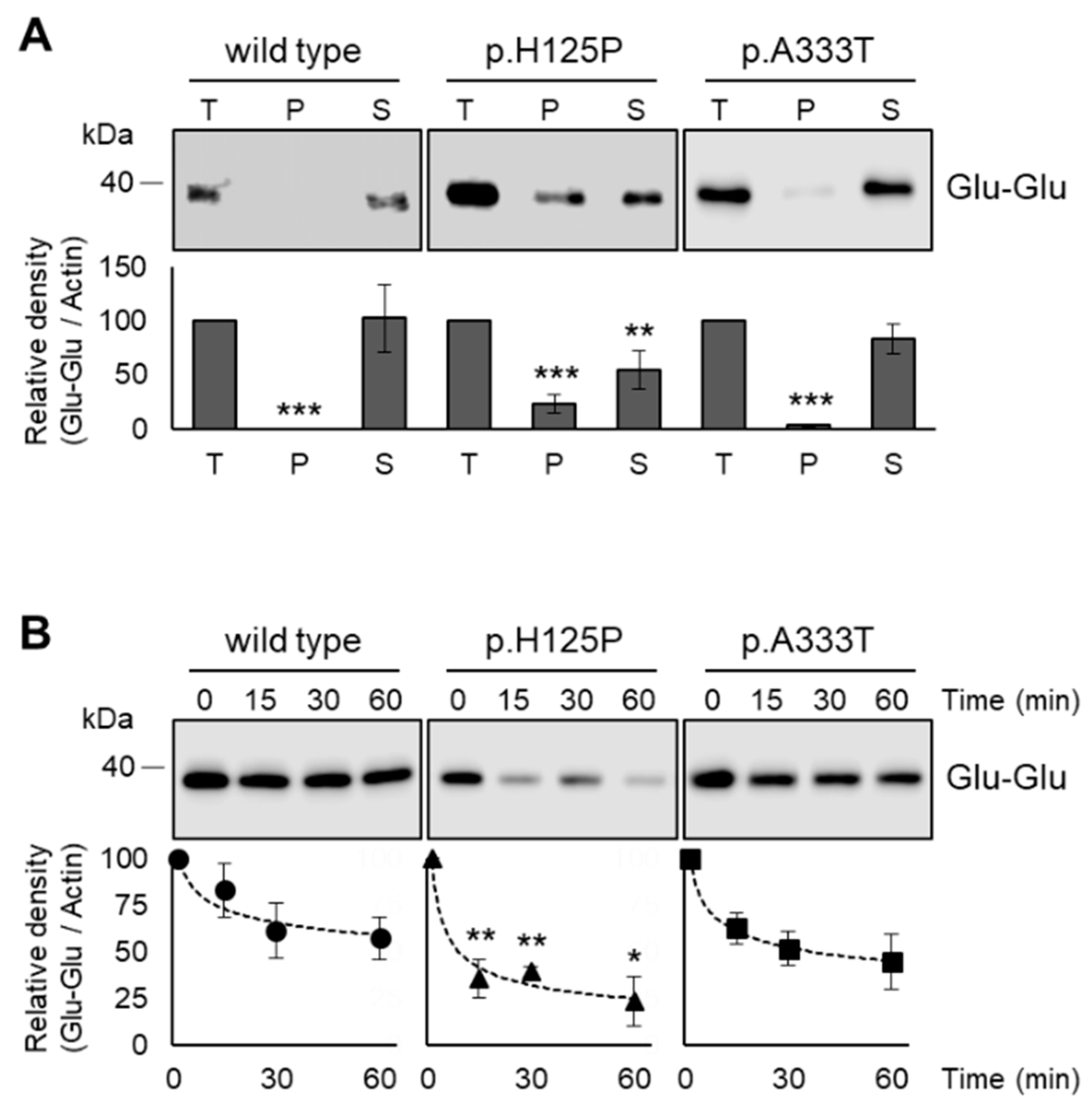
3.6. Increased Intracellular Aggregation and Protease Sensitivity of p.H125P and p.A333T SCD1 Variants
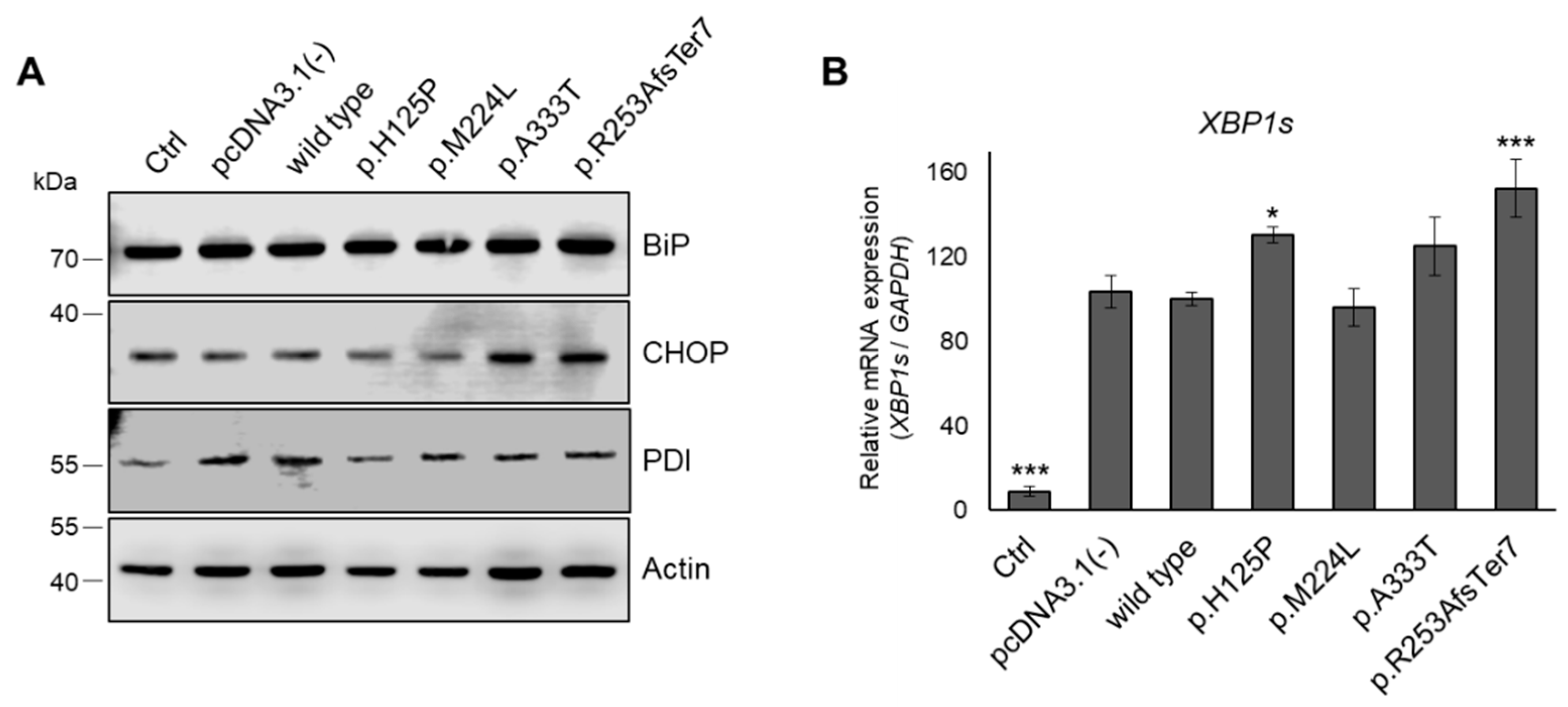
3.7. ER Stress in HEK293T Cells Expressing Genetic Variants of SCD1
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Shimabukuro, M.; Zhou, Y.T.; Levi, M.; Unger, R.H. Fatty acid-induced beta cell apoptosis: A link between obesity and diabetes. Proc. Natl. Acad. Sci. USA 1998, 95, 2498–2502. [Google Scholar] [CrossRef] [PubMed]
- Han, J.; Kaufman, R.J. The role of ER stress in lipid metabolism and lipotoxicity. J. Lipid Res. 2016, 57, 1329–1338. [Google Scholar] [CrossRef] [PubMed]
- Yazıcı, D.; Sezer, H. Insulin Resistance, Obesity and Lipotoxicity. Adv. Exp. Med. Biol. 2017, 960, 277–304. [Google Scholar] [CrossRef] [PubMed]
- Colvin, B.N.; Longtine, M.S.; Chen, B.; Costa, M.L.; Nelson, D.M. Oleate attenuates palmitate-induced endoplasmic reticulum stress and apoptosis in placental trophoblasts. Reproduction 2017, 153, 369–380. [Google Scholar] [CrossRef] [PubMed]
- Sarnyai, F.; Somogyi, A.; Gór-Nagy, Z.; Zámbó, V.; Szelényi, P.; Mátyási, J.; Simon-Szabó, L.; Kereszturi, É.; Tóth, B.; Csala, M. Effect of cis- and trans-Monounsaturated Fatty Acids on Palmitate Toxicity and on Palmitate-induced Accumulation of Ceramides and Diglycerides. Int. J. Mol. Sci. 2020, 21, 2626. [Google Scholar] [CrossRef]
- Maedler, K.; Spinas, G.A.; Dyntar, D.; Moritz, W.; Kaiser, N.; Donath, M.Y. Distinct effects of saturated and monounsaturated fatty acids on beta-cell turnover and function. Diabetes 2001, 50, 69–76. [Google Scholar] [CrossRef]
- Chen, X.; Li, L.; Liu, X.; Luo, R.; Liao, G.; Li, L.; Liu, J.; Cheng, J.; Lu, Y.; Chen, Y. Oleic acid protects saturated fatty acid mediated lipotoxicity in hepatocytes and rat of non-alcoholic steatohepatitis. Life Sci. 2018, 203, 291–304. [Google Scholar] [CrossRef]
- Bogie, J.F.J.; Haidar, M.; Kooij, G.; Hendriks, J.J.A. Fatty acid metabolism in the progression and resolution of CNS disorders. Adv. Drug Deliv. Rev. 2020, 159, 198–213. [Google Scholar] [CrossRef]
- Turolo, S.; Edefonti, A.; Syren, M.L.; Marangoni, F.; Morello, W.; Agostoni, C.; Montini, G. Fatty Acids in Nephrotic Syndrome and Chronic Kidney Disease. J. Ren. Nutr. 2018, 28, 145–155. [Google Scholar] [CrossRef]
- Yan, D.; Ye, S.; He, Y.; Wang, S.; Xiao, Y.; Xiang, X.; Deng, M.; Luo, W.; Chen, X.; Wang, X. Fatty acids and lipid mediators in inflammatory bowel disease: From mechanism to treatment. Front. Immunol. 2023, 14, 1286667. [Google Scholar] [CrossRef]
- Ipsen, D.H.; Lykkesfeldt, J.; Tveden-Nyborg, P. Molecular mechanisms of hepatic lipid accumulation in non-alcoholic fatty liver disease. Cell. Mol. Life Sci. 2018, 75, 3313–3327. [Google Scholar] [CrossRef]
- Perna, M.; Hewlings, S. Saturated Fatty Acid Chain Length and Risk of Cardiovascular Disease: A Systematic Review. Nutrients 2022, 15, 30. [Google Scholar] [CrossRef]
- Fretts, A.M.; Jensen, P.N.; Hoofnagle, A.N.; McKnight, B.; Howard, B.V.; Umans, J.; Sitlani, C.M.; Siscovick, D.S.; King, I.B.; Djousse, L.; et al. Plasma ceramides containing saturated fatty acids are associated with risk of type 2 diabetes. J. Lipid Res. 2021, 62, 100119. [Google Scholar] [CrossRef] [PubMed]
- Harrison, S.; Couture, P.; Lamarche, B. Diet Quality, Saturated Fat and Metabolic Syndrome. Nutrients 2020, 12, 3232. [Google Scholar] [CrossRef] [PubMed]
- Jin, Z.; Chai, Y.D.; Hu, S. Fatty Acid Metabolism and Cancer. Adv. Exp. Med. Biol. 2021, 1280, 231–241. [Google Scholar] [CrossRef] [PubMed]
- Enoch, H.G.; Catalá, A.; Strittmatter, P. Mechanism of rat liver microsomal stearyl-CoA desaturase. Studies of the substrate specificity, enzyme-substrate interactions, and the function of lipid. J. Biol. Chem. 1976, 251, 5095–5103. [Google Scholar] [CrossRef]
- Zambo, V.; Simon-Szabo, L.; Sarnyai, F.; Matyasi, J.; Gor-Nagy, Z.; Somogyi, A.; Szelenyi, P.; Kereszturi, E.; Toth, B.; Csala, M. Investigation of the putative rate-limiting role of electron transfer in fatty acid desaturation using transfected HEK293T cells. FEBS Lett 2020, 594, 530–539. [Google Scholar] [CrossRef]
- Mauvoisin, D.; Mounier, C. Hormonal and nutritional regulation of SCD1 gene expression. Biochimie 2011, 93, 78–86. [Google Scholar] [CrossRef]
- Zulkifli, R.M.; Parr, T.; Salter, A.M.; Brameld, J.M. Regulation of ovine and porcine stearoyl coenzyme A desaturase gene promoters by fatty acids and sterols. J. Anim. Sci. 2010, 88, 2565–2575. [Google Scholar] [CrossRef]
- Yao, D.W.; Luo, J.; He, Q.Y.; Li, J.; Wang, H.; Shi, H.B.; Xu, H.F.; Wang, M.; Loor, J.J. Characterization of the liver X receptor-dependent regulatory mechanism of goat stearoyl-coenzyme A desaturase 1 gene by linoleic acid. J. Dairy Sci. 2016, 99, 3945–3957. [Google Scholar] [CrossRef]
- Zhang, L.; Ge, L.; Tran, T.; Stenn, K.; Prouty, S.M. Isolation and characterization of the human stearoyl-CoA desaturase gene promoter: Requirement of a conserved CCAAT cis-element. Biochem. J. 2001, 357, 183–193. [Google Scholar] [CrossRef] [PubMed]
- Kato, H.; Sakaki, K.; Mihara, K. Ubiquitin-proteasome-dependent degradation of mammalian ER stearoyl-CoA desaturase. J. Cell Sci. 2006, 119, 2342–2353. [Google Scholar] [CrossRef] [PubMed]
- Tibori, K.; Orosz, G.; Zámbó, V.; Szelényi, P.; Sarnyai, F.; Tamási, V.; Rónai, Z.; Mátyási, J.; Tóth, B.; Csala, M.; et al. Molecular Mechanisms Underlying the Elevated Expression of a Potentially Type 2 Diabetes Mellitus Associated SCD1 Variant. Int. J. Mol. Sci. 2022, 23, 6221. [Google Scholar] [CrossRef] [PubMed]
- Tibori, K.; Zámbó, V.; Orosz, G.; Szelényi, P.; Sarnyai, F.; Tamási, V.; Rónai, Z.; Csala, M.; Kereszturi, É. Allele-specific effect of various dietary fatty acids and ETS1 transcription factor on SCD1 expression. Sci. Rep. 2024, 14, 177. [Google Scholar] [CrossRef]
- Liu, Z.; Yin, X.; Mai, H.; Li, G.; Lin, Z.; Jie, W.; Li, K.; Zhou, H.; Wei, S.; Hu, L.; et al. SCD rs41290540 single-nucleotide polymorphism modifies miR-498 binding and is associated with a decreased risk of coronary artery disease. Mol. Genet. Genom. Med. 2020, 8, e1136. [Google Scholar] [CrossRef]
- Pan, G.; Cavalli, M.; Wadelius, C. Polymorphisms rs55710213 and rs56334587 regulate SCD1 expression by modulating HNF4A binding. Biochim. Et Biophys. Acta. Gene Regul. Mech. 2021, 1864, 194724. [Google Scholar] [CrossRef]
- Warensjö, E.; Ingelsson, E.; Lundmark, P.; Lannfelt, L.; Syvänen, A.C.; Vessby, B.; Risérus, U. Polymorphisms in the SCD1 gene: Associations with body fat distribution and insulin sensitivity. Obes. (Silver Spring Md.) 2007, 15, 1732–1740. [Google Scholar] [CrossRef]
- Martín-Núñez, G.M.; Cabrera-Mulero, R.; Rojo-Martínez, G.; Gómez-Zumaquero, J.M.; Chaves, F.J.; de Marco, G.; Soriguer, F.; Castaño, L.; Morcillo, S. Polymorphisms in the SCD1 gene are associated with indices of stearoyl CoA desaturase activity and obesity: A prospective study. Mol. Nutr. Food Res. 2013, 57, 2177–2184. [Google Scholar] [CrossRef]
- Stryjecki, C.; Roke, K.; Clarke, S.; Nielsen, D.; Badawi, A.; El-Sohemy, A.; Ma, D.W.; Mutch, D.M. Enzymatic activity and genetic variation in SCD1 modulate the relationship between fatty acids and inflammation. Mol. Genet. Metab. 2012, 105, 421–427. [Google Scholar] [CrossRef]
- Ng, P.C.; Henikoff, S. Predicting deleterious amino acid substitutions. Genome Res. 2001, 11, 863–874. [Google Scholar] [CrossRef]
- Adzhubei, I.A.; Schmidt, S.; Peshkin, L.; Ramensky, V.E.; Gerasimova, A.; Bork, P.; Kondrashov, A.S.; Sunyaev, S.R. A method and server for predicting damaging missense mutations. Nat. Methods 2010, 7, 248–249. [Google Scholar] [CrossRef] [PubMed]
- Schubach, M.; Maass, T.; Nazaretyan, L.; Röner, S.; Kircher, M. CADD v1.7: Using protein language models, regulatory CNNs and other nucleotide-level scores to improve genome-wide variant predictions. Nucleic Acids Res. 2024, 52, D1143–D1154. [Google Scholar] [CrossRef] [PubMed]
- Ioannidis, N.M.; Rothstein, J.H.; Pejaver, V.; Middha, S.; McDonnell, S.K.; Baheti, S.; Musolf, A.; Li, Q.; Holzinger, E.; Karyadi, D.; et al. REVEL: An Ensemble Method for Predicting the Pathogenicity of Rare Missense Variants. Am. J. Hum. Genet. 2016, 99, 877–885. [Google Scholar] [CrossRef] [PubMed]
- Dong, C.; Wei, P.; Jian, X.; Gibbs, R.; Boerwinkle, E.; Wang, K.; Liu, X. Comparison and integration of deleteriousness prediction methods for nonsynonymous SNVs in whole exome sequencing studies. Hum. Mol. Genet. 2015, 24, 2125–2137. [Google Scholar] [CrossRef] [PubMed]
- Reva, B.; Antipin, Y.; Sander, C. Predicting the functional impact of protein mutations: Application to cancer genomics. Nucleic Acids Res. 2011, 39, e118. [Google Scholar] [CrossRef]
- Choi, Y.; Chan, A.P. PROVEAN web server: A tool to predict the functional effect of amino acid substitutions and indels. Bioinformatics 2015, 31, 2745–2747. [Google Scholar] [CrossRef]
- Pejaver, V.; Urresti, J.; Lugo-Martinez, J.; Pagel, K.A.; Lin, G.N.; Nam, H.J.; Mort, M.; Cooper, D.N.; Sebat, J.; Iakoucheva, L.M.; et al. Inferring the molecular and phenotypic impact of amino acid variants with MutPred2. Nat. Commun. 2020, 11, 5918. [Google Scholar] [CrossRef]
- Schwarz, J.M.; Cooper, D.N.; Schuelke, M.; Seelow, D. MutationTaster2: Mutation prediction for the deep-sequencing age. Nat. Methods 2014, 11, 361–362. [Google Scholar] [CrossRef]
- Jagadeesh, K.A.; Wenger, A.M.; Berger, M.J.; Guturu, H.; Stenson, P.D.; Cooper, D.N.; Bernstein, J.A.; Bejerano, G. M-CAP eliminates a majority of variants of uncertain significance in clinical exomes at high sensitivity. Nat. Genet. 2016, 48, 1581–1586. [Google Scholar] [CrossRef]
- Wang, H.; Klein, M.G.; Zou, H.; Lane, W.; Snell, G.; Levin, I.; Li, K.; Sang, B.C. Crystal structure of human stearoyl-coenzyme A desaturase in complex with substrate. Nat. Struct. Mol. Biol. 2015, 22, 581–585. [Google Scholar] [CrossRef]
- Yang, J.; Zhang, Y. I-TASSER server: New development for protein structure and function predictions. Nucleic Acids Res. 2015, 43, W174–W181. [Google Scholar] [CrossRef] [PubMed]
- Lepage, G.; Roy, C.C. Direct transesterification of all classes of lipids in a one-step reaction. J. Lipid Res. 1986, 27, 114–120. [Google Scholar] [CrossRef] [PubMed]
- Ecker, J.; Scherer, M.; Schmitz, G.; Liebisch, G. A rapid GC-MS method for quantification of positional and geometric isomers of fatty acid methyl esters. J. Chromatogr. B Anal. Technol. Biomed. Life Sci. 2012, 897, 98–104. [Google Scholar] [CrossRef] [PubMed]
- Kálmán, F.S.; Lizák, B.; Nagy, S.K.; Mészáros, T.; Zámbó, V.; Mandl, J.; Csala, M.; Kereszturi, E. Natural mutations lead to enhanced proteasomal degradation of human Ncb5or, a novel flavoheme reductase. Biochimie 2013, 95, 1403–1410. [Google Scholar] [CrossRef] [PubMed]
- Marciniak, S.J.; Ron, D. Endoplasmic reticulum stress signaling in disease. Physiol. Rev. 2006, 86, 1133–1149. [Google Scholar] [CrossRef]
- Rudkowska, I.; Julien, P.; Couture, P.; Lemieux, S.; Tchernof, A.; Barbier, O.; Vohl, M.C. Cardiometabolic risk factors are influenced by Stearoyl-CoA Desaturase (SCD) -1 gene polymorphisms and n-3 polyunsaturated fatty acid supplementation. Mol. Nutr. Food Res. 2014, 58, 1079–1086. [Google Scholar] [CrossRef]
- Gong, J.; Campos, H.; McGarvey, S.; Wu, Z.; Goldberg, R.; Baylin, A. Genetic variation in stearoyl-CoA desaturase 1 is associated with metabolic syndrome prevalence in Costa Rican adults. J. Nutr. 2011, 141, 2211–2218. [Google Scholar] [CrossRef]
- Staňková, B.; Macášek, J.; Zeman, M.; Vecka, M.; Tvrzická, E.; Jáchymová, M.; Slabý, A.; Žák, A. Polymorphisms rs2167444 and rs508384 in the SCD1 Gene Are Linked with High ApoB-48 Levels and Adverse Profile of Cardiometabolic Risk Factors. Folia Biol. 2019, 65, 159–169. [Google Scholar] [CrossRef]
- Guo, Y.; Xiong, Z.; Su, M.; Huang, L.; Liao, J.; Xiao, H.; Huang, X.; Xiong, Z. Positive association of SCD1 genetic variation and metabolic syndrome in dialysis patients in China. Pers. Med. 2020, 17, 111–119. [Google Scholar] [CrossRef]
- Shameer, K.; Tripathi, L.P.; Kalari, K.R.; Dudley, J.T.; Sowdhamini, R. Interpreting functional effects of coding variants: Challenges in proteome-scale prediction, annotation and assessment. Brief. Bioinform. 2016, 17, 841–862. [Google Scholar] [CrossRef]
- Rodrigues, C.; Santos-Silva, A.; Costa, E.; Bronze-da-Rocha, E. Performance of In Silico Tools for the Evaluation of UGT1A1 Missense Variants. Hum. Mutat. 2015, 36, 1215–1225. [Google Scholar] [CrossRef] [PubMed]
- Ding, X.; Singh, P.; Schimenti, K.; Tran, T.N.; Fragoza, R.; Hardy, J.; Orwig, K.E.; Olszewska, M.; Kurpisz, M.K.; Yatsenko, A.N.; et al. In vivo versus in silico assessment of potentially pathogenic missense variants in human reproductive genes. Proc. Natl. Acad. Sci. USA 2023, 120, e2219925120. [Google Scholar] [CrossRef] [PubMed]
- Tabet, D.; Parikh, V.; Mali, P.; Roth, F.P.; Claussnitzer, M. Scalable Functional Assays for the Interpretation of Human Genetic Variation. Annu. Rev. Genet. 2022, 56, 441–465. [Google Scholar] [CrossRef] [PubMed]
- Teigen, M.; Ølnes, Å.S.; Bjune, K.; Leren, T.P.; Bogsrud, M.P.; Strøm, T.B. Functional characterization of missense variants affecting the extracellular domains of ABCA1 using a fluorescence-based assay. J. Lipid Res. 2024, 65, 100482. [Google Scholar] [CrossRef] [PubMed]
- Choi, H.; Andersen, J.P.; Molday, R.S. Expression and functional characterization of missense mutations in ATP8A2 linked to severe neurological disorders. Hum. Mutat. 2019, 40, 2353–2364. [Google Scholar] [CrossRef] [PubMed]
- Kereszturi, E.; Szmola, R.; Kukor, Z.; Simon, P.; Weiss, F.U.; Lerch, M.M.; Sahin-Tóth, M. Hereditary pancreatitis caused by mutation-induced misfolding of human cationic trypsinogen: A novel disease mechanism. Hum. Mutat. 2009, 30, 575–582. [Google Scholar] [CrossRef]
- Talmud, P.J.; Humphries, S.E. Gene:environment interaction in lipid metabolism and effect on coronary heart disease risk. Curr. Opin. Lipidol. 2002, 13, 149–154. [Google Scholar] [CrossRef]
- Cole, C.B.; Nikpay, M.; McPherson, R. Gene-environment interaction in dyslipidemia. Curr. Opin. Lipidol. 2015, 26, 133–138. [Google Scholar] [CrossRef]
- Ordovás, J.M.; Robertson, R.; Cléirigh, E.N. Gene-gene and gene-environment interactions defining lipid-related traits. Curr. Opin. Lipidol. 2011, 22, 129–136. [Google Scholar] [CrossRef]

Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Susán, H.K.; Orosz, G.; Zámbó, V.; Csala, M.; Kereszturi, É. Severity Ranking of Missense and Frameshift Genetic Variants in SCD1 by In Silico and In Vitro Functional Analysis. Nutrients 2024, 16, 3259. https://doi.org/10.3390/nu16193259
Susán HK, Orosz G, Zámbó V, Csala M, Kereszturi É. Severity Ranking of Missense and Frameshift Genetic Variants in SCD1 by In Silico and In Vitro Functional Analysis. Nutrients. 2024; 16(19):3259. https://doi.org/10.3390/nu16193259
Chicago/Turabian StyleSusán, Hanna K., Gabriella Orosz, Veronika Zámbó, Miklós Csala, and Éva Kereszturi. 2024. "Severity Ranking of Missense and Frameshift Genetic Variants in SCD1 by In Silico and In Vitro Functional Analysis" Nutrients 16, no. 19: 3259. https://doi.org/10.3390/nu16193259