
Oral Trehalose Intake Modulates the Microbiota–Gut–Brain Axis and Is Neuroprotective in a Synucleinopathy Mouse Model

 , , ,
, , ,  and
and
Abstract
:1. Introduction
2. Materials and Methods
2.1. Animals
2.2. Groups and Treatments
2.3. In Vivo Tests
2.3.1. Open Field Test
2.3.2. Stool Harvest for Microbiota Profiling
2.3.3. Nesting Test
2.4. Microbiota Analysis
2.4.1. DNA Extraction
2.4.2. 16S rRNA Gene Sequencing and Sequence Analysis
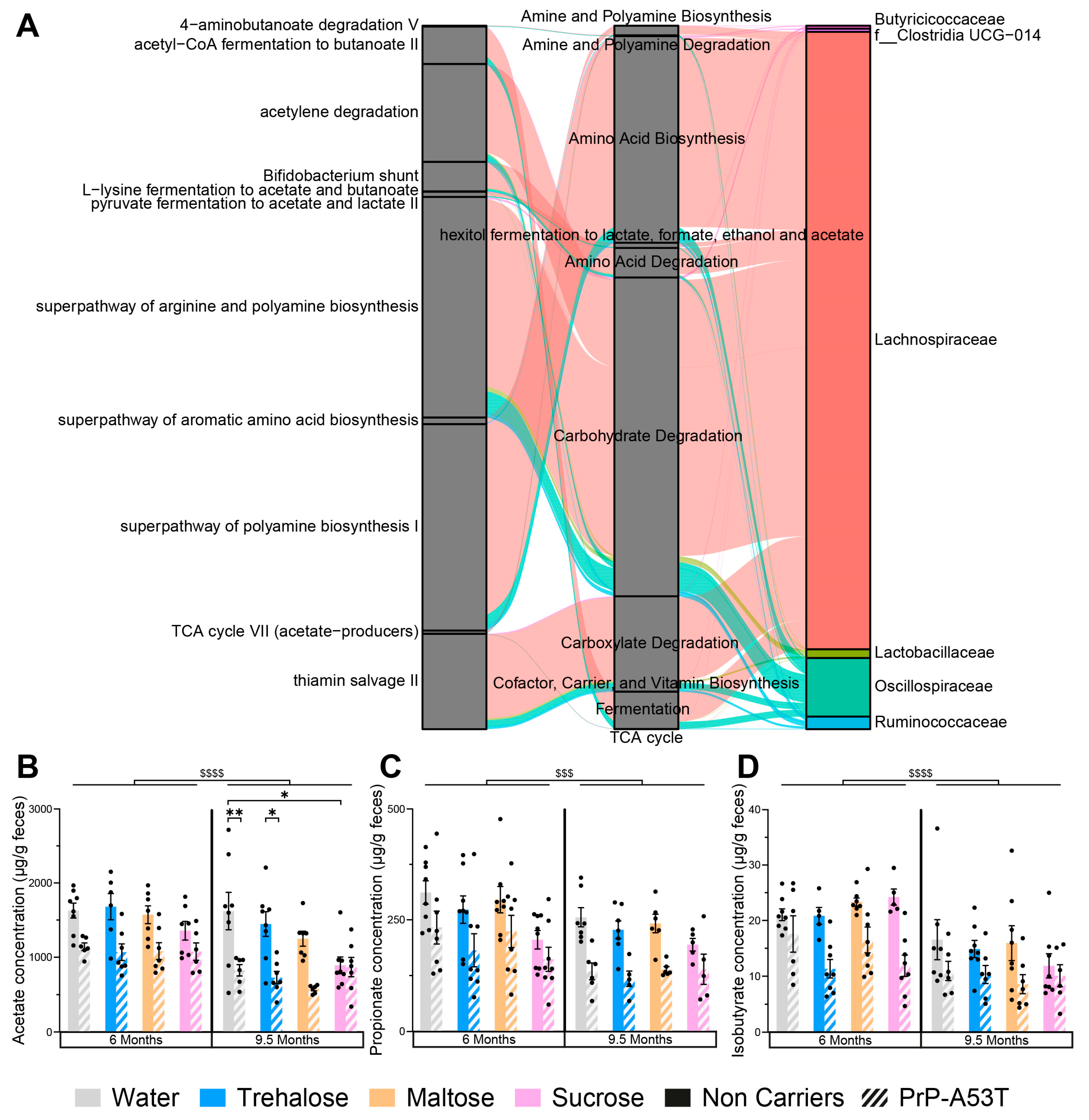
2.5. Short Chain Fatty Acids Analysis
2.6. Tissue Preparation
2.7. Immunofluorescence Analysis
2.8. Statistical Analysis
3. Results
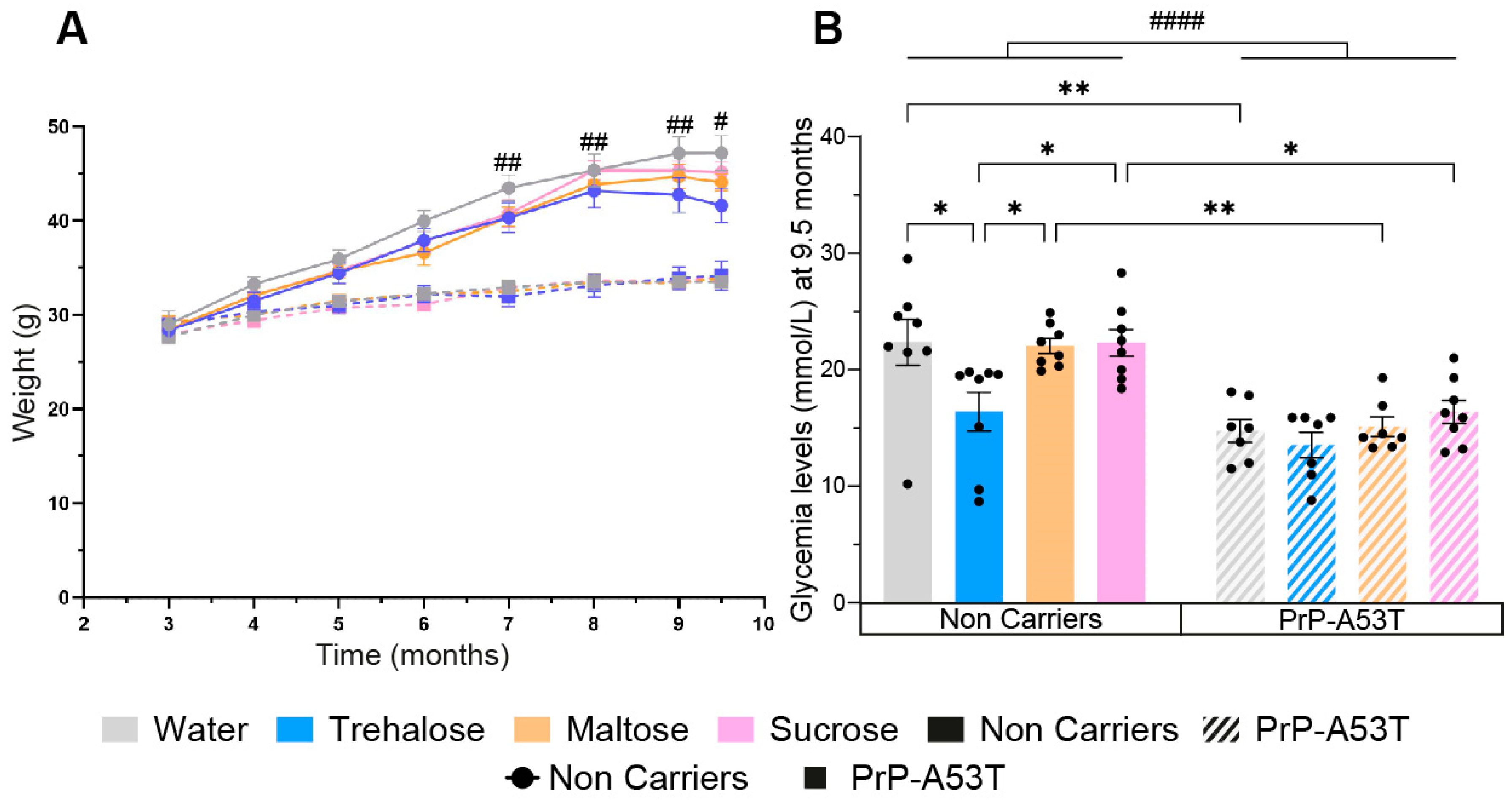
3.1. Effect of Genotype and Sugar-Enriched Diet on Mouse Weights and Glycemia
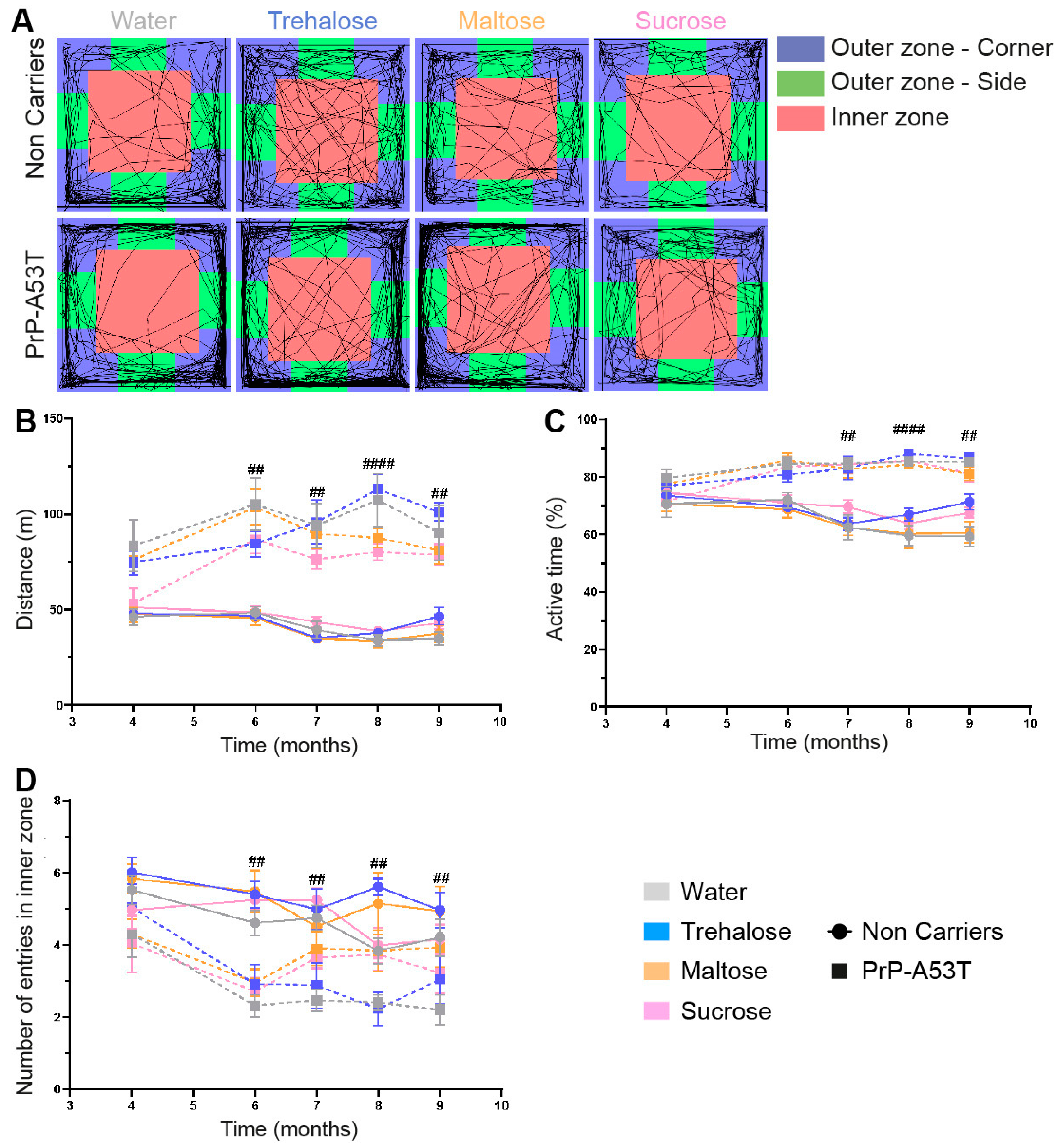
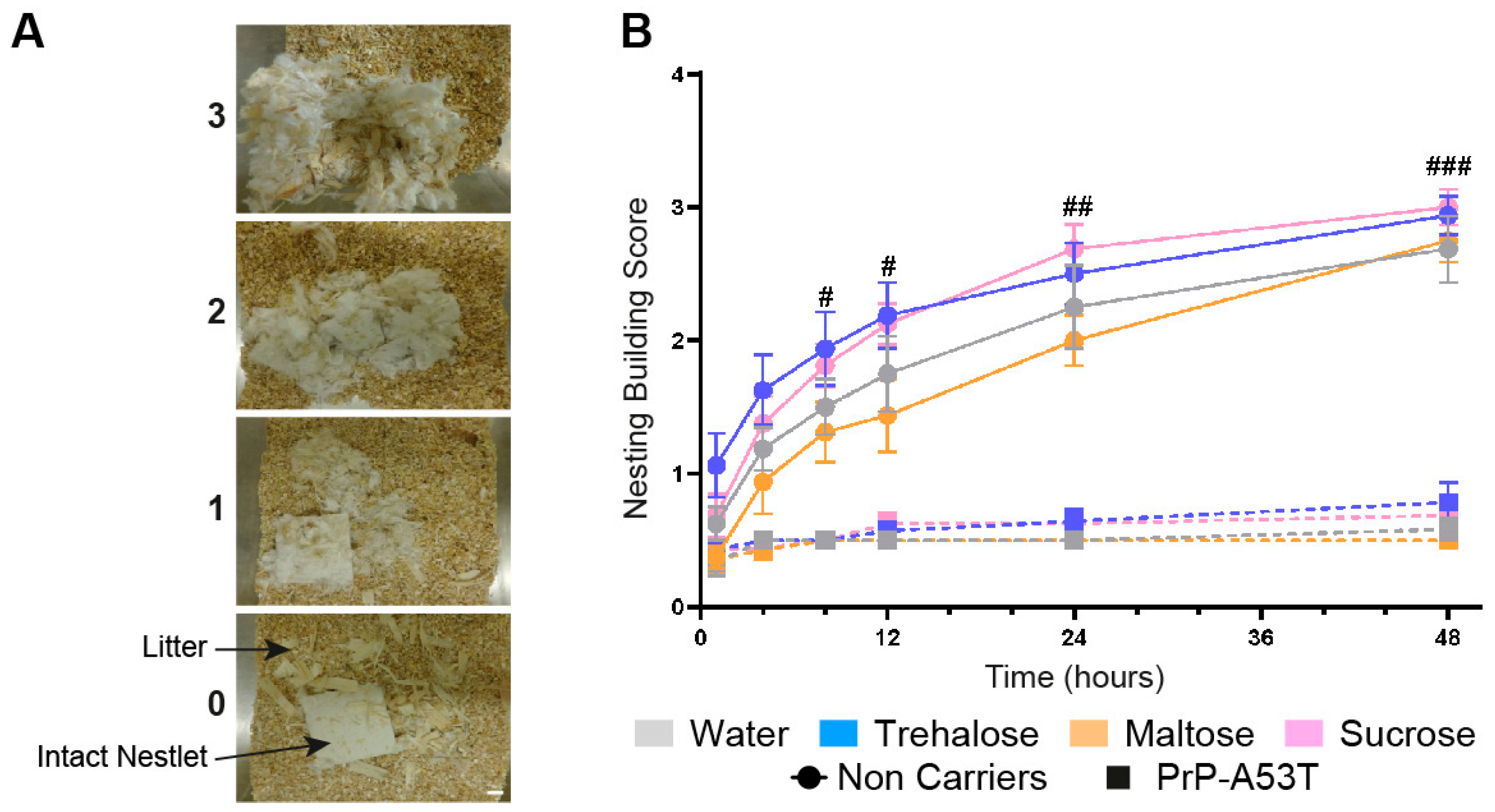
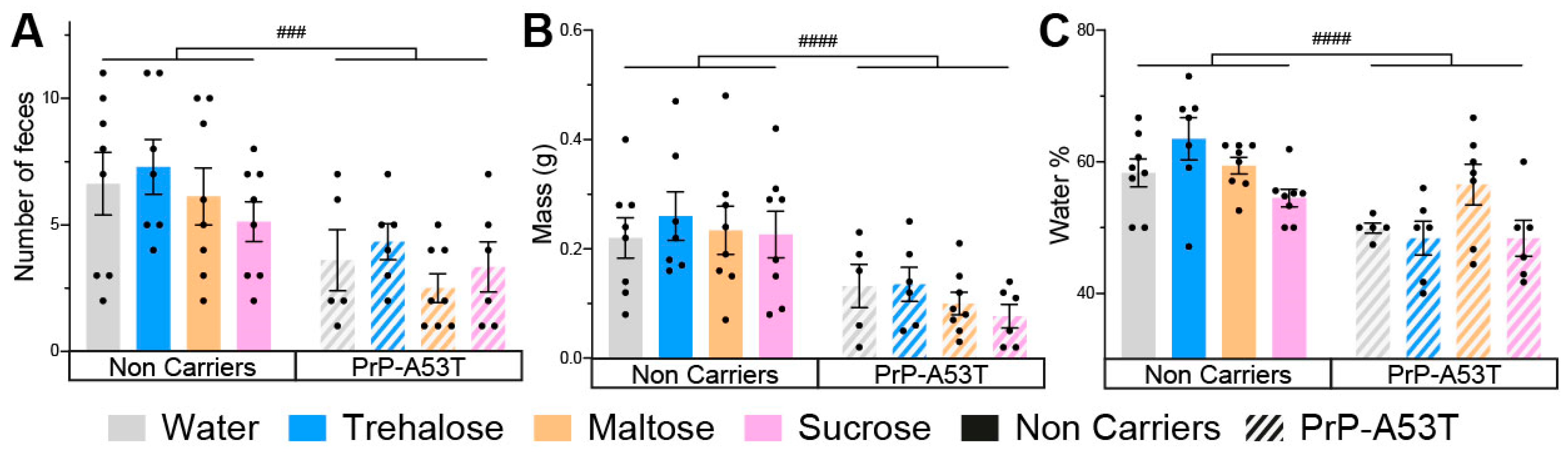
3.2. Effect of Genotype and Sugar-Enriched Diet on Motor and Non-Motor Symptoms
3.3. Accumulation of Human α-Syn in PrP-A53T Mice
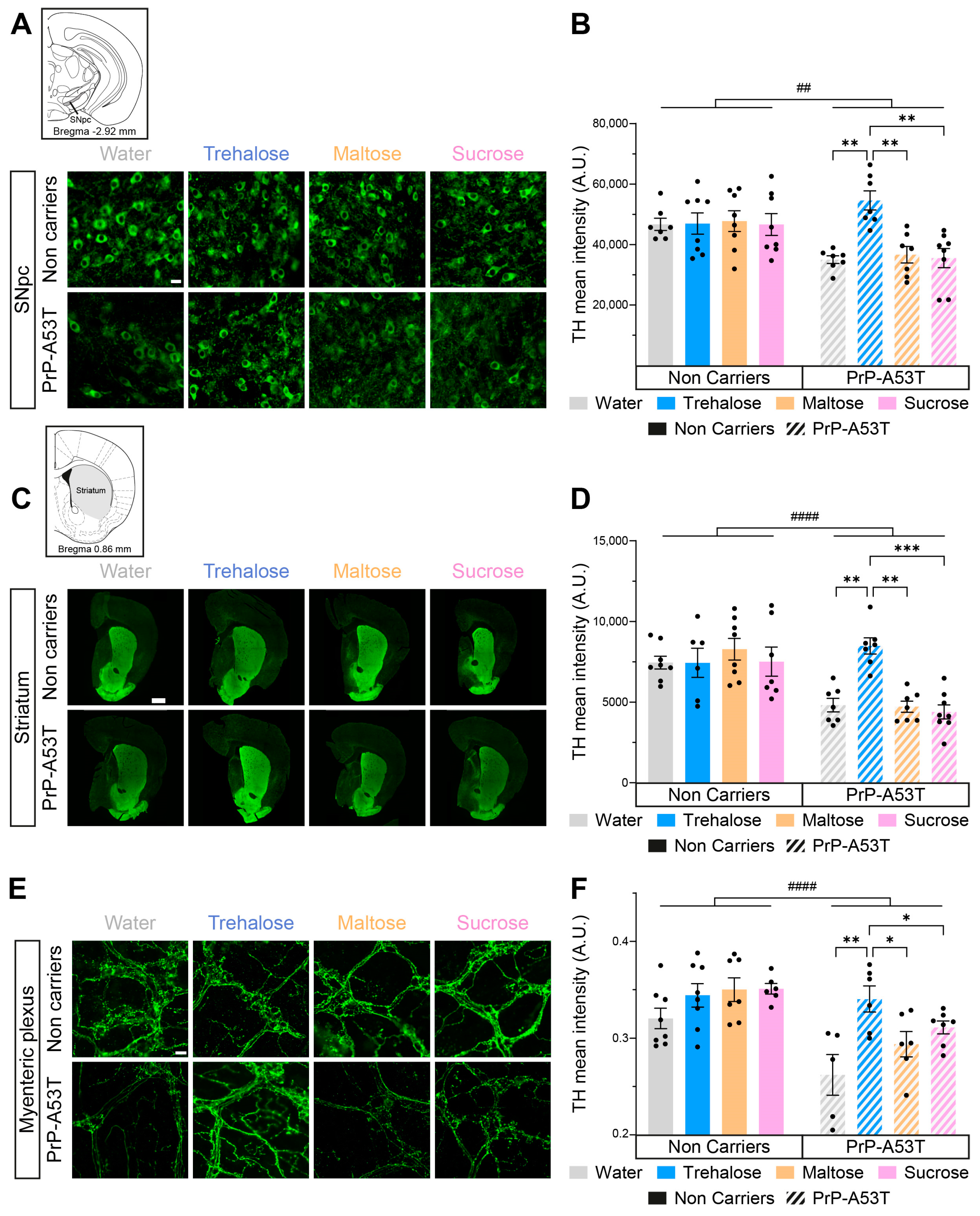
3.4. Neurodegeneration in PrP-A53T Mice and Neuroprotective Effect of Trehalose
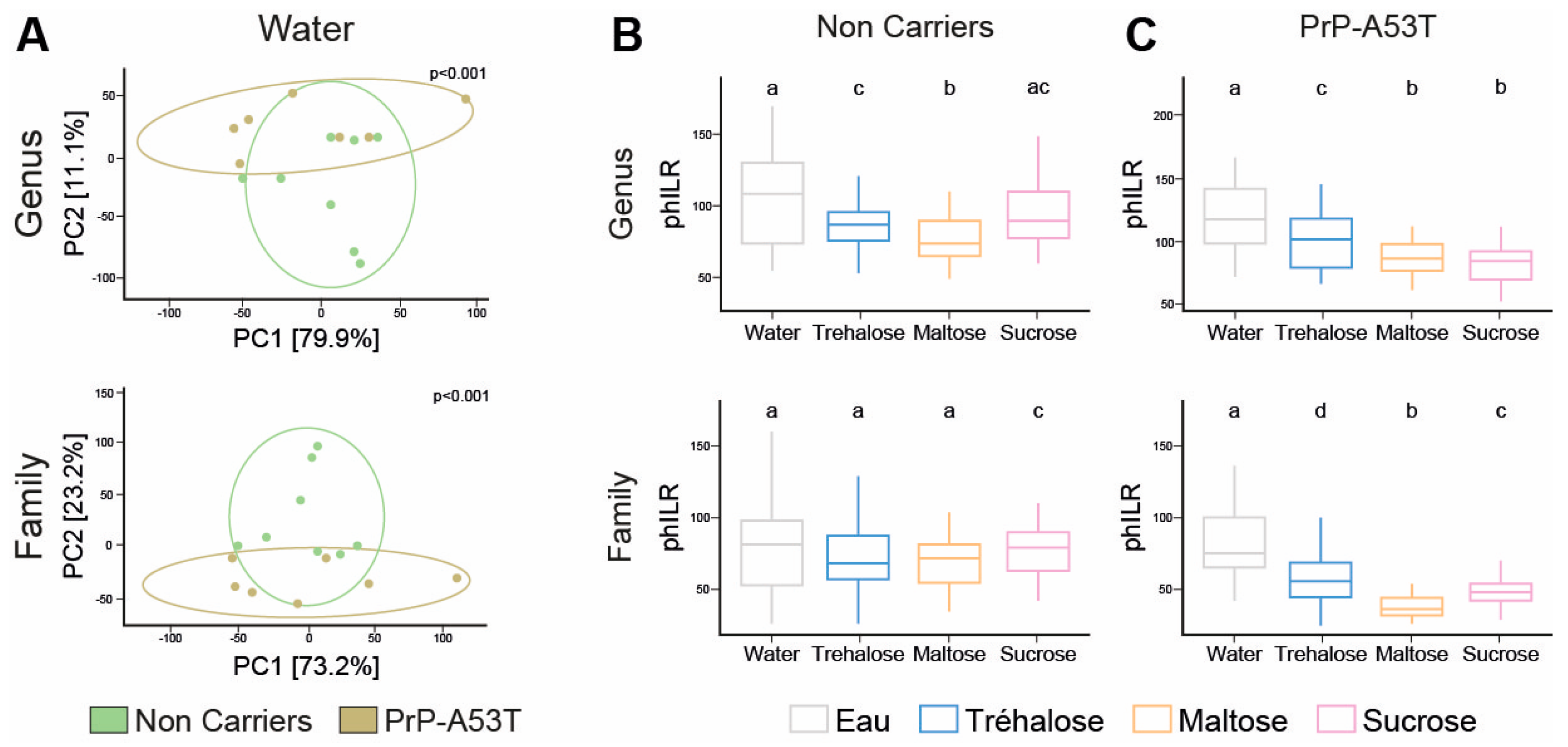
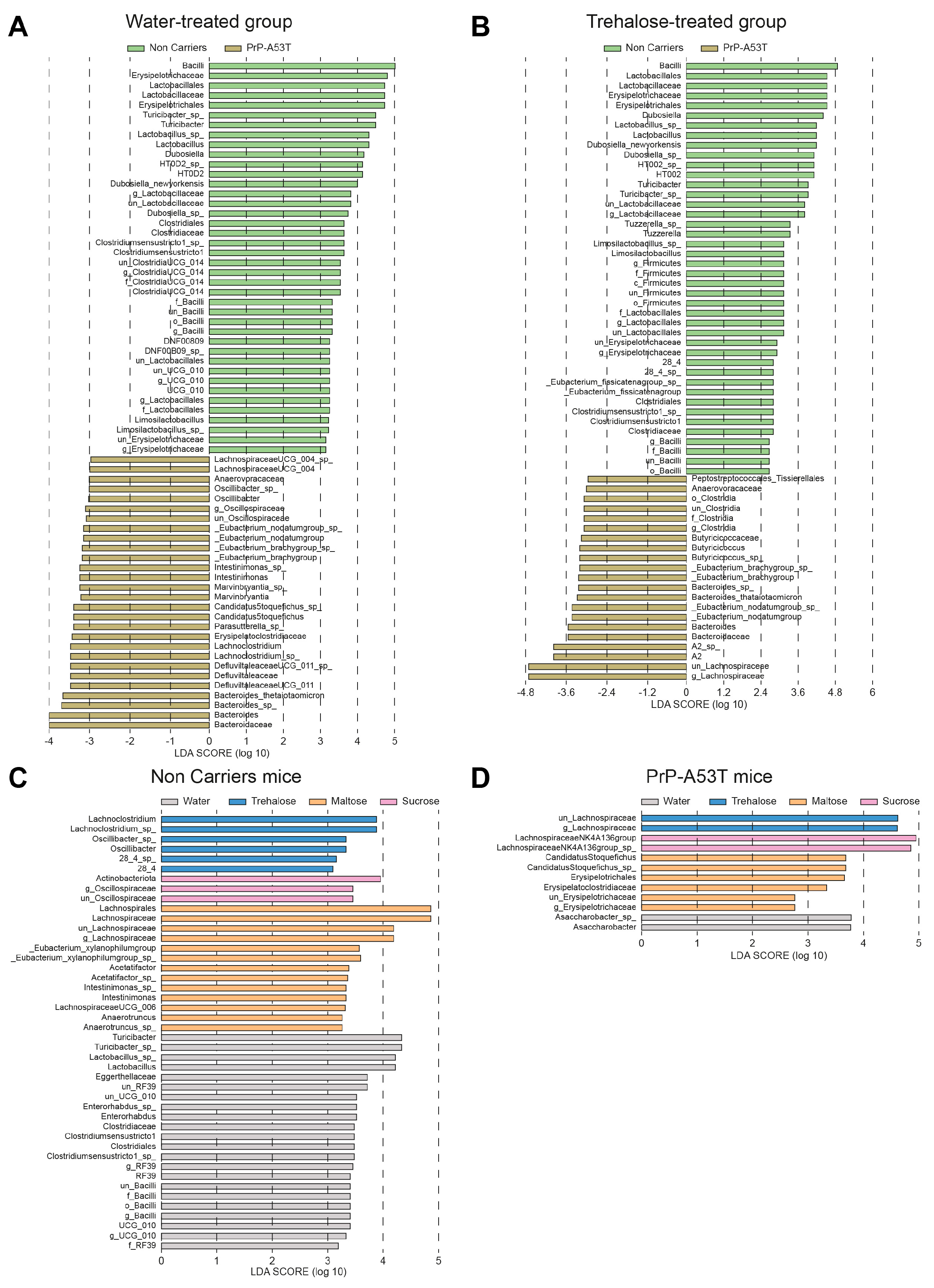
3.5. Effect of Genotype and Sugar-Enriched Diet on the Structure and Composition of the Mice Gut Microbiota
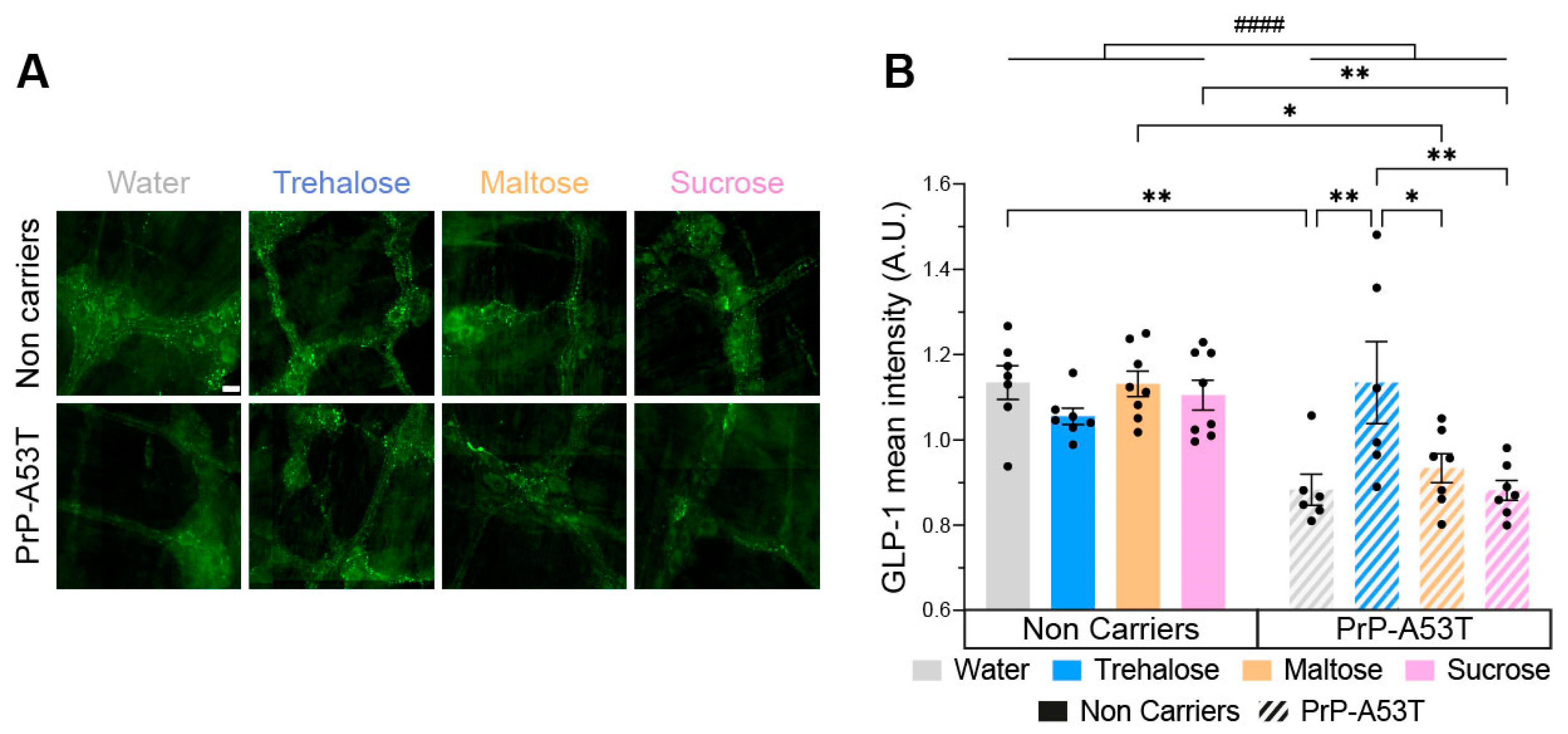
3.6. Trehalose Restores Enteric Glucagon-like Peptide 1 (GLP-1) Immunoreactivity
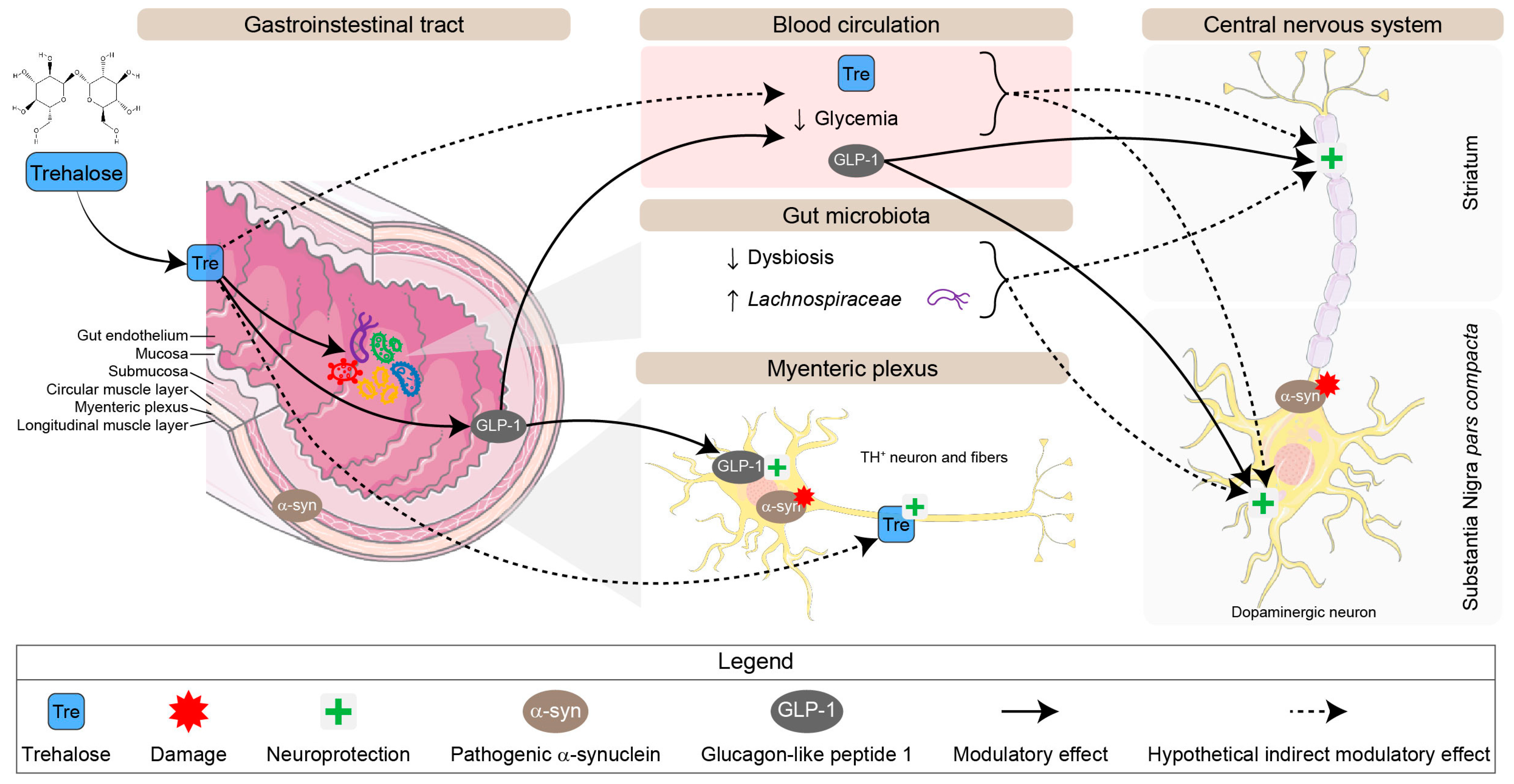
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Ou, Z.; Pan, J.; Tang, S.; Duan, D.; Yu, D.; Nong, H.; Wang, Z. Global Trends in the Incidence, Prevalence, and Years Lived with Disability of Parkinson’s Disease in 204 Countries/Territories From 1990 to 2019. Front. Public Health 2021, 9, 776847. [Google Scholar] [CrossRef] [PubMed]
- Klingelhoefer, L.; Reichmann, H. Pathogenesis of Parkinson Disease--the Gut-Brain Axis and Environmental Factors. Nat. Rev. Neurol. 2015, 11, 625–636. [Google Scholar] [CrossRef] [PubMed]
- Wirdefeldt, K.; Adami, H.-O.; Cole, P.; Trichopoulos, D.; Mandel, J. Epidemiology and Etiology of Parkinson’s Disease: A Review of the Evidence. Eur. J. Epidemiol. 2011, 26 (Suppl. 1), S1–S58. [Google Scholar] [CrossRef]
- Rocha, E.M.; De Miranda, B.; Sanders, L.H. Alphα-synuclein: Pathology, Mitochondrial Dysfunction and Neuroinflammation in Parkinson’s Disease. Neurobiol. Dis. 2018, 109, 249–257. [Google Scholar] [CrossRef] [PubMed]
- Schapira, A.H.; Jenner, P. Etiology and Pathogenesis of Parkinson’s Disease. Mov. Disord. Off. J. Mov. Disord. Soc. 2011, 26, 1049–1055. [Google Scholar] [CrossRef] [PubMed]
- Savica, R.; Rocca, W.A.; Ahlskog, J.E. When Does Parkinson Disease Start? Arch. Neurol. 2010, 67, 798–801. [Google Scholar] [CrossRef] [PubMed]
- Singaram, C.; Ashraf, W.; Gaumnitz, E.A.; Torbey, C.; Sengupta, A.; Pfeiffer, R.; Quigley, E.M. Dopaminergic Defect of Enteric Nervous System in Parkinson’s Disease Patients with Chronic Constipation. Lancet 1995, 346, 861–864. [Google Scholar] [CrossRef]
- Petrov, V.A.; Saltykova, I.V.; Zhukova, I.A.; Alifirova, V.M.; Zhukova, N.G.; Dorofeeva, Y.B.; Tyakht, A.V.; Kovarsky, B.A.; Alekseev, D.G.; Kostryukova, E.S.; et al. Analysis of Gut Microbiota in Patients with Parkinson’s Disease. Bull. Exp. Biol. Med. 2017, 162, 734–737. [Google Scholar] [CrossRef]
- Rietdijk, C.D.; Perez-Pardo, P.; Garssen, J.; van Wezel, R.J.A.; Kraneveld, A.D. Exploring Braak’s Hypothesis of Parkinson’s Disease. Front. Neurol. 2017, 8, 37. [Google Scholar] [CrossRef]
- Scheperjans, F.; Aho, V.; Pereira, P.A.B.; Koskinen, K.; Paulin, L.; Pekkonen, E.; Haapaniemi, E.; Kaakkola, S.; Eerola-Rautio, J.; Pohja, M.; et al. Gut Microbiota Are Related to Parkinson’s Disease and Clinical Phenotype. Mov. Disord. Off. J. Mov. Disord. Soc. 2015, 30, 350–358. [Google Scholar] [CrossRef]
- Li, W.; Wu, X.; Hu, X.; Wang, T.; Liang, S.; Duan, Y.; Jin, F.; Qin, B. Structural Changes of Gut Microbiota in Parkinson’s Disease and Its Correlation with Clinical Features. Sci. China Life Sci. 2017, 60, 1223–1233. [Google Scholar] [CrossRef] [PubMed]
- Unger, M.M.; Spiegel, J.; Dillmann, K.-U.; Grundmann, D.; Philippeit, H.; Bürmann, J.; Faßbender, K.; Schwiertz, A.; Schäfer, K.-H. Short Chain Fatty Acids and Gut Microbiota Differ between Patients with Parkinson’s Disease and Age-Matched Controls. Park. Relat. Disord. 2016, 32, 66–72. [Google Scholar] [CrossRef] [PubMed]
- Braak, H.; de Vos, R.A.I.; Bohl, J.; Del Tredici, K. Gastric Alphα-synuclein Immunoreactive Inclusions in Meissner’s and Auerbach’s Plexuses in Cases Staged for Parkinson’s Disease-Related Brain Pathology. Neurosci. Lett. 2006, 396, 67–72. [Google Scholar] [CrossRef]
- Zheng, D.; Liwinski, T.; Elinav, E. Interaction between Microbiota and Immunity in Health and Disease. Cell Res. 2020, 30, 492–506. [Google Scholar] [CrossRef] [PubMed]
- Bruno, M.A. and B. Brain-Gut-Microbiota Axis in Parkinson’s Disease. World J. Gastroenterol. WJG 2015, 21, 10609–10620. [Google Scholar]
- Abdel-Haq, R.; Schlachetzki, J.C.M.; Glass, C.K.; Mazmanian, S.K. Microbiome–Microglia Connections via the Gut–Brain Axis. J. Exp. Med. 2019, 216, 41–59. [Google Scholar] [CrossRef] [PubMed]
- Fung, T.C.; Olson, C.A.; Hsiao, E.Y. Interactions between the Microbiota, Immune and Nervous Systems in Health and Disease. Nat. Neurosci. 2017, 20, 145–155. [Google Scholar] [CrossRef]
- Forsythe, P.; Bienenstock, J.; Kunze, W.A. Vagal Pathways for Microbiome-Brain-Gut Axis Communication. Adv. Exp. Med. Biol. 2014, 817, 115–133. [Google Scholar] [CrossRef]
- Dighriri, I.M.; Alsubaie, A.M.; Hakami, F.M.; Hamithi, D.M.; Alshekh, M.M.; Khobrani, F.A.; Dalak, F.E.; Hakami, A.A.; Alsueaadi, E.H.; Alsaawi, L.S.; et al. Effects of Omega-3 Polyunsaturated Fatty Acids on Brain Functions: A Systematic Review. Cureus 2022, 14, e30091. [Google Scholar] [CrossRef]
- Tramontano, M.; Andrejev, S.; Pruteanu, M.; Klünemann, M.; Kuhn, M.; Galardini, M.; Jouhten, P.; Zelezniak, A.; Zeller, G.; Bork, P.; et al. Nutritional Preferences of Human Gut Bacteria Reveal Their Metabolic Idiosyncrasies. Nat. Microbiol. 2018, 3, 514–522. [Google Scholar] [CrossRef]
- Lei, E.; Vacy, K.; Boon, W.C. Fatty Acids and Their Therapeutic Potential in Neurological Disorders. Neurochem. Int. 2016, 95, 75–84. [Google Scholar] [CrossRef] [PubMed]
- Dalile, B.; Van Oudenhove, L.; Vervliet, B.; Verbeke, K. The Role of Short-Chain Fatty Acids in Microbiota-Gut-Brain Communication. Nat. Rev. Gastroenterol. Hepatol. 2019, 16, 461–478. [Google Scholar] [CrossRef] [PubMed]
- Nankova, B.B.; Agarwal, R.; MacFabe, D.F.; La Gamma, E.F. Enteric Bacterial Metabolites Propionic and Butyric Acid Modulate Gene Expression, Including CREB-Dependent Catecholaminergic Neurotransmission, in PC12 Cells—Possible Relevance to Autism Spectrum Disorders. PLoS ONE 2014, 9, e103740. [Google Scholar] [CrossRef] [PubMed]
- Cacabelos, R. Parkinson’s Disease: From Pathogenesis to Pharmacogenomics. Int. J. Mol. Sci. 2017, 18, 551. [Google Scholar] [CrossRef] [PubMed]
- Dong, J.; Beard, J.D.; Umbach, D.M.; Park, Y.; Huang, X.; Blair, A.; Kamel, F.; Chen, H. Dietary Fat Intake and Risk for Parkinson’s Disease. Mov. Disord. Off. J. Mov. Disord. Soc. 2014, 29, 1623–1630. [Google Scholar] [CrossRef] [PubMed]
- Marras, C.; Canning, C.G.; Goldman, S.M. Environment, Lifestyle, and Parkinson’s Disease: Implications for Prevention in the next Decade. Mov. Disord. Off. J. Mov. Disord. Soc. 2019, 34, 801–811. [Google Scholar] [CrossRef]
- Paul, M.J.; Primavesi, L.F.; Jhurreea, D.; Zhang, Y. Trehalose Metabolism and Signaling. Annu. Rev. Plant Biol. 2008, 59, 417–441. [Google Scholar] [CrossRef] [PubMed]
- Yu, W.-B.; Jiang, T.; Lan, D.-M.; Lu, J.-H.; Yue, Z.-Y.; Wang, J.; Zhou, P. Trehalose Inhibits Fibrillation of A53T Mutant Alphα-synuclein and Disaggregates Existing Fibrils. Arch. Biochem. Biophys. 2012, 523, 144–150. [Google Scholar] [CrossRef]
- Sarkar, S.; Chigurupati, S.; Raymick, J.; Mann, D.; Bowyer, J.F.; Schmitt, T.; Beger, R.D.; Hanig, J.P.; Schmued, L.C.; Paule, M.G. Neuroprotective Effect of the Chemical Chaperone, Trehalose in a Chronic MPTP-Induced Parkinson’s Disease Mouse Model. Neurotoxicology 2014, 44, 250–262. [Google Scholar] [CrossRef]
- Lee, H.-J.; Yoon, Y.-S.; Lee, S.-J. Mechanism of Neuroprotection by Trehalose: Controversy Surrounding Autophagy Induction. Cell Death Dis. 2018, 9, 1–12. [Google Scholar] [CrossRef]
- Di Rienzi, S.C.; Britton, R.A. Adaptation of the Gut Microbiota to Modern Dietary Sugars and Sweeteners. Adv. Nutr. 2020, 11, 616–629. [Google Scholar] [CrossRef] [PubMed]
- Ruhal, R.; Kataria, R.; Choudhury, B. Trends in Bacterial Trehalose Metabolism and Significant Nodes of Metabolic Pathway in the Direction of Trehalose Accumulation. Microb. Biotechnol. 2013, 6, 493–502. [Google Scholar] [CrossRef] [PubMed]
- The Jackson Laboratory 006823—Hualphα-syn(A53T) Transgenic Line G2-3 Strain Details. Available online: https://www.jax.org/strain/006823 (accessed on 24 May 2023).
- Lee, M.K.; Stirling, W.; Xu, Y.; Xu, X.; Qui, D.; Mandir, A.S.; Dawson, T.M.; Copeland, N.G.; Jenkins, N.A.; Price, D.L. Human Alphα-synuclein-Harboring Familial Parkinson’s Disease-Linked Ala-53 → Thr Mutation Causes Neurodegenerative Disease with Alphα-synuclein Aggregation in Transgenic Mice. Proc. Natl. Acad. Sci. USA 2002, 99, 8968–8973. [Google Scholar] [CrossRef] [PubMed]
- Howson, P.A.; Johnston, T.H.; Ravenscroft, P.; Hill, M.P.; Su, J.; Brotchie, J.M.; Koprich, J.B. Beneficial Effects of Trehalose on Striatal Dopaminergic Deficits in Rodent and Primate Models of Synucleinopathy in Parkinson’s Disease. J. Pharmacol. Exp. Ther. 2019, 369, 364–374. [Google Scholar] [CrossRef]
- Barone, P.; Antonini, A.; Colosimo, C.; Marconi, R.; Morgante, L.; Avarello, T.P.; Bottacchi, E.; Cannas, A.; Ceravolo, G.; Ceravolo, R.; et al. The PRIAMO Study: A Multicenter Assessment of Nonmotor Symptoms and Their Impact on Quality of Life in Parkinson’s Disease. Mov. Disord. Off. J. Mov. Disord. Soc. 2009, 24, 1641–1649. [Google Scholar] [CrossRef]
- Campos, F.L.; Carvalho, M.M.; Cristovão, A.C.; Je, G.; Baltazar, G.; Salgado, A.J.; Kim, Y.-S.; Sousa, N. Rodent Models of Parkinson’s Disease: Beyond the Motor Symptomatology. Front. Behav. Neurosci. 2013, 7, 175. [Google Scholar] [CrossRef] [PubMed]
- Lamontagne-Proulx, J.; Coulombe, K.; Morissette, M.; Rieux, M.; Calon, F.; Di Paolo, T.; Soulet, D. Sex and Age Differences in a Progressive Synucleinopathy Mouse Model. Biomolecules 2023, 13, 977. [Google Scholar] [CrossRef] [PubMed]
- Kraeuter, A.-K.; Guest, P.C.; Sarnyai, Z. The Nest Building Test in Mice for Assessment of General Well-Being. Methods Mol. Biol. Clifton NJ 2019, 1916, 87–91. [Google Scholar] [CrossRef]
- Deacon, R.M. Assessing Nest Building in Mice. Nat. Protoc. 2006, 1, 1117–1119. [Google Scholar] [CrossRef]
- Klindworth, A.; Pruesse, E.; Schweer, T.; Peplies, J.; Quast, C.; Horn, M.; Glöckner, F.O. Evaluation of General 16S Ribosomal RNA Gene PCR Primers for Classical and Next-Generation Sequencing-Based Diversity Studies. Nucleic Acids Res. 2013, 41, e1. [Google Scholar] [CrossRef]
- Martin, M. Cutadapt Removes Adapter Sequences from High-Throughput Sequencing Reads. EMBnet. J. 2011, 17, 10–12. [Google Scholar] [CrossRef]
- Callahan, B.J.; McMurdie, P.J.; Rosen, M.J.; Han, A.W.; Johnson, A.J.A.; Holmes, S.P. DADA2: High-Resolution Sample Inference from Illumina Amplicon Data. Nat. Methods 2016, 13, 581–583. [Google Scholar] [CrossRef] [PubMed]
- Wright, E.S. DECIPHER: Harnessing Local Sequence Context to Improve Protein Multiple Sequence Alignment. BMC Bioinform. 2015, 16, 322. [Google Scholar] [CrossRef]
- Schliep, K.P. Phangorn: Phylogenetic Analysis in R. Bioinformatics 2011, 27, 592–593. [Google Scholar] [CrossRef]
- McMurdie, P.J.; Holmes, S. Phyloseq: An R Package for Reproducible Interactive Analysis and Graphics of Microbiome Census Data. PLoS ONE 2013, 8, e61217. [Google Scholar] [CrossRef] [PubMed]
- Gloor, G.B.; Macklaim, J.M.; Pawlowsky-Glahn, V.; Egozcue, J.J. Microbiome Datasets Are Compositional: And This Is Not Optional. Front. Microbiol. 2017, 8, 2224. [Google Scholar] [CrossRef]
- Silverman, J.D.; Washburne, A.D.; Mukherjee, S.; David, L.A. A Phylogenetic Transform Enhances Analysis of Compositional Microbiota Data. eLife 2017, 6, e21887. [Google Scholar] [CrossRef] [PubMed]
- Roussel, C.; Chabaud, S.; Lessard-Lord, J.; Cattero, V.; Pellerin, F.-A.; Feutry, P.; Bochard, V.; Bolduc, S.; Desjardins, Y. UPEC Colonic-Virulence and Urovirulence Are Blunted by Proanthocyanidins-Rich Cranberry Extract Microbial Metabolites in a Gut Model and a 3D Tissue-Engineered Urothelium. Microbiol. Spectr. 2022, 10, e0243221. [Google Scholar] [CrossRef] [PubMed]
- Côté, M.; Drouin-Ouellet, J.; Cicchetti, F.; Soulet, D. The Critical Role of the MyD88-Dependent Pathway in Non-CNS MPTP-Mediated Toxicity. Brain. Behav. Immun. 2011, 25, 1143–1152. [Google Scholar] [CrossRef] [PubMed]
- Côté, M.; Poirier, A.-A.; Aubé, B.; Jobin, C.; Lacroix, S.; Soulet, D. Partial Depletion of the Proinflammatory Monocyte Population Is Neuroprotective in the Myenteric Plexus but Not in the Basal Ganglia in a MPTP Mouse Model of Parkinson’s Disease. Brain. Behav. Immun. 2015, 46, 154–167. [Google Scholar] [CrossRef]
- Segata, N.; Izard, J.; Waldron, L.; Gevers, D.; Miropolsky, L.; Garrett, W.S.; Huttenhower, C. Metagenomic Biomarker Discovery and Explanation. Genome Biol. 2011, 12, R60. [Google Scholar] [CrossRef] [PubMed]
- Douglas, G.M.; Maffei, V.J.; Zaneveld, J.R.; Yurgel, S.N.; Brown, J.R.; Taylor, C.M.; Huttenhower, C.; Langille, M.G.I. PICRUSt2 for Prediction of Metagenome Functions. Nat. Biotechnol. 2020, 38, 685–688. [Google Scholar] [CrossRef] [PubMed]
- Lamontagne-Proulx, J.; Coulombe, K.; Dahhani, F.; Côté, M.; Guyaz, C.; Tremblay, C.; Di Marzo, V.; Flamand, N.; Calon, F.; Soulet, D. Effect of Docosahexaenoic Acid (DHA) at the Enteric Level in a Synucleinopathy Mouse Model. Nutrients 2021, 13, 4218. [Google Scholar] [CrossRef] [PubMed]
- Rota, L.; Pellegrini, C.; Benvenuti, L.; Antonioli, L.; Fornai, M.; Blandizzi, C.; Cattaneo, A.; Colla, E. Constipation, Deficit in Colon Contractions and Alphα-synuclein Inclusions within the Colon Precede Motor Abnormalities and Neurodegeneration in the Central Nervous System in a Mouse Model of Alphα-synucleinopathy. Transl. Neurodegener. 2019, 8, 5. [Google Scholar] [CrossRef] [PubMed]
- Blanco, A.; Blanco, G. Chapter 4—Carbohydrates. In Medical Biochemistry; Blanco, A., Blanco, G., Eds.; Academic Press: Cambridge, MA, USA, 2017; pp. 73–97. ISBN 978-0-12-803550-4. [Google Scholar]
- Mizunoe, Y.; Kobayashi, M.; Sudo, Y.; Watanabe, S.; Yasukawa, H.; Natori, D.; Hoshino, A.; Negishi, A.; Okita, N.; Komatsu, M.; et al. Trehalose Protects against Oxidative Stress by Regulating the Keap1–Nrf2 and Autophagy Pathways. Redox Biol. 2017, 15, 115–124. [Google Scholar] [CrossRef] [PubMed]
- Isenbrandt, A.; Morissette, M.; Bourque, M.; Lamontagne-Proulx, J.; Coulombe, K.; Soulet, D.; Di Paolo, T. Effect of Sex and Gonadectomy on Brain MPTP Toxicity and Response to Dutasteride Treatment in Mice. Neuropharmacology 2021, 201, 108784. [Google Scholar] [CrossRef] [PubMed]
- Coulombe, K.; Kerdiles, O.; Tremblay, C.; Emond, V.; Lebel, M.; Boulianne, A.-S.; Plourde, M.; Cicchetti, F.; Calon, F. Impact of DHA Intake in a Mouse Model of Synucleinopathy. Exp. Neurol. 2018, 301, 39–49. [Google Scholar] [CrossRef] [PubMed]
- Damier, P.; Hirsch, E.C.; Agid, Y.; Graybiel, A.M. The Substantia Nigra of the Human Brain. II. Patterns of Loss of Dopamine-Containing Neurons in Parkinson’s Disease. Brain J. Neurol. 1999, 122, 1437–1448. [Google Scholar] [CrossRef]
- Nordström, U.; Beauvais, G.; Ghosh, A.; Pulikkaparambil Sasidharan, B.C.; Lundblad, M.; Fuchs, J.; Joshi, R.L.; Lipton, J.W.; Roholt, A.; Medicetty, S.; et al. Progressive Nigrostriatal Terminal Dysfunction and Degeneration in the Engrailed1 Heterozygous Mouse Model of Parkinson’s Disease. Neurobiol. Dis. 2015, 73, 70–82. [Google Scholar] [CrossRef]
- Pupyshev, A.B.; Korolenko, T.A.; Akopyan, A.A.; Amstislavskaya, T.G.; Tikhonova, M.A. Suppression of Autophagy in the Brain of Transgenic Mice with Overexpression of A53T-Mutant α-Synuclein as an Early Event at Synucleinopathy Progression. Neurosci. Lett. 2018, 672, 140–144. [Google Scholar] [CrossRef]
- Ma, S.-X.; Seo, B.A.; Kim, D.; Xiong, Y.; Kwon, S.-H.; Brahmachari, S.; Kim, S.; Kam, T.-I.; Nirujogi, R.S.; Kwon, S.H.; et al. Complement and Coagulation Cascades Are Potentially Involved in Dopaminergic Neurodegeneration in α-Synuclein-Based Mouse Models of Parkinson’s Disease. J. Proteome Res. 2021, 20, 3428–3443. [Google Scholar] [CrossRef] [PubMed]
- Khalifeh, M.; Barreto, G.E.; Sahebkar, A. Trehalose as a Promising Therapeutic Candidate for the Treatment of Parkinson’s Disease. Br. J. Pharmacol. 2019, 176, 1173–1189. [Google Scholar] [CrossRef] [PubMed]
- Unger, E.L.; Eve, D.J.; Perez, X.A.; Reichenbach, D.K.; Xu, Y.; Lee, M.K.; Andrews, A.M. Locomotor Hyperactivity and Alterations in Dopamine Neurotransmission Are Associated with Overexpression of A53T Mutant Human Alphα-synuclein in Mice. Neurobiol. Dis. 2006, 21, 431–443. [Google Scholar] [CrossRef] [PubMed]
- Deusser, J.; Schmidt, S.; Ettle, B.; Plötz, S.; Huber, S.; Müller, C.P.; Masliah, E.; Winkler, J.; Kohl, Z. Serotonergic Dysfunction in the A53T Alphα-synuclein Mouse Model of Parkinson’s Disease. J. Neurochem. 2015, 135, 589–597. [Google Scholar] [CrossRef]
- Erro, R.; Pappatà, S.; Amboni, M.; Vicidomini, C.; Longo, K.; Santangelo, G.; Picillo, M.; Vitale, C.; Moccia, M.; Giordano, F.; et al. Anxiety Is Associated with Striatal Dopamine Transporter Availability in Newly Diagnosed Untreated Parkinson’s Disease Patients. Park. Relat. Disord. 2012, 18, 1034–1038. [Google Scholar] [CrossRef] [PubMed]
- Weintraub, D.; Newberg, A.B.; Cary, M.S.; Siderowf, A.D.; Moberg, P.J.; Kleiner-Fisman, G.; Duda, J.E.; Stern, M.B.; Mozley, D.; Katz, I.R. Striatal Dopamine Transporter Imaging Correlates with Anxiety and Depression Symptoms in Parkinson’s Disease. J. Nucl. Med. Off. Publ. Soc. Nucl. Med. 2005, 46, 227–232. [Google Scholar]
- Salamone, J.D.; Correa, M. The Mysterious Motivational Functions of Mesolimbic Dopamine. Neuron 2012, 76, 470–485. [Google Scholar] [CrossRef]
- Keshavarzian, A.; Green, S.J.; Engen, P.A.; Voigt, R.M.; Naqib, A.; Forsyth, C.B.; Mutlu, E.; Shannon, K.M. Colonic Bacterial Composition in Parkinson’s Disease. Mov. Disord. Off. J. Mov. Disord. Soc. 2015, 30, 1351–1360. [Google Scholar] [CrossRef] [PubMed]
- Hill-Burns, E.M.; Debelius, J.W.; Morton, J.T.; Wissemann, W.T.; Lewis, M.R.; Wallen, Z.D.; Peddada, S.D.; Factor, S.A.; Molho, E.; Zabetian, C.P.; et al. Parkinson’s Disease and Parkinson’s Disease Medications Have Distinct Signatures of the Gut Microbiome. Mov. Disord. Off. J. Mov. Disord. Soc. 2017, 32, 739–749. [Google Scholar] [CrossRef]
- Ulluwishewa, D.; Anderson, R.C.; McNabb, W.C.; Moughan, P.J.; Wells, J.M.; Roy, N.C. Regulation of Tight Junction Permeability by Intestinal Bacteria and Dietary Components. J. Nutr. 2011, 141, 769–776. [Google Scholar] [CrossRef]
- Knudsen, K.; Krogh, K.; Østergaard, K.; Borghammer, P. Constipation in Parkinson’s Disease: Subjective Symptoms, Objective Markers, and New Perspectives. Mov. Disord. Off. J. Mov. Disord. Soc. 2017, 32, 94–105. [Google Scholar] [CrossRef] [PubMed]
- Sampson, T.R.; Debelius, J.W.; Thron, T.; Janssen, S.; Shastri, G.G.; Ilhan, Z.E.; Challis, C.; Schretter, C.E.; Rocha, S.; Gradinaru, V.; et al. Gut Microbiota Regulate Motor Deficits and Neuroinflammation in a Model of Parkinson’s Disease. Cell 2016, 167, 1469–1480.e12. [Google Scholar] [CrossRef] [PubMed]
- Ho, L.; Ono, K.; Tsuji, M.; Mazzola, P.; Singh, R.; Pasinetti, G.M. Protective Roles of Intestinal Microbiota Derived Short Chain Fatty Acids in Alzheimer’s Disease-Type Beta-Amyloid Neuropathological Mechanisms. Expert Rev. Neurother. 2018, 18, 83–90. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Christophersen, C.T.; Sorich, M.J.; Gerber, J.P.; Angley, M.T.; Conlon, M.A. Elevated Fecal Short Chain Fatty Acid and Ammonia Concentrations in Children with Autism Spectrum Disorder. Dig. Dis. Sci. 2012, 57, 2096–2102. [Google Scholar] [CrossRef]
- Grider, J.R.; Piland, B.E. The Peristaltic Reflex Induced by Short-Chain Fatty Acids Is Mediated by Sequential Release of 5-HT and Neuronal CGRP but Not BDNF. Am. J. Physiol. Gastrointest. Liver Physiol. 2007, 292, G429–G437. [Google Scholar] [CrossRef] [PubMed]
- Soret, R.; Chevalier, J.; De Coppet, P.; Poupeau, G.; Derkinderen, P.; Segain, J.P.; Neunlist, M. Short-Chain Fatty Acids Regulate the Enteric Neurons and Control Gastrointestinal Motility in Rats. Gastroenterology 2010, 138, 1772–1782. [Google Scholar] [CrossRef] [PubMed]
- Salazar, N.; Arboleya, S.; Fernández-Navarro, T.; de los Reyes-Gavilán, C.G.; Gonzalez, S.; Gueimonde, M. Age-Associated Changes in Gut Microbiota and Dietary Components Related with the Immune System in Adulthood and Old Age: A Cross-Sectional Study. Nutrients 2019, 11, 1765. [Google Scholar] [CrossRef] [PubMed]
- Barichella, M.; Severgnini, M.; Cilia, R.; Cassani, E.; Bolliri, C.; Caronni, S.; Ferri, V.; Cancello, R.; Ceccarani, C.; Faierman, S.; et al. Unraveling Gut Microbiota in Parkinson’s Disease and Atypical Parkinsonism. Mov. Disord. Off. J. Mov. Disord. Soc. 2019, 34, 396–405. [Google Scholar] [CrossRef]
- Vacca, M.; Celano, G.; Calabrese, F.M.; Portincasa, P.; Gobbetti, M.; De Angelis, M. The Controversial Role of Human Gut Lachnospiraceae. Microorganisms 2020, 8, 573. [Google Scholar] [CrossRef]
- Chmiela, T.; Węgrzynek, J.; Kasprzyk, A.; Waksmundzki, D.; Wilczek, D.; Gorzkowska, A. If Not Insulin Resistance so What?—Comparison of Fasting Glycemia in Idiopathic Parkinson’s Disease and Atypical Parkinsonism. Diabetes Metab. Syndr. Obes. Targets Ther. 2022, 15, 1451–1460. [Google Scholar] [CrossRef]
- Labandeira, C.M.; Fraga-Bau, A.; Arias Ron, D.; Muñoz, A.; Alonso-Losada, G.; Koukoulis, A.; Romero-Lopez, J.; Rodriguez-Perez, A.I. Diabetes, Insulin and New Therapeutic Strategies for Parkinson’s Disease: Focus on Glucagon-like Peptide-1 Receptor Agonists. Front. Neuroendocrinol. 2021, 62, 100914. [Google Scholar] [CrossRef] [PubMed]
- Meng, L.; Li, Y.; Liu, C.; Zhang, G.; Chen, J.; Xiong, M.; Pan, L.; Zhang, X.; Chen, G.; Xiong, J.; et al. Islet Amyloid Polypeptide Triggers α-Synuclein Pathology in Parkinson’s Disease. Prog. Neurobiol. 2023, 226, 102462. [Google Scholar] [CrossRef] [PubMed]
- Carvelli, L.; Morón, J.A.; Kahlig, K.M.; Ferrer, J.V.; Sen, N.; Lechleiter, J.D.; Leeb-Lundberg, L.M.F.; Merrill, G.; Lafer, E.M.; Ballou, L.M.; et al. PI 3-Kinase Regulation of Dopamine Uptake. J. Neurochem. 2002, 81, 859–869. [Google Scholar] [CrossRef]
- McGarry, A.; Rosanbalm, S.; Leinonen, M.; Olanow, C.W.; To, D.; Bell, A.; Lee, D.; Chang, J.; Dubow, J.; Dhall, R.; et al. Safety, Tolerability, and Efficacy of NLY01 in Early Untreated Parkinson’s Disease: A Randomised, Double-Blind, Placebo-Controlled Trial. Lancet Neurol. 2024, 23, 37–45. [Google Scholar] [CrossRef] [PubMed]
- Oku, T.; Nakamura, S. Estimation of Intestinal Trehalase Activity from a Laxative Threshold of Trehalose and Lactulose on Healthy Female Subjects. Eur. J. Clin. Nutr. 2000, 54, 783–788. [Google Scholar] [CrossRef] [PubMed]
- Yoshizane, C.; Mizote, A.; Yamada, M.; Arai, N.; Arai, S.; Maruta, K.; Mitsuzumi, H.; Ariyasu, T.; Ushio, S.; Fukuda, S. Glycemic, Insulinemic and Incretin Responses after Oral Trehalose Ingestion in Healthy Subjects. Nutr. J. 2017, 16, 9. [Google Scholar] [CrossRef]
- Hashemian, S.; Shojaei, M.; Radbakhsh, S.; Ashari, S.; Matbou Riahi, M.; Shateri Amiri, Z.; Atkin, S.L.; Banach, M.; Bagherniya, M.; Emami, S.A.; et al. The Effects of Oral Trehalose on Glycaemia, Inflammation, and Quality of Life in Patients with Type 2 Diabetes: A Pilot Randomized Controlled Trial. Arch. Med. Sci. AMS 2023, 19, 1693–1700. [Google Scholar] [CrossRef] [PubMed]
- DeBosch, B.J.; Heitmeier, M.R.; Mayer, A.L.; Higgins, C.B.; Crowley, J.R.; Kraft, T.E.; Chi, M.; Newberry, E.P.; Chen, Z.; Finck, B.N.; et al. Trehalose Inhibits Solute Carrier 2A (SLC2A) Proteins to Induce Autophagy and Prevent Hepatic Steatosis. Sci. Signal. 2016, 9, ra21. [Google Scholar] [CrossRef]
- Bodnaruc, A.M.; Prud’homme, D.; Blanchet, R.; Giroux, I. Nutritional Modulation of Endogenous Glucagon-like Peptide-1 Secretion: A Review. Nutr. Metab. 2016, 13, 92. [Google Scholar] [CrossRef]
- Baggio, L.L.; Drucker, D.J. Biology of Incretins: GLP-1 and GIP. Gastroenterology 2007, 132, 2131–2157. [Google Scholar] [CrossRef]
- Hayes, M.R.; Leichner, T.M.; Zhao, S.; Lee, G.S.; Chowansky, A.; Zimmer, D.; De Jonghe, B.C.; Kanoski, S.E.; Grill, H.J.; Bence, K.K. Intracellular Signals Mediating the Food Intake-Suppressive Effects of Hindbrain Glucagon-like Peptide-1 Receptor Activation. Cell Metab. 2011, 13, 320–330. [Google Scholar] [CrossRef]
- Marathe, C.S.; Rayner, C.K.; Jones, K.L.; Horowitz, M. Relationships Between Gastric Emptying, Postprandial Glycemia, and Incretin Hormones. Diabetes Care 2013, 36, 1396–1405. [Google Scholar] [CrossRef] [PubMed]
- Amato, A.; Cinci, L.; Rotondo, A.; Serio, R.; Faussone-Pellegrini, M.S.; Vannucchi, M.G.; Mulè, F. Peripheral Motor Action of Glucagon-like Peptide-1 through Enteric Neuronal Receptors. Neurogastroenterol. Motil. 2010, 22, 664-e203. [Google Scholar] [CrossRef] [PubMed]
- Anderberg, R.H.; Anefors, C.; Bergquist, F.; Nissbrandt, H.; Skibicka, K.P. Dopamine Signaling in the Amygdala, Increased by Food Ingestion and GLP-1, Regulates Feeding Behavior. Physiol. Behav. 2014, 136, 135–144. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.-F.; Liu, J.-J.; Xia, J.; Liu, J.; Mirabella, V.; Pang, Z.P. Endogenous Glucagon-like Peptide-1 Suppresses High-Fat Food Intake by Reducing Synaptic Drive onto Mesolimbic Dopamine Neurons. Cell Rep. 2015, 12, 726–733. [Google Scholar] [CrossRef] [PubMed]
- Perry, T.; Haughey, N.J.; Mattson, M.P.; Egan, J.M.; Greig, N.H. Protection and Reversal of Excitotoxic Neuronal Damage by Glucagon-like Peptide-1 and Exendin-4. J. Pharmacol. Exp. Ther. 2002, 302, 881–888. [Google Scholar] [CrossRef]
- Li, Y.; Perry, T.; Kindy, M.S.; Harvey, B.K.; Tweedie, D.; Holloway, H.W.; Powers, K.; Shen, H.; Egan, J.M.; Sambamurti, K.; et al. GLP-1 Receptor Stimulation Preserves Primary Cortical and Dopaminergic Neurons in Cellular and Rodent Models of Stroke and Parkinsonism. Proc. Natl. Acad. Sci. USA 2009, 106, 1285–1290. [Google Scholar] [CrossRef]
- Harkavyi, A.; Abuirmeileh, A.; Lever, R.; Kingsbury, A.E.; Biggs, C.S.; Whitton, P.S. Glucagon-like Peptide 1 Receptor Stimulation Reverses Key Deficits in Distinct Rodent Models of Parkinson’s Disease. J. Neuroinflamm. 2008, 5, 19. [Google Scholar] [CrossRef]
- Liu, W.; Jalewa, J.; Sharma, M.; Li, G.; Li, L.; Hölscher, C. Neuroprotective Effects of Lixisenatide and Liraglutide in the 1-Methyl-4-Phenyl-1,2,3,6-Tetrahydropyridine Mouse Model of Parkinson’s Disease. Neuroscience 2015, 303, 42–50. [Google Scholar] [CrossRef]
- Athauda, D.; Maclagan, K.; Skene, S.S.; Bajwa-Joseph, M.; Letchford, D.; Chowdhury, K.; Hibbert, S.; Budnik, N.; Zampedri, L.; Dickson, J.; et al. Exenatide Once Weekly versus Placebo in Parkinson’s Disease: A Randomised, Double-Blind, Placebo-Controlled Trial. Lancet Lond. Engl. 2017, 390, 1664–1675. [Google Scholar] [CrossRef]
- Deacon, C.F.; Johnsen, A.H.; Holst, J.J. Degradation of Glucagon-like Peptide-1 by Human Plasma in Vitro Yields an N-Terminally Truncated Peptide That Is a Major Endogenous Metabolite in Vivo. J. Clin. Endocrinol. Metab. 1995, 80, 952–957. [Google Scholar] [CrossRef] [PubMed]
- Muscogiuri, G.; DeFronzo, R.A.; Gastaldelli, A.; Holst, J.J. Glucagon-like Peptide-1 and the Central/Peripheral Nervous System: Crosstalk in Diabetes. Trends Endocrinol. Metab. TEM 2017, 28, 88–103. [Google Scholar] [CrossRef] [PubMed]
- Athauda, D.; Foltynie, T. The Glucagon-like Peptide 1 (GLP) Receptor as a Therapeutic Target in Parkinson’s Disease: Mechanisms of Action. Drug Discov. Today 2016, 21, 802–818. [Google Scholar] [CrossRef] [PubMed]
- Bang-Berthelsen, C.H.; Holm, T.L.; Pyke, C.; Simonsen, L.; Søkilde, R.; Pociot, F.; Heller, R.S.; Folkersen, L.; Kvist, P.H.; Jackerott, M.; et al. GLP-1 Induces Barrier Protective Expression in Brunner’s Glands and Regulates Colonic Inflammation. Inflamm. Bowel Dis. 2016, 22, 2078–2097. [Google Scholar] [CrossRef] [PubMed]
- Chen, A.; Gibney, P.A. Dietary Trehalose as a Bioactive Nutrient. Nutrients 2023, 15, 1393. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; DeBosch, B.J. Microbial and Metabolic Impacts of Trehalose and Trehalose Analogues. Gut Microbes 2020, 11, 1475–1482. [Google Scholar] [CrossRef]
- Richards, A.B.; Krakowka, S.; Dexter, L.B.; Schmid, H.; Wolterbeek, A.P.M.; Waalkens-Berendsen, D.H.; Shigoyuki, A.; Kurimoto, M. Trehalose: A Review of Properties, History of Use and Human Tolerance, and Results of Multiple Safety Studies. Food Chem. Toxicol. Int. J. Publ. Br. Ind. Biol. Res. Assoc. 2002, 40, 871–898. [Google Scholar] [CrossRef] [PubMed]
- Collins, J.; Danhof, H.; Britton, R.A. The Role of Trehalose in the Global Spread of Epidemic Clostridium Difficile. Gut Microbes 2018, 10, 204–209. [Google Scholar] [CrossRef]
- Eyre, D.W.; Didelot, X.; Buckley, A.M.; Freeman, J.; Moura, I.B.; Crook, D.W.; Peto, T.E.A.; Walker, A.S.; Wilcox, M.H.; Dingle, K.E. Clostridium Difficile Trehalose Metabolism Variants Are Common and Not Associated with Adverse Patient Outcomes When Variably Present in the Same Lineage. EBioMedicine 2019, 43, 347–355. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Primer | Sequence |
|---|---|
| V3-V4_F_341F (first PCR) | ACACTCTTTCCCTACACGACGCTCTTCCGATCTCCTACGGGNGGCWGCAG |
| V3-V4_R_805R (first PCR) | GTGACTGGAGTTCAGACGTGTGCTCTTCCGATCTGACTACHVGGGTATCTAATC |
| Generic forward second-PCR primer | AATGATACGGCGACCACCGAGATCTACAC[index1]ACACTCTTTCCCTACACGAC |
| Generic reverse second-PCR primer | CAAGCAGAAGACGGCATACGAGAT[index2]GTGACTGGAGTTCAGACGTGT |
| Primary antibodies | Host | Company | Catalog # | Dilution |
| S129 phosphorylated α-synuclein | Rabbit | Abcam | Ab51253 | 1/200 |
| Tyrosine Hydroxylase | Sheep | Invitrogen | PA1-4679 | 1/1000 |
| Glucagon like peptide-1 | Rabbit | Invitrogen | PA5-79303 | 1/750 |
| Anti-Choline Acetyltransferase | Goat | Sigma-Aldrich | AB144P | 1/100 |
| Secondary antibodies | Host | Company | Catalog # | Dilution |
| Anti-Rabbit IgG Alexa FluorTM 647 | Donkey | Life technologies | A31573 | 1/1000 |
| Anti-Sheep IgG Alexa FluorTM 647 | Donkey | Invitrogen | A21448 | 1/1000 |
| Anti-Rabbit IgG Alexa FluorTM 488 | Donkey | Life Technologies | A21206 | 1/1000 |
| Anti-Goat IgG Alexa FluorTM 633 | Donkey | Life Technologies | A21082 | 1/1000 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Pradeloux, S.; Coulombe, K.; Ouamba, A.J.K.; Isenbrandt, A.; Calon, F.; Roy, D.; Soulet, D. Oral Trehalose Intake Modulates the Microbiota–Gut–Brain Axis and Is Neuroprotective in a Synucleinopathy Mouse Model. Nutrients 2024, 16, 3309. https://doi.org/10.3390/nu16193309
Pradeloux S, Coulombe K, Ouamba AJK, Isenbrandt A, Calon F, Roy D, Soulet D. Oral Trehalose Intake Modulates the Microbiota–Gut–Brain Axis and Is Neuroprotective in a Synucleinopathy Mouse Model. Nutrients. 2024; 16(19):3309. https://doi.org/10.3390/nu16193309
Chicago/Turabian StylePradeloux, Solène, Katherine Coulombe, Alexandre Jules Kennang Ouamba, Amandine Isenbrandt, Frédéric Calon, Denis Roy, and Denis Soulet. 2024. "Oral Trehalose Intake Modulates the Microbiota–Gut–Brain Axis and Is Neuroprotective in a Synucleinopathy Mouse Model" Nutrients 16, no. 19: 3309. https://doi.org/10.3390/nu16193309
APA StylePradeloux, S., Coulombe, K., Ouamba, A. J. K., Isenbrandt, A., Calon, F., Roy, D., & Soulet, D. (2024). Oral Trehalose Intake Modulates the Microbiota–Gut–Brain Axis and Is Neuroprotective in a Synucleinopathy Mouse Model. Nutrients, 16(19), 3309. https://doi.org/10.3390/nu16193309