
Intermittent Fasting as a Neuroprotective Strategy: Gut–Brain Axis Modulation and Metabolic Reprogramming in Neurodegenerative Disorders

 ,
, 
 , , , and
, , , and
Abstract
1. Introduction
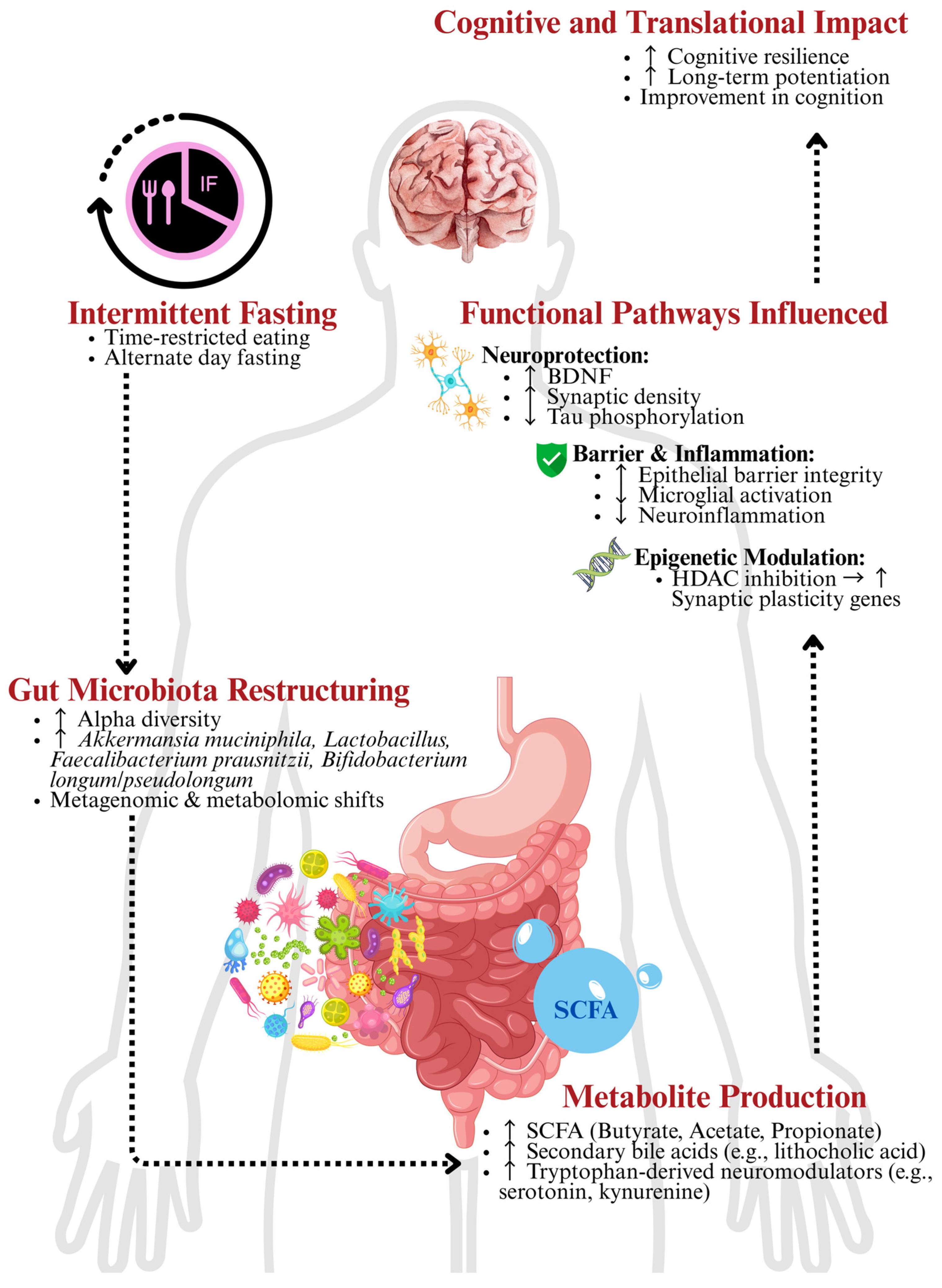
2. IF and the GBA
2.1. Gut Microbiota and Roles of SCFA
2.2. Modulation of Neuro-Immuno-Inflammation
2.3. Circadian Rhythm and Chrononutrition
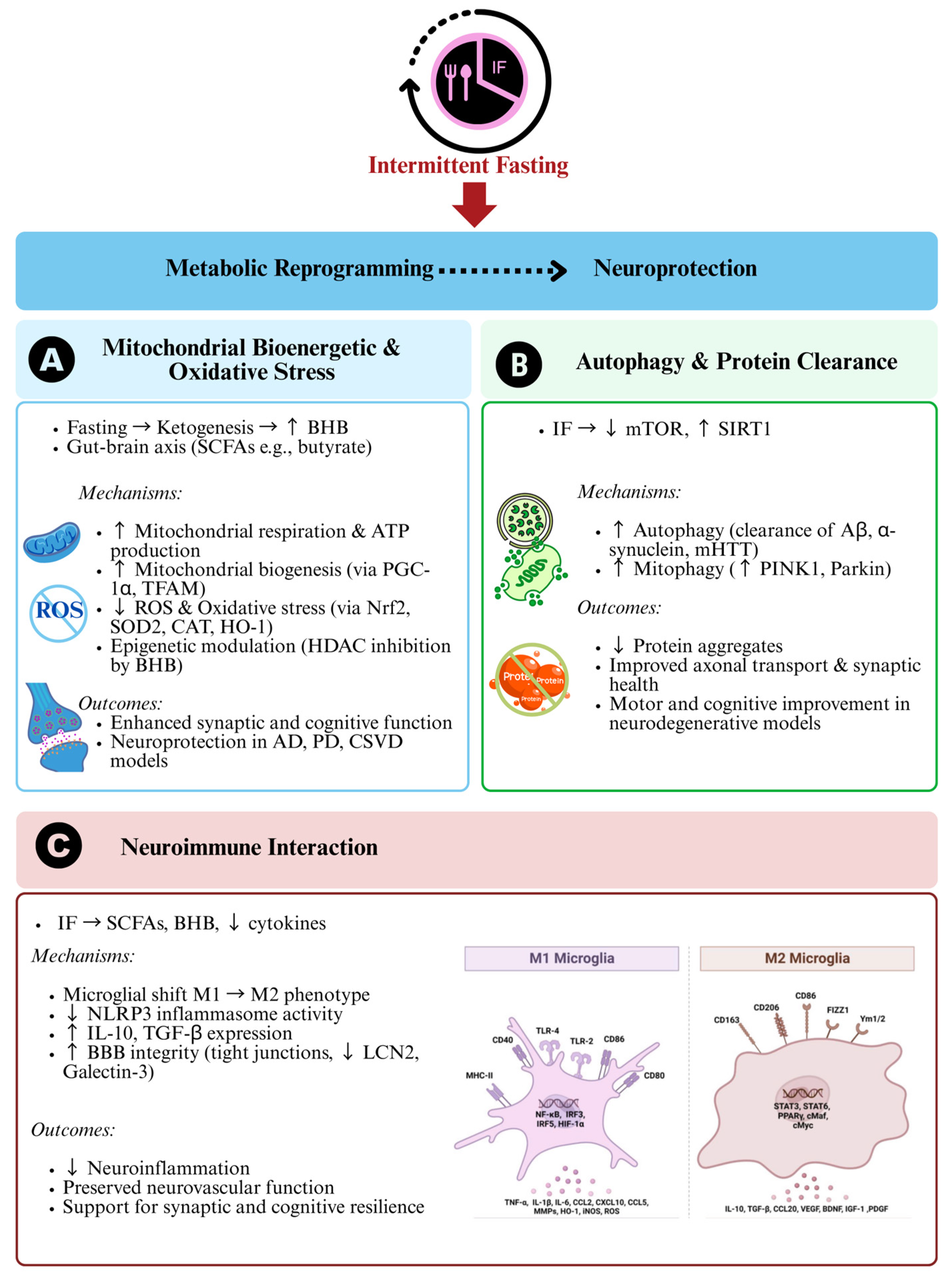
3. Metabolic Reprogramming and Neuroprotection
3.1. Mitochondrial Bioenergetics and Oxidative Stress
3.2. Autophagy and Protein Clearance
3.3. Neuroimmune Interactions
4. Disease-Specific Evidence
4.1. Alzheimer’s Disease (AD)
4.2. Parkinson’s Disease (PD)
4.3. Huntington’s Disease (HD)
4.4. Amyotrophic Lateral Sclerosis (ALS)
5. Clinical Translation and Future Directions
5.1. Safety, Adherence, and Ethical Considerations
5.2. Precision Nutrition: Toward Biomarker-Guided, Individualized Fasting
5.3. Synergistic Therapeutic Combinations: A Systems-Level Strategy
6. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| AD | Alzheimer’s disease |
| AhR | Aryl hydrocarbon receptor |
| ALS | Amyotrophic lateral sclerosis |
| AMPK | Adenosine monophosphate-activated protein kinase |
| ATP | Adenosine triphosphate |
| Aβ | Amyloid-β |
| BDNF | Brain-derived neurotrophic factor |
| BHB | β-hydroxybutyrate |
| CAT | Catalase |
| CNS | Central nervous system |
| CRP | C-reactive protein |
| CSF | Cerebrospinal fluid |
| CSVD | Cerebral small vessel disease |
| DAT-SPECT | Dopamine transporter single photon emission computed tomography |
| DMN | Default mode network |
| DXA | Dual-energy X-ray absorptiometry |
| EEG | Electroencephalography |
| FMD | Fasting-mimicking diet |
| FOXO3 | Forkhead Box O3 |
| GBA | Gut–brain axis |
| HD | Huntington’s disease |
| HDAC | Histone deacetylase |
| HO-1 | Heme oxygenase-1 |
| IF | Intermittent fasting |
| IGF-1 | Insulin-like growth factor 1 |
| IL | Interleukins |
| iNOS | Inducible nitric oxide synthase |
| LPS | lipopolysaccharides |
| MCI | Mild cognitive impairment |
| mHTT | Mutant huntingtin |
| MoCA | Montreal cognitive assessment |
| MPTP | 1-Methyl-4-phenyl-1,2,3,6-tetrahydropyridine |
| mTOR | Mechanistic target of rapamycin |
| MUNE | Motor unit number estimation |
| NfL | Neurofilament light chain |
| NF-κB | Nuclear factor-kappa B |
| NLRP3 | NOD-, LRR- and pyrin domain-containing protein 3 |
| Nrf2 | Nuclear factor erythroid 2–related factor 2 |
| PD | Parkinson’s disease |
| PET | Positron emission tomography |
| PGC-1α | Peroxisome proliferator-activated receptor-γ coactivator 1-α |
| PINK1 | PTEN-induced kinase 1 |
| ROS | Reactive oxygen species |
| SCFA | Short-chain fatty acids |
| SIRT1 | Sirtuin |
| SOD | superoxide dismutase |
| STAT3 | Signal transducer and activator of transcription 3 |
| TFAM | Mitochondrial transcription factor A |
| TGF-β | Transforming growth factor beta |
| TLR4 | Toll-like receptor 4 |
| TNF-α | Tumour necrosis factor alpha |
| UPDRS | Unified Parkinson’s disease rating scale |
References
- García-González, N.; Gonçalves-Sánchez, J.; Gómez-Nieto, R.; Gonçalves-Estella, J.M.; López, D.E. Advances and Challenges in Gene Therapy for Neurodegenerative Diseases: A Systematic Review. Int. J. Mol. Sci. 2024, 25, 12485. [Google Scholar] [CrossRef]
- Kormas, P.; Moutzouri, A. Current Psychological Approaches in Neurodegenerative Diseases. In Handbook of Computational Neurodegeneration; Vlamos, P., Kotsireas, I.S., Tarnanas, I., Eds.; Springer: Cham, Switzerland, 2022. [Google Scholar] [CrossRef]
- Wang, S.; Jiang, Y.; Yang, A.; Meng, F.; Zhang, J. The Expanding Burden of Neurodegenerative Diseases: An Unmet Medical and Social Need. Aging Dis. 2025, 16, 1–16. [Google Scholar] [CrossRef]
- Lu, S.; Zhao, Q.; Guan, Y.; Sun, Z.; Li, W.; Guo, S.; Zhang, A. The communication mechanism of the gut-brain axis and its effect on central nervous system diseases: A systematic review. Biomed. Pharmacother. 2024, 178, 117207. [Google Scholar] [CrossRef]
- Zheng, Y.; Bonfili, L.; Wei, T.; Eleuteri, A.M. Understanding the Gut–Brain Axis and Its Therapeutic Implications for Neurodegenerative Disorders. Nutrients 2023, 15, 4631. [Google Scholar] [CrossRef]
- Loh, J.S.; Mak, W.Q.; Tan, L.K.S.; Ng, C.X.; Chan, H.H.; Yeow, S.H.; Foo, J.B.; Ong, Y.S.; How, C.W.; Khaw, K.Y. Microbiota–gut–brain axis and its therapeutic applications in neurodegenerative diseases. Signal Transduct. Target. Ther. 2024, 9, 37. [Google Scholar] [CrossRef]
- Silva, Y.P.; Bernardi, A.; Frozza, R.L. The Role of Short-Chain Fatty Acids from Gut Microbiota in Gut-Brain Communication. Front. Endocrinol. 2020, 11, 25. [Google Scholar] [CrossRef]
- Vasim, I.; Majeed, C.N.; DeBoer, M.D. Intermittent Fasting and Metabolic Health. Nutrients 2022, 14, 631. [Google Scholar] [CrossRef]
- Pascual, P.E.; Rolands, M.R.; Eldridge, A.L.; Kassis, A.; Mainardi, F.; Lê, K.; Karagounis, L.G.; Gut, P.; Varady, K.A. A meta-analysis comparing the effectiveness of alternate day fasting, the 5:2 diet, and time-restricted eating for weight loss. Obesity 2022, 31, 9–21. [Google Scholar] [CrossRef]
- Anton, S.D.; Moehl, K.; Donahoo, W.T.; Marosi, K.; Lee, S.A.; Mainous, A.G., 3rd; Leeuwenburgh, C.; Mattson, M.P. Flipping the Metabolic Switch: Understanding and Applying the Health Benefits of Fasting. Obesity 2018, 26, 254–268. [Google Scholar] [CrossRef]
- Smith, R.L.; Soeters, M.R.; Wüst, R.C.I.; Houtkooper, R.H. Metabolic Flexibility as an Adaptation to Energy Resources and Requirements in Health and Disease. Endocr. Rev. 2018, 39, 489–517. [Google Scholar] [CrossRef]
- Guevara-Cruz, M.; Hernández-Gómez, K.G.; Condado-Huerta, C.; González-Salazar, L.E.; Peña-Flores, A.K.; Pichardo-Ontiveros, E.; Serralde-Zúñiga, A.E.; Sánchez-Tapia, M.; Maya, O.; Medina-Vera, I.; et al. Intermittent fasting, calorie restriction, and a ketogenic diet improve mitochondrial function by reducing lipopolysaccharide signaling in monocytes during obesity: A randomized clinical trial. Clin. Nutr. 2024, 43, 1914–1928. [Google Scholar] [CrossRef]
- Shabkhizan, R.; Haiaty, S.; Moslehian, M.S.; Bazmani, A.; Sadeghsoltani, F.; Bagheri, H.S.; Rahbarghazi, R.; Sakhinia, E. The Beneficial and Adverse Effects of Autophagic Response to Caloric Restriction and Fasting. Adv. Nutr. Int. Rev. J. 2023, 14, 1211–1225. [Google Scholar] [CrossRef]
- Tavakoli, A.; Akhgarjand, C.; Ansar, H.; Houjaghani, H.; Khormani, A.; Djafarian, K.; Rostamian, A.; Ranjbar, M.; Farsani, G.M. The effects of intermittent fasting on antioxidant and inflammatory markers and liver enzymes in postmenopausal, overweight and obese women with rheumatoid arthritis: A randomized controlled trial. Sci. Rep. 2025, 15, 2357. [Google Scholar] [CrossRef]
- Diab, R.; Dimachkie, L.; Zein, O.; Dakroub, A.; Eid, A.H. Intermittent Fasting Regulates Metabolic Homeostasis and Improves Cardiovascular Health. Cell Biochem. Biophys. 2024, 82, 1583–1597. [Google Scholar] [CrossRef]
- O’rIordan, K.J.; Moloney, G.M.; Keane, L.; Clarke, G.; Cryan, J.F. The gut microbiota-immune-brain axis: Therapeutic implications. Cell Rep. Med. 2025, 6, 101982. [Google Scholar] [CrossRef]
- Nassir, C.M.N.C.M.; Ramli, M.D.C.; Ghazali, M.M.; Jaffer, U.; Hamid, H.A.; Mehat, M.Z.; Hein, Z.M. The Microbiota–Gut–Brain Axis: Key Mechanisms Driving Glymphopathy and Cerebral Small Vessel Disease. Life 2024, 15, 3. [Google Scholar] [CrossRef]
- Hou, K.; Wu, Z.-X.; Chen, X.-Y.; Wang, J.-Q.; Zhang, D.; Xiao, C.; Zhu, D.; Koya, J.B.; Wei, L.; Li, J.; et al. Microbiota in health and diseases. Signal Transduct. Target. Ther. 2022, 7, 135. [Google Scholar] [CrossRef]
- Khan, S.I.; Rana, M.I.; Ayyaz, A.; Khan, M.Y.; Imran, M. Intermittent fasting positively modulates human gut microbial diversity and ameliorates blood lipid profile. Front. Microbiol. 2022, 13, 922727. [Google Scholar] [CrossRef]
- Zeb, F.; Osaili, T.; Obaid, R.S.; Naja, F.; Radwan, H.; Ismail, L.C.; Hasan, H.; Hashim, M.; Alam, I.; Sehar, B.; et al. Gut Microbiota and Time-Restricted Feeding/Eating: A Targeted Biomarker and Approach in Precision Nutrition. Nutrients 2023, 15, 259. [Google Scholar] [CrossRef]
- Meyers, G.R.; Samouda, H.; Bohn, T. Short Chain Fatty Acid Metabolism in Relation to Gut Microbiota and Genetic Variability. Nutrients 2022, 14, 5361. [Google Scholar] [CrossRef]
- Xu, R.-C.; Miao, W.-T.; Xu, J.-Y.; Xu, W.-X.; Liu, M.-R.; Ding, S.-T.; Jian, Y.-X.; Lei, Y.-H.; Yan, N.; Liu, H.-D. Neuroprotective Effects of Sodium Butyrate and Monomethyl Fumarate Treatment through GPR109A Modulation and Intestinal Barrier Restoration on PD Mice. Nutrients 2022, 14, 4163. [Google Scholar] [CrossRef]
- Sun, J.; Lu, L.; Lian, Y.; Xu, S.; Zhu, Y.; Wu, Y.; Lin, Q.; Hou, J.; Li, Y.; Yu, Z. Sodium butyrate attenuates microglia-mediated neuroinflammation by modulating the TLR4/MyD88/NF-κB pathway and microbiome-gut-brain axis in cardiac arrest mice. Mol. Brain 2025, 18, 13. [Google Scholar] [CrossRef]
- Bayazid, A.B.; Kim, J.G.; Azam, S.; Jeong, S.A.; Kim, D.H.; Park, C.W.; Lim, B.O. Sodium butyrate ameliorates neurotoxicity and exerts anti-inflammatory effects in high fat diet-fed mice. Food Chem. Toxicol. 2022, 159, 112743. [Google Scholar] [CrossRef]
- Zou, F.; Qiu, Y.; Huang, Y.; Zou, H.; Cheng, X.; Niu, Q.; Luo, A.; Sun, J. Effects of short-chain fatty acids in inhibiting HDAC and activating p38 MAPK are critical for promoting B10 cell generation and function. Cell Death Dis. 2021, 12, 582. [Google Scholar] [CrossRef]
- Church, J.S.; Bannish, J.A.M.; Adrian, L.A.; Martinez, K.R.; Henshaw, A.; Schwartzer, J.J. Serum short chain fatty acids mediate hippocampal BDNF and correlate with decreasing neuroinflammation following high pectin fiber diet in mice. Front. Neurosci. 2023, 17, 1134080. [Google Scholar] [CrossRef]
- Cao, T.; Zhou, X.; Zheng, X.; Cui, Y.; Tsien, J.Z.; Li, C.; Wang, H. Histone Deacetylase Inhibitor Alleviates the Neurodegenerative Phenotypes and Histone Dysregulation in Presenilins-Deficient Mice. Front. Aging Neurosci. 2018, 10, 137. [Google Scholar] [CrossRef]
- Zhao, Y.; Jia, M.; Ding, C.; Bao, B.; Li, H.; Ma, J.; Dong, W.; Gao, R.; Chen, X.; Chen, J.; et al. Time-restricted feeding mitigates Alzheimer’s disease-associated cognitive impairments via a B. pseudolongum-propionic acid-FFAR3 axis. iMeta 2025, 4, e70006. [Google Scholar] [CrossRef]
- Ye, Y.; Fu, C.; Li, Y.; Sun, J.; Li, X.; Chai, S.; Li, S.; Hou, M.; Cai, H.; Wang, Z.; et al. Alternate-day fasting improves cognitive and brain energy deficits by promoting ketone metabolism in the 3xTg mouse model of Alzheimer’s disease. Exp. Neurol. 2024, 381, 114920. [Google Scholar] [CrossRef]
- Angoorani, P.; Ejtahed, H.-S.; Hasani-Ranjbar, S.; Siadat, S.D.; Soroush, A.R.; Larijani, B. Gut microbiota modulation as a possible mediating mechanism for fasting-induced alleviation of metabolic complications: A systematic review. Nutr. Metab. 2021, 18, 105. [Google Scholar] [CrossRef]
- Tu, J.; Zhang, J.; Chen, G. Higher dietary butyrate intake is associated with better cognitive function in older adults: Evidence from a cross-sectional study. Front. Aging Neurosci. 2025, 17, 1522498. [Google Scholar] [CrossRef]
- Wu, F.; Guo, Y.; Wang, Y.; Sui, X.; Wang, H.; Zhang, H.; Xin, B.; Yang, C.; Zhang, C.; Jiang, S.; et al. Effects of Long-Term Fasting on Gut Microbiota, Serum Metabolome, and Their Association in Male Adults. Nutrients 2024, 17, 35. [Google Scholar] [CrossRef]
- Wu, J.; Man, D.; Shi, D.; Wu, W.; Wang, S.; Wang, K.; Li, Y.; Yang, L.; Bian, X.; Wang, Q.; et al. Intermittent Fasting Alleviates Risk Markers in a Murine Model of Ulcerative Colitis by Modulating the Gut Microbiome and Metabolome. Nutrients 2022, 14, 5311. [Google Scholar] [CrossRef] [PubMed]
- Ferrucci, L.; Fabbri, E. Inflammageing: Chronic inflammation in ageing, cardiovascular disease, and frailty. Nat. Rev. Cardiol. 2018, 15, 505–522. [Google Scholar] [CrossRef]
- Caldarelli, M.; Rio, P.; Marrone, A.; Giambra, V.; Gasbarrini, A.; Gambassi, G.; Cianci, R. Inflammaging: The Next Challenge—Exploring the Role of Gut Microbiota, Environmental Factors, and Sex Differences. Biomedicines 2024, 12, 1716. [Google Scholar] [CrossRef]
- Di Vincenzo, F.; Del Gaudio, A.; Petito, V.; Lopetuso, L.R.; Scaldaferri, F. Gut microbiota, intestinal permeability, and systemic inflammation: A narrative review. Intern. Emerg. Med. 2023, 19, 275–293. [Google Scholar] [CrossRef]
- Violi, F.; Cammisotto, V.; Bartimoccia, S.; Pignatelli, P.; Carnevale, R.; Nocella, C. Gut-derived low-grade endotoxaemia, atherothrombosis and cardiovascular disease. Nat. Rev. Cardiol. 2022, 20, 24–37. [Google Scholar] [CrossRef]
- Kalyan, M.; Tousif, A.H.; Sonali, S.; Vichitra, C.; Sunanda, T.; Praveenraj, S.S.; Ray, B.; Gorantla, V.R.; Rungratanawanich, W.; Mahalakshmi, A.M.; et al. Role of Endogenous Lipopolysaccharides in Neurological Disorders. Cells 2022, 11, 4038. [Google Scholar] [CrossRef]
- Zhang, Y.; Zhu, X.; Yu, X.; Novák, P.; Gui, Q.; Yin, K. Enhancing intestinal barrier efficiency: A novel metabolic diseases therapy. Front. Nutr. 2023, 10, 1120168. [Google Scholar] [CrossRef]
- Lee, C.; Kim, B.G.; Kim, J.H.; Chun, J.; Im, J.P.; Kim, J.S. Sodium butyrate inhibits the NF-kappa B signaling pathway and histone deacetylation, and attenuates experimental colitis in an IL-10 independent manner. Int. Immunopharmacol. 2017, 51, 47–56. [Google Scholar] [CrossRef]
- Wang, W.; Dernst, A.; Martin, B.; Lorenzi, L.; Cadefau-Fabregat, M.; Phulphagar, K.; Wagener, A.; Budden, C.; Stair, N.; Wagner, T.; et al. Butyrate and propionate are microbial danger signals that activate the NLRP3 inflammasome in human macrophages upon TLR stimulation. Cell Rep. 2024, 43, 114736. [Google Scholar] [CrossRef]
- Wang, X.; Yang, Q.; Liao, Q.; Li, M.; Zhang, P.; Santos, H.O.; Kord-Varkaneh, H.; Abshirini, M. Effects of intermittent fasting diets on plasma concentrations of inflammatory biomarkers: A systematic review and meta-analysis of randomized controlled trials. Nutrition 2020, 79–80, 110974. [Google Scholar] [CrossRef]
- Trepanowski, J.F.; Kroeger, C.M.; Barnosky, A.; Klempel, M.; Bhutani, S.; Hoddy, K.K.; Rood, J.; Ravussin, E.; Varady, K.A. Effects of alternate-day fasting or daily calorie restriction on body composition, fat distribution, and circulating adipokines: Secondary analysis of a randomized controlled trial. Clin. Nutr. 2018, 37, 1871–1878. [Google Scholar] [CrossRef] [PubMed]
- Jordan, S.; Tung, N.; Casanova-Acebes, M.; Chang, C.; Cantoni, C.; Zhang, D.; Wirtz, T.H.; Naik, S.; Rose, S.A.; Brocker, C.N.; et al. Dietary Intake Regulates the Circulating Inflammatory Monocyte Pool. Cell 2019, 178, 1102–1114.e17. [Google Scholar] [CrossRef]
- Rangan, P.; Lobo, F.; Parrella, E.; Rochette, N.; Morselli, M.; Stephen, T.-L.; Cremonini, A.L.; Tagliafico, L.; Persia, A.; Caffa, I.; et al. Fasting-mimicking diet cycles reduce neuroinflammation to attenuate cognitive decline in Alzheimer’s models. Cell Rep. 2022, 40, 111417. [Google Scholar] [CrossRef] [PubMed]
- Wu, L.; Zhao, Y.; Gong, X.; Liang, Z.; Yu, J.; Wang, J.; Zhang, Y.; Wang, X.; Shu, X.; Bao, J. Intermittent Fasting Ameliorates β-Amyloid Deposition and Cognitive Impairment Accompanied by Decreased Lipid Droplet Aggregation Within Microglia in an Alzheimer’s Disease Model. Mol. Nutr. Food Res. 2025, 69, e202400660. [Google Scholar] [CrossRef]
- Whittaker, D.S.; Akhmetova, L.; Carlin, D.; Romero, H.; Welsh, D.K.; Colwell, C.S.; Desplats, P. Circadian modulation by time-restricted feeding rescues brain pathology and improves memory in mouse models of Alzheimer’s disease. Cell Metab. 2023, 35, 1704–1721.e6. [Google Scholar] [CrossRef] [PubMed]
- Salminen, A. Activation of aryl hydrocarbon receptor (AhR) in Alzheimer’s disease: Role of tryptophan metabolites generated by gut host-microbiota. J. Mol. Med. 2023, 101, 201–222. [Google Scholar] [CrossRef]
- Ayyar, V.S.; Sukumaran, S. Circadian rhythms: Influence on physiology, pharmacology, and therapeutic interventions. J. Pharmacokinet. Pharmacodyn. 2021, 48, 321–338. [Google Scholar] [CrossRef]
- Nassan, M.; Videnovic, A. Circadian rhythms in neurodegenerative disorders. Nat. Rev. Neurol. 2021, 18, 7–24. [Google Scholar] [CrossRef]
- Zhao, E.; Tait, C.; Minacapelli, C.D.; Catalano, C.; Rustgi, V.K. Circadian Rhythms, the Gut Microbiome, and Metabolic Disorders. Gastro Hep Adv. 2022, 1, 93–105. [Google Scholar] [CrossRef]
- Charlot, A.; Hutt, F.; Sabatier, E.; Zoll, J. Beneficial Effects of Early Time-Restricted Feeding on Metabolic Diseases: Importance of Aligning Food Habits with the Circadian Clock. Nutrients 2021, 13, 1405. [Google Scholar] [CrossRef] [PubMed]
- Jamshed, H.; Beyl, R.A.; della Manna, D.L.; Yang, E.S.; Ravussin, E.; Peterson, C.M. Early Time-Restricted Feeding Improves 24-Hour Glucose Levels and Affects Markers of the Circadian Clock, Aging, and Autophagy in Humans. Nutrients 2019, 11, 1234. [Google Scholar] [CrossRef] [PubMed]
- Ye, Y.; Xu, H.; Xie, Z.; Wang, L.; Sun, Y.; Yang, H.; Hu, D.; Mao, Y. Time-Restricted Feeding Reduces the Detrimental Effects of a High-Fat Diet, Possibly by Modulating the Circadian Rhythm of Hepatic Lipid Metabolism and Gut Microbiota. Front. Nutr. 2020, 7, 596285. [Google Scholar] [CrossRef]
- Manoogian, E.N.C.; Chow, L.S.; Taub, P.R.; Laferrère, B.; Panda, S. Time-restricted Eating for the Prevention and Management of Metabolic Diseases. Endocr. Rev. 2021, 43, 405–436. [Google Scholar] [CrossRef]
- Chawla, S.; Beretoulis, S.; Deere, A.; Radenkovic, D. The Window Matters: A Systematic Review of Time Restricted Eating Strategies in Relation to Cortisol and Melatonin Secretion. Nutrients 2021, 13, 2525. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Y.; Jia, M.; Chen, W.; Liu, Z. The neuroprotective effects of intermittent fasting on brain aging and neurodegenerative diseases via regulating mitochondrial function. Free. Radic. Biol. Med. 2022, 182, 206–218. [Google Scholar] [CrossRef]
- Qian, L.; Zhu, Y.; Deng, C.; Liang, Z.; Chen, J.; Chen, Y.; Wang, X.; Liu, Y.; Tian, Y.; Yang, Y. Peroxisome proliferator-activated receptor gamma coactivator-1 (PGC-1) family in physiological and pathophysiological process and diseases. Signal Transduct. Target. Ther. 2024, 9, 50. [Google Scholar] [CrossRef]
- Mehramiz, M.; Porter, T.; O’bRien, E.K.; Rainey-Smith, S.R.; Laws, S.M.; Atwood, C. A Potential Role for Sirtuin-1 in Alzheimer’s Disease: Reviewing the Biological and Environmental Evidence. J. Alzheimer’s Dis. Rep. 2023, 7, 823–843. [Google Scholar] [CrossRef]
- Wang, W.; Zhao, F.; Ma, X.; Perry, G.; Zhu, X. Mitochondria dysfunction in the pathogenesis of Alzheimer’s disease: Recent advances. Mol. Neurodegener. 2020, 15, 30. [Google Scholar] [CrossRef]
- Patai, R.; Patel, K.; Csik, B.; Gulej, R.; Nagaraja, R.Y.; Nagy, D.; Chandragiri, S.S.; Shanmugarama, S.; Kordestan, K.V.; Nagykaldi, M.; et al. Aging, mitochondrial dysfunction, and cerebral microhemorrhages: A preclinical evaluation of SS-31 (elamipretide) and development of a high-throughput machine learning-driven imaging pipeline for cerebromicrovascular protection therapeutic screening. GeroScience 2025, 47, 4871–4887. [Google Scholar] [CrossRef]
- Morais, L.H.; Stiles, L.; Freeman, M.; Oguienko, A.D.; Hoang, J.D.; Jones, J.; Quan, B.; Devine, J.; Bois, J.S.; Chou, T.F.; et al. The gut microbiome promotes mitochondrial respiration in the brain of a Parkinson’s disease mouse model. bioRxiv 2024. [Google Scholar] [CrossRef]
- Hernandez, A.; Truckenbrod, L.; Federico, Q.; Campos, K.; Moon, B.; Ferekides, N.; Hoppe, M.; D’aGostino, D.; Burke, S. Metabolic switching is impaired by aging and facilitated by ketosis independent of glycogen. Aging 2020, 12, 7963–7984. [Google Scholar] [CrossRef] [PubMed]
- Newman, J.C.; Verdin, E. β-Hydroxybutyrate: A Signaling Metabolite. Annu. Rev. Nutr. 2017, 37, 51–76. [Google Scholar] [CrossRef] [PubMed]
- Shimazu, T.; Hirschey, M.D.; Newman, J.; He, W.; Shirakawa, K.; Le Moan, N.; Grueter, C.A.; Lim, H.; Saunders, L.R.; Stevens, R.D.; et al. Suppression of oxidative stress by β-hydroxybutyrate, an endogenous histone deacetylase inhibitor. Science 2013, 339, 211–214. [Google Scholar] [CrossRef]
- Wu, Y.; Gong, Y.; Luan, Y.; Li, Y.; Liu, J.; Yue, Z.; Yuan, B.; Sun, J.; Xie, C.; Li, L.; et al. BHBA treatment improves cognitive function by targeting pleiotropic mechanisms in transgenic mouse model of Alzheimer’s disease. FASEB J. 2019, 34, 1412–1429. [Google Scholar] [CrossRef]
- Tieu, K.; Perier, C.; Caspersen, C.; Teismann, P.; Wu, D.C.; Yan, S.D.; Naini, A.; Vila, M.; Jackson-Lewis, V.; Ramasamy, R.; et al. D-beta-hydroxybutyrate rescues mitochondrial respiration and mitigates features of Parkinson disease. J. Clin. Investig. 2003, 112, 892–901. [Google Scholar] [CrossRef]
- Leclercq, S.; Le Roy, T.; Furgiuele, S.; Coste, V.; Bindels, L.B.; Leyrolle, Q.; Neyrinck, A.M.; Quoilin, C.; Amadieu, C.; Petit, G.; et al. Gut Microbiota-Induced Changes in β-Hydroxybutyrate Metabolism Are Linked to Altered Sociability and Depression in Alcohol Use Disorder. Cell Rep. 2020, 33, 108238. [Google Scholar] [CrossRef]
- Zhang, H.; Tao, Y.; Leng, S.X. Ketogenic Diet: An Effective Treatment Approach for Neurodegenerative Diseases. Curr. Neuropharmacol. 2022, 20, 2303–2319. [Google Scholar] [CrossRef]
- Waldman, H.S.; McAllister, M.J. Exogenous Ketones as Therapeutic Signaling Molecules in High-Stress Occupations: Implications for Mitigating Oxidative Stress and Mitochondrial Dysfunction in Future Research. Nutr. Metab. Insights 2020, 13, 1178638820979029. [Google Scholar] [CrossRef]
- Gudden, J.; Vasquez, A.A.; Bloemendaal, M. The Effects of Intermittent Fasting on Brain and Cognitive Function. Nutrients 2021, 13, 3166. [Google Scholar] [CrossRef]
- Park, H.; Kang, J.-H.; Lee, S. Autophagy in Neurodegenerative Diseases: A Hunter for Aggregates. Int. J. Mol. Sci. 2020, 21, 3369. [Google Scholar] [CrossRef] [PubMed]
- Boeckholt, T. Intermittent Fasting (IF) Promotes Longevity Through Alterations of the Mammalian Target of Rapamycin (mTOR) and the Epigenome. Master’s Thesis, South Dakota State University, Brookings, SD, USA, 2020. Available online: https://openprairie.sdstate.edu/cgi/viewcontent.cgi?article=1012&context=biomicro_plan-b (accessed on 20 March 2025).
- Tagliafico, L.; Nencioni, A.; Monacelli, F. Fasting and Cognitive Impairment. Nutrients 2023, 15, 5108. [Google Scholar] [CrossRef]
- Szegő, É.M.; Höfs, L.; Antoniou, A.; Dinter, E.; Bernhardt, N.; Schneider, A.; Di Monte, D.A.; Falkenburger, B.H. Intermittent fasting reduces alpha-synuclein pathology and functional decline in a mouse model of Parkinson’s disease. Nat. Commun. 2025, 16, 4470. [Google Scholar] [CrossRef]
- Liang, Y.; Sigrist, S. Autophagy and proteostasis in the control of synapse aging and disease. Curr. Opin. Neurobiol. 2018, 48, 113–121. [Google Scholar] [CrossRef] [PubMed]
- Zeng, K.; Yu, X.; Mahaman, Y.A.R.; Wang, J.-Z.; Liu, R.; Li, Y.; Wang, X. Defective mitophagy and the etiopathogenesis of Alzheimer’s disease. Transl. Neurodegener. 2022, 11, 32. [Google Scholar] [CrossRef]
- Braun, M.M.; Puglielli, L. Defective PTEN-induced kinase 1/Parkin mediated mitophagy and neurodegenerative diseases. Front. Cell. Neurosci. 2022, 16, 1031153. [Google Scholar] [CrossRef]
- Ma, L.; Chen, Y.-Q.; You, Z.-J.; Jiang, Z.-S.; Fang, Y.; Dong, L. Intermittent fasting attenuates lipopolysaccharide-induced acute lung injury in mice by modulating macrophage polarization. J. Nutr. Biochem. 2022, 110, 109133. [Google Scholar] [CrossRef] [PubMed]
- Zhang, D.; Jian, Y.-P.; Zhang, Y.-N.; Li, Y.; Gu, L.-T.; Sun, H.-H.; Liu, M.-D.; Zhou, H.-L.; Wang, Y.-S.; Xu, Z.-X. Short-chain fatty acids in diseases. Cell Commun. Signal. 2023, 21, 212. [Google Scholar] [CrossRef]
- Youm, Y.-H.; Nguyen, K.Y.; Grant, R.W.; Goldberg, E.L.; Bodogai, M.; Kim, D.; D’Agostino, D.; Planavsky, N.; Lupfer, C.; Kanneganti, T.-D.; et al. The ketone metabolite β-hydroxybutyrate blocks NLRP3 inflammasome–mediated inflammatory disease. Nat. Med. 2015, 21, 263–269. [Google Scholar] [CrossRef]
- Han, S.-C.; Kang, J.-I.; Choi, Y.K.; Boo, H.-J.; Yoon, W.-J.; Kang, H.-K.; Yoo, E.-S. Intermittent Fasting Modulates Immune Response by Generating Tregs via TGF-β Dependent Mechanisms in Obese Mice with Allergic Contact Dermatitis. Biomol. Ther. 2023, 32, 136–145. [Google Scholar] [CrossRef]
- Lee, J.; An, H.S.; Shin, H.J.; Jang, H.M.; Im, C.O.; Jeong, Y.; Eum, K.; Yoon, S.; Lee, S.J.; Jeong, E.A.; et al. Intermittent Fasting Reduces Neuroinflammation and Cognitive Impairment in High-Fat Diet-Fed Mice by Downregulating Lipocalin-2 and Galectin-3. Nutrients 2024, 16, 159. [Google Scholar] [CrossRef] [PubMed]
- Peng, X.; Fan, R.; Xie, L.; Shi, X.; Wang, F.; Xu, W.; Dong, K.; Zhang, S.; Ma, D.; Yu, X.; et al. Time-restricted feeding rescues circadian disruption-aggravated progression of Alzheimer’s disease in diabetic mice. J. Nutr. Biochem. 2022, 110, 109128. [Google Scholar] [CrossRef] [PubMed]
- Jeong, S.; Davis, C.K.; Vemuganti, R. Mechanisms of time-restricted feeding-induced neuroprotection and neuronal plasticity in ischemic stroke as a function of circadian rhythm. Exp. Neurol. 2024, 383, 115045. [Google Scholar] [CrossRef] [PubMed]
- Komen, J.C.; Thorburn, D.R. Turn up the power—Pharmacological activation of mitochondrial biogenesis in mouse models. Br. J. Pharmacol. 2014, 171, 1818–1836. [Google Scholar] [CrossRef]
- Dai, S.; Wei, J.; Zhang, H.; Luo, P.; Yang, Y.; Jiang, X.; Fei, Z.; Liang, W.; Jiang, J.; Li, X. Intermittent fasting reduces neuroinflammation in intracerebral hemorrhage through the Sirt3/Nrf2/HO-1 pathway. J. Neuroinflammation 2022, 19, 1–15. [Google Scholar] [CrossRef]
- Anton, S.D.; Lee, S.A.; Donahoo, W.T.; McLaren, C.; Manini, T.; Leeuwenburgh, C.; Pahor, M. The Effects of Time Restricted Feeding on Overweight, Older Adults: A Pilot Study. Nutrients 2019, 11, 1500. [Google Scholar] [CrossRef]
- Elias, A.; Padinjakara, N.; Lautenschlager, N.T. Effects of intermittent fasting on cognitive health and Alzheimer’s disease. Nutr. Rev. 2023, 81, 1225–1233. [Google Scholar] [CrossRef]
- Daas, M.; de Roos, N. Intermittent fasting contributes to aligned circadian rhythms through interactions with the gut microbiome. Benef. Microbes 2021, 12, 147–162. [Google Scholar] [CrossRef]
- Gędek, A.; Koziorowski, D.; Szlufik, S. Assessment of factors influencing glymphatic activity and implications for clinical medicine. Front. Neurol. 2023, 14, 1232304. [Google Scholar] [CrossRef]
- Reddy, O.C.; van der Werf, Y.D. The Sleeping Brain: Harnessing the Power of the Glymphatic System through Lifestyle Choices. Brain Sci. 2020, 10, 868. [Google Scholar] [CrossRef]
- Yang, C.; Zhang, T.; Wang, W.; Xiang, Y.; Huang, Q.; Xie, C.; Zhao, L.; Zheng, H.; Yang, Y.; Gao, H. Brain-Region Specific Metabolic Abnormalities in Parkinson’s Disease and Levodopa-Induced Dyskinesia. Front. Aging Neurosci. 2020, 12, 75. [Google Scholar] [CrossRef] [PubMed]
- Jiang, Z.; Yin, X.; Wang, M.; Wang, Y.; Li, F.; Gao, Y.; Han, G.; Gao, Z.; Wang, Z. β-Hydroxybutyrate alleviates pyroptosis in MPP+/MPTP-induced Parkinson’s disease models via inhibiting STAT3/NLRP3/GSDMD pathway. Int. Immunopharmacol. 2022, 113, 109451. [Google Scholar] [CrossRef]
- Mierziak, J.; Burgberger, M.; Wojtasik, W. 3-Hydroxybutyrate as a Metabolite and a Signal Molecule Regulating Processes of Living Organisms. Biomolecules 2021, 11, 402. [Google Scholar] [CrossRef] [PubMed]
- Chui, Z.S.W.; Chan, L.M.L.; Zhang, E.W.H.; Liang, S.; Choi, E.P.H.; Lok, K.Y.W.; Tun, H.M.; Kwok, J.Y.Y. Effects of microbiome-based interventions on neurodegenerative diseases: A systematic review and meta-analysis. Sci. Rep. 2024, 14, 9558. [Google Scholar] [CrossRef] [PubMed]
- Pramono, A.; Ardiaria, M.; Limijadi, E.K.S.; Noer, E.R.; Lestari, E.S.; Siswanto, F.M. Intermittent fasting modulates human gut microbiota diversity in a phenotype-dependent manner: A systematic review. Biosci. Microbiota Food Health 2024, 43, 170–182. [Google Scholar] [CrossRef]
- Fock, E.; Parnova, R. Mechanisms of Blood–Brain Barrier Protection by Microbiota-Derived Short-Chain Fatty Acids. Cells 2023, 12, 657. [Google Scholar] [CrossRef]
- Neth, B.J.; Bauer, B.A.; Benarroch, E.E.; Savica, R. The Role of Intermittent Fasting in Parkinson’s Disease. Front. Neurol. 2021, 12, 682184. [Google Scholar] [CrossRef]
- Kamel, W.; Al Hashel, J.; Damier, P. How do Parkinson’s disease patients manage Ramadan fasting? An observational study. Rev. Neurol. 2019, 175, 560–563. [Google Scholar] [CrossRef]
- Akdemir, Ü.Ö.; Tokçaer, A.B.; Atay, L.Ö. Dopamine transporter SPECT imaging in Parkinson’s disease and parkinsonian disorders. Turk. J. Med. Sci. 2021, 51, 400–410. [Google Scholar] [CrossRef]
- Wells, R.G.; Neilson, L.E.; McHill, A.W.; Hiller, A.L. Dietary fasting and time-restricted eating in Huntington’s disease: Therapeutic potential and underlying mechanisms. Transl. Neurodegener. 2024, 13, 17. [Google Scholar] [CrossRef]
- Ehrnhoefer, D.E.; Martin, D.D.O.; Schmidt, M.E.; Qiu, X.; Ladha, S.; Caron, N.S.; Skotte, N.H.; Nguyen, Y.T.N.; Vaid, K.; Southwell, A.L.; et al. Preventing mutant huntingtin proteolysis and intermittent fasting promote autophagy in models of Huntington disease. Acta Neuropathol. Commun. 2018, 6, 16. [Google Scholar] [CrossRef]
- Khan, W.; Alusi, S.; Tawfik, H.; Hussain, A.; Gadekallu, T.R. The relationship between non-motor features and weight-loss in the premanifest stage of Huntington’s disease. PLoS ONE 2021, 16, e0253817. [Google Scholar] [CrossRef] [PubMed]
- Phillips, M.C.L.; McManus, E.J.; Brinkhuis, M.; Romero-Ferrando, B. Time-Restricted Ketogenic Diet in Huntington’s Disease: A Case Study. Front. Behav. Neurosci. 2022, 16, 931636. [Google Scholar] [CrossRef]
- Wells, R.G.; Neilson, L.E.; McHill, A.W.; Hiller, A.L.; Sipilä, J. Time-restricted eating in early-stage Huntington’s disease: A 12-week interventional clinical trial protocol. PLoS ONE 2025, 20, e0319253. [Google Scholar] [CrossRef] [PubMed]
- Scaricamazza, S.; Salvatori, I.; Amadio, S.; Nesci, V.; Torcinaro, A.; Giacovazzo, G.; Primiano, A.; Gloriani, M.; Candelise, N.; Pieroni, L.; et al. Repurposing of Trimetazidine for amyotrophic lateral sclerosis: A study in SOD1G93Amice. Br. J. Pharmacol. 2022, 179, 1732–1752. [Google Scholar] [CrossRef]
- Mehrabani, S.; Bagherniya, M.; Askari, G.; Read, M.I.; Sahebkar, A. The effect of fasting or calorie restriction on mitophagy induction: A literature review. J. Cachex- Sarcopenia Muscle 2020, 11, 1447–1458. [Google Scholar] [CrossRef]
- Beard, E.; Lengacher, S.; Dias, S.; Magistretti, P.J.; Finsterwald, C. Astrocytes as Key Regulators of Brain Energy Metabolism: New Therapeutic Perspectives. Front. Physiol. 2022, 12, 825816. [Google Scholar] [CrossRef] [PubMed]
- Phillips, M.C.L.; Johnston, S.E.; Simpson, P.; Chang, D.K.; Mather, D.; Dick, R.J. Time-restricted ketogenic diet in amyotrophic lateral sclerosis: A case study. Front. Neurol. 2024, 14, 1329541. [Google Scholar] [CrossRef]
- Kovács, Z.; Brunner, B.; Ari, C. Beneficial Effects of Exogenous Ketogenic Supplements on Aging Processes and Age-Related Neurodegenerative Diseases. Nutrients 2021, 13, 2197. [Google Scholar] [CrossRef]
- Verde, F.; Otto, M.; Silani, V. Neurofilament Light Chain as Biomarker for Amyotrophic Lateral Sclerosis and Frontotemporal Dementia. Front. Neurosci. 2021, 15, 679199. [Google Scholar] [CrossRef]
- De Marchi, F.; Munitic, I.; Amedei, A.; Berry, J.D.; Feldman, E.L.; Aronica, E.; Nardo, G.; Van Weehaeghe, D.; Niccolai, E.; Prtenjaca, N.; et al. Interplay between immunity and amyotrophic lateral sclerosis: Clinical impact. Neurosci. Biobehav. Rev. 2021, 127, 958–978. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Z.-L.; Jia, X.-B.; Sun, M.-F.; Zhu, Y.-L.; Qiao, C.-M.; Zhang, B.-P.; Zhao, L.-P.; Yang, Q.; Cui, C.; Chen, X.; et al. Neuroprotection of Fasting Mimicking Diet on MPTP-Induced Parkinson’s Disease Mice via Gut Microbiota and Metabolites. Neurotherapeutics 2019, 16, 741–760. [Google Scholar] [CrossRef] [PubMed]
- Norman, K.; Haß, U.; Pirlich, M. Malnutrition in Older Adults—Recent Advances and Remaining Challenges. Nutrients 2021, 13, 2764. [Google Scholar] [CrossRef]
- Sharifi, S.; Rostami, F.; Khorzoughi, K.B.; Rahmati, M. Effect of time-restricted eating and intermittent fasting on cognitive function and mental health in older adults: A systematic review. Prev. Med. Rep. 2024, 42, 102757. [Google Scholar] [CrossRef] [PubMed]
- Henderson, Y.O.; Bithi, N.; Link, C.; Yang, J.; Schugar, R.; Llarena, N.; Brown, J.M.; Hine, C. Late-life intermittent fasting decreases aging-related frailty and increases renal hydrogen sulfide production in a sexually dimorphic manner. GeroScience 2021, 43, 1527–1554. [Google Scholar] [CrossRef]
- Clayton, D.J.; Varley, I.; Papageorgiou, M. Intermittent fasting and bone health: A bone of contention? Br. J. Nutr. 2023, 130, 1487–1499. [Google Scholar] [CrossRef]
- O’COnnor, S.G.; Boyd, P.; Bailey, C.P.; Nebeling, L.; Reedy, J.; Czajkowski, S.M.; Shams-White, M.M. A qualitative exploration of facilitators and barriers of adherence to time-restricted eating. Appetite 2022, 178, 106266. [Google Scholar] [CrossRef]
- Wang, P.; Tadeo, X.; Chew, H.S.J.; Sapanel, Y.; Ong, Y.H.; Leung, N.Y.T.; Chow, E.K.-H.; Ho, D.; Faghih, R. N-of-1 health optimization: Digital monitoring of biomarker dynamics to gamify adherence to metabolic switching. PNAS Nexus 2024, 3, pgae214. [Google Scholar] [CrossRef]
- Kim, B.H.; Joo, Y.; Kim, M.-S.; Choe, H.K.; Tong, Q.; Kwon, O. Effects of Intermittent Fasting on the Circulating Levels and Circadian Rhythms of Hormones. Endocrinol. Metab. 2021, 36, 745–756. [Google Scholar] [CrossRef]
- Bahammam, A.S.; Almeneessier, A.S.; Sharif, M.M.; Bahammam, S.; Nashwan, S.Z.; Perumal, S.R.P.; Cardinali, D.P.; Alzoghaibi, M. The influence of intermittent fasting on the circadian pattern of melatonin while controlling for caloric intake, energy expenditure, light exposure, and sleep schedules: A preliminary report. Ann. Thorac. Med. 2017, 12, 183–190. [Google Scholar] [CrossRef]
- Gabel, K.; Hamm, A.; Czyzewski, O.; Perez, J.S.; Fought-Boudaia, A.; Motl, R.W.; Hibbing, P.R. A Narrative Review of Intermittent Fasting with Exercise. J. Acad. Nutr. Diet. 2024, 125, 153–171. [Google Scholar] [CrossRef] [PubMed]
- Khalafi, M.; Symonds, M.E.; Maleki, A.H.; Sakhaei, M.H.; Ehsanifar, M.; Rosenkranz, S.K. Combined versus independent effects of exercise training and intermittent fasting on body composition and cardiometabolic health in adults: A systematic review and meta-analysis. Nutr. J. 2024, 23, 7. [Google Scholar] [CrossRef] [PubMed]
- Tay, A.; Pringle, H.; Penning, E.; Plank, L.D.; Murphy, R. PROFAST: A Randomized Trial Assessing the Effects of Intermittent Fasting and Lacticaseibacillus rhamnosus Probiotic among People with Prediabetes. Nutrients 2020, 12, 3530. [Google Scholar] [CrossRef] [PubMed]
- Ma, X.; Shin, Y.-J.; Jang, H.-M.; Joo, M.-K.; Yoo, J.-W.; Kim, D.-H. Lactobacillus rhamnosus and Bifidobacterium longum alleviate colitis and cognitive impairment in mice by regulating IFN-γ to IL-10 and TNF-α to IL-10 expression ratios. Sci. Rep. 2021, 11, 20659. [Google Scholar] [CrossRef]
- Lee, H.-J.; Lee, K.-E.; Kim, J.-K.; Kim, D.-H. Suppression of gut dysbiosis by Bifidobacterium longum alleviates cognitive decline in 5XFAD transgenic and aged mice. Sci. Rep. 2019, 9, 11814. [Google Scholar] [CrossRef]
- Blagosklonny, M.V. Fasting and rapamycin: Diabetes versus benevolent glucose intolerance. Cell Death Dis. 2019, 10, 607. [Google Scholar] [CrossRef]

{kind=link}
{kind=link}
| Mediator/Pathway | Site of Action | Effect of IF | Neurodegenerative Relevance | Ref. |
|---|---|---|---|---|
| TLR4 |
|
| Reduces microglial activation and neuroinflammation | [37,38] |
| NF-κB |
|
| Limits transcription of pro-inflammatory cytokines | [40] |
| NLRP3 Inflammasome |
|
| Reduces IL-1β secretion and neurotoxicity | [41] |
| Tight junction proteins (e.g., Occludin, Claudin-1) |
|
| Prevents systemic inflammation via endotoxin leakage | [39] |
| SCFA (e.g., Butyrate, Propionate) |
|
| Enhances BDNF, reduces oxidative and inflammatory damage | [21,24,25] |
| IL-6, TNF-α, CRP |
|
| Reduced systemic-to-CNS inflammatory signalling | [42,43] |
| AhR |
|
| Regulates mucosal immunity and neuroimmune crosstalk | [48] |
| Disease | Study Type | Model/Population | IF Protocol | Key Findings |
|---|---|---|---|---|
| Alzheimer’s Disease (AD) | Preclinical [29] | 3xTg-AD mice | ADF for 3 months |
|
| Clinical [88] | Elderly individuals with subjective cognitive decline | 16:8 TRE for 12 weeks |
| |
| Clinical [89] | MCI patients | 16:8 TRE for 12 weeks |
| |
| Parkinson’s Disease (PD) | Preclinical [114] | MPTP-induced PD mice | fasting mimicking diet (FMD), fasting 3 days followed by 4 days of refeeding for three 1-week cycles |
|
| Clinical [99,100] | PD patients practising Ramadan fasting | ~14-h daily fast for 30 days |
| |
| Huntington’s Disease (HD) | Preclinical [102,103] | R6/2 transgenic mice | ADF for 8 weeks |
|
| Clinical [106] | Prodromal HD (NCT06490367) | TRE (10-h feeding) for 12 weeks |
| |
| Amyotrophic Lateral Sclerosis (ALS) | Preclinical [107] | SOD1-G93A transgenic mice | ADF starting pre-symptomatically |
|
| Clinical [110] | ALS patients attempting modified fasting | IF with nutritional support |
|
| Patient Profile | Suggested IF Strategy | Potential Risks | Mechanistic Rationale | Monitoring Biomarkers |
|---|---|---|---|---|
| Elderly with mild cognitive impairment | 12:12 progressing to 14:10 TRF |
|
|
|
| Parkinson’s disease (early-stage) | 16:8 TRF or alternate day fasting |
|
|
|
| Genetic risk carriers (e.g., APOE4+) | 14:10 TRF with Mediterranean meals |
|
|
|
| Metabolically obese with neuroinflammation | Alternate-day or 5:2 fasting |
|
|
|
| ALS with weight loss risk | 12:12 mild TRF with caloric support |
|
|
|
| Circadian misaligned (e.g., night-shift workers or AD) | Chronotype-adjusted 10:14 TRF |
|
|
|
| HD patients (early symptomatic) | 14:10 TRF with high-protein support |
|
|
|
| High-performing adults seeking neuroprotection | 16:8 TRF with exercise pairing |
|
|
|
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hein, Z.M.; Arbain, M.F.F.; Kumar, S.; Mehat, M.Z.; Hamid, H.A.; Che Ramli, M.D.; Che Mohd Nassir, C.M.N. Intermittent Fasting as a Neuroprotective Strategy: Gut–Brain Axis Modulation and Metabolic Reprogramming in Neurodegenerative Disorders. Nutrients 2025, 17, 2266. https://doi.org/10.3390/nu17142266
Hein ZM, Arbain MFF, Kumar S, Mehat MZ, Hamid HA, Che Ramli MD, Che Mohd Nassir CMN. Intermittent Fasting as a Neuroprotective Strategy: Gut–Brain Axis Modulation and Metabolic Reprogramming in Neurodegenerative Disorders. Nutrients. 2025; 17(14):2266. https://doi.org/10.3390/nu17142266
Chicago/Turabian StyleHein, Zaw Myo, Muhammad Faqhrul Fahmy Arbain, Suresh Kumar, Muhammad Zulfadli Mehat, Hafizah Abdul Hamid, Muhammad Danial Che Ramli, and Che Mohd Nasril Che Mohd Nassir. 2025. "Intermittent Fasting as a Neuroprotective Strategy: Gut–Brain Axis Modulation and Metabolic Reprogramming in Neurodegenerative Disorders" Nutrients 17, no. 14: 2266. https://doi.org/10.3390/nu17142266
APA StyleHein, Z. M., Arbain, M. F. F., Kumar, S., Mehat, M. Z., Hamid, H. A., Che Ramli, M. D., & Che Mohd Nassir, C. M. N. (2025). Intermittent Fasting as a Neuroprotective Strategy: Gut–Brain Axis Modulation and Metabolic Reprogramming in Neurodegenerative Disorders. Nutrients, 17(14), 2266. https://doi.org/10.3390/nu17142266