Selenium for the Prevention of Cutaneous Melanoma

Abstract
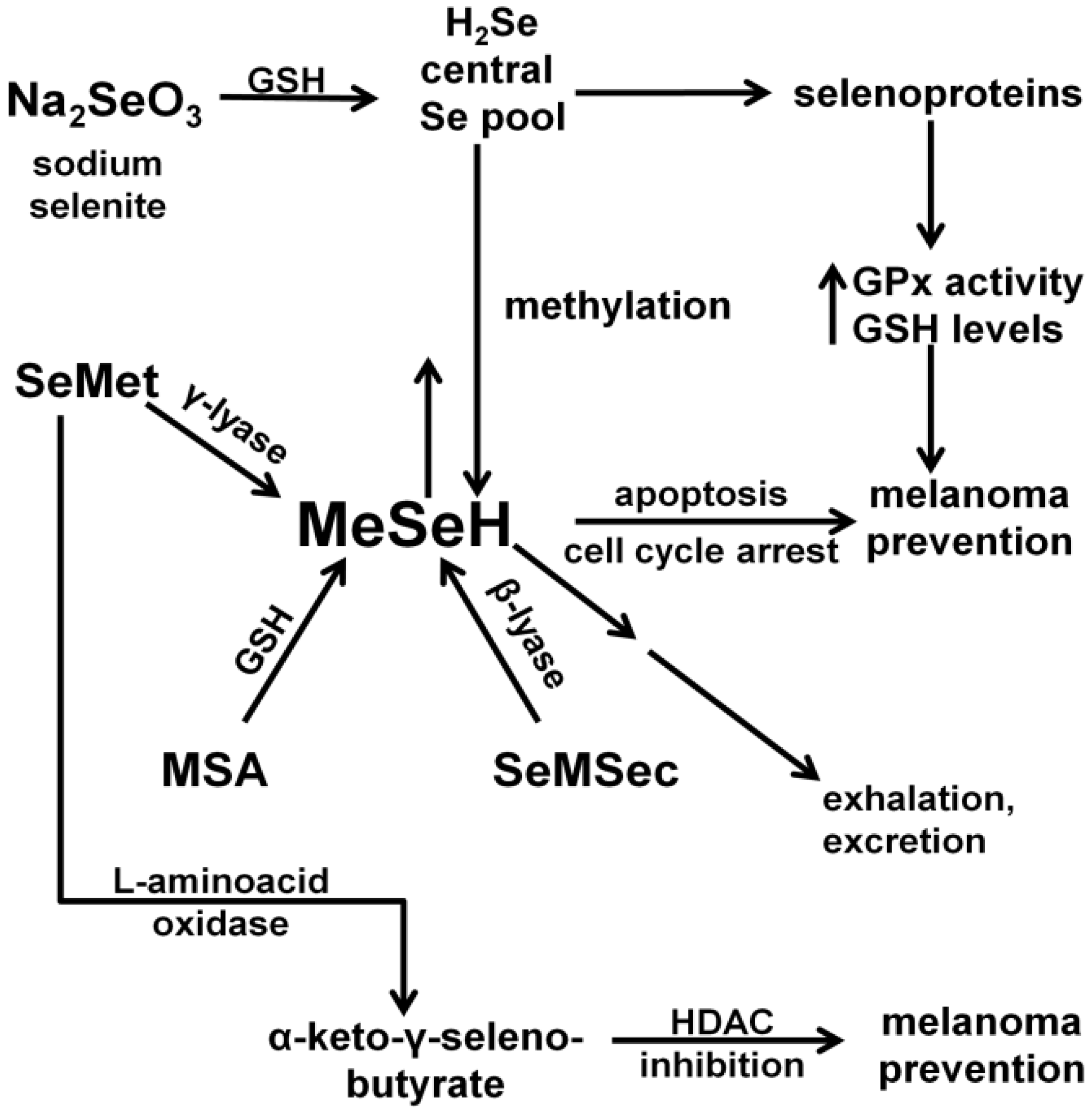
:1. Introduction

2. Experimental Section
2.1. Se Compounds and Other Reagents
2.2. Cell Culture
2.3. XBP1 Splicing Analysis
2.4. Normal Human Melanocytes
2.5. UV Irradiation
2.6. GPx Activity Assay
2.7. Glutathione Measurement
2.8. Cell Cycle Analysis
2.9. Cell Viability Assay
2.10. Caspase Assay
2.11. Topical SeMet Treatment
2.12. Total Se Quantification of Tissue Samples by ICP-DRC-MS
2.13. Animals
2.14. Statistical Analysis
2.15. Immunochemical Analysis (Western Blot)
3. Results
3.1. Se Restores Selenoprotein Activity and GSH Levels in UV-Irradiated Melanocytes

3.2. Topical SeMet Delays Onset of UV-Induced Melanoma


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Predictor | Coefficient | Hazard Ratio | Lower 95% CI | Upper 95% CI | Z | p-value |
|---|---|---|---|---|---|---|
| Group = SeMet | −0.5182 | 0.5956 | 0.3668 | 0.9672 | −2.095 | 0.036 |
| Sex = m | 0.2364 | 1.2667 | 0.7840 | 2.0464 | 0.966 | 0.33 |
| Tumor number: Multiple Linear Regression of Slopes for Individual Lines | ||||
| Parameter | Estimate | SE | t | p-value |
| (Intercept) | 0.15196 | 0.03326 | 4.57 | 2.05 × 10−5 |
| SeMet | 0.08455 | 0.03421 | 2.472 | 0.0159 |
| Male | −0.06901 | 0.03431 | −2.01 | 0.0482 |
| Tumor area: Multiple Linear Regression of Slopes for Individual Lines | ||||
| Parameter | Estimate | SE | t | p-value |
| (Intercept) | 0.27541 | 0.03819 | 7.212 | 5.13 × 10−10 |
| SeMet | 0.08307 | 0.03928 | 2.115 | 0.038 |
| Male | −0.05537 | 0.03940 | −1.405 | 0.164 |
3.3. The Small Molecule Metabolite MeSeH Causes Cell Cycle Arrest and Induces Apoptosis in Melanoma Cells

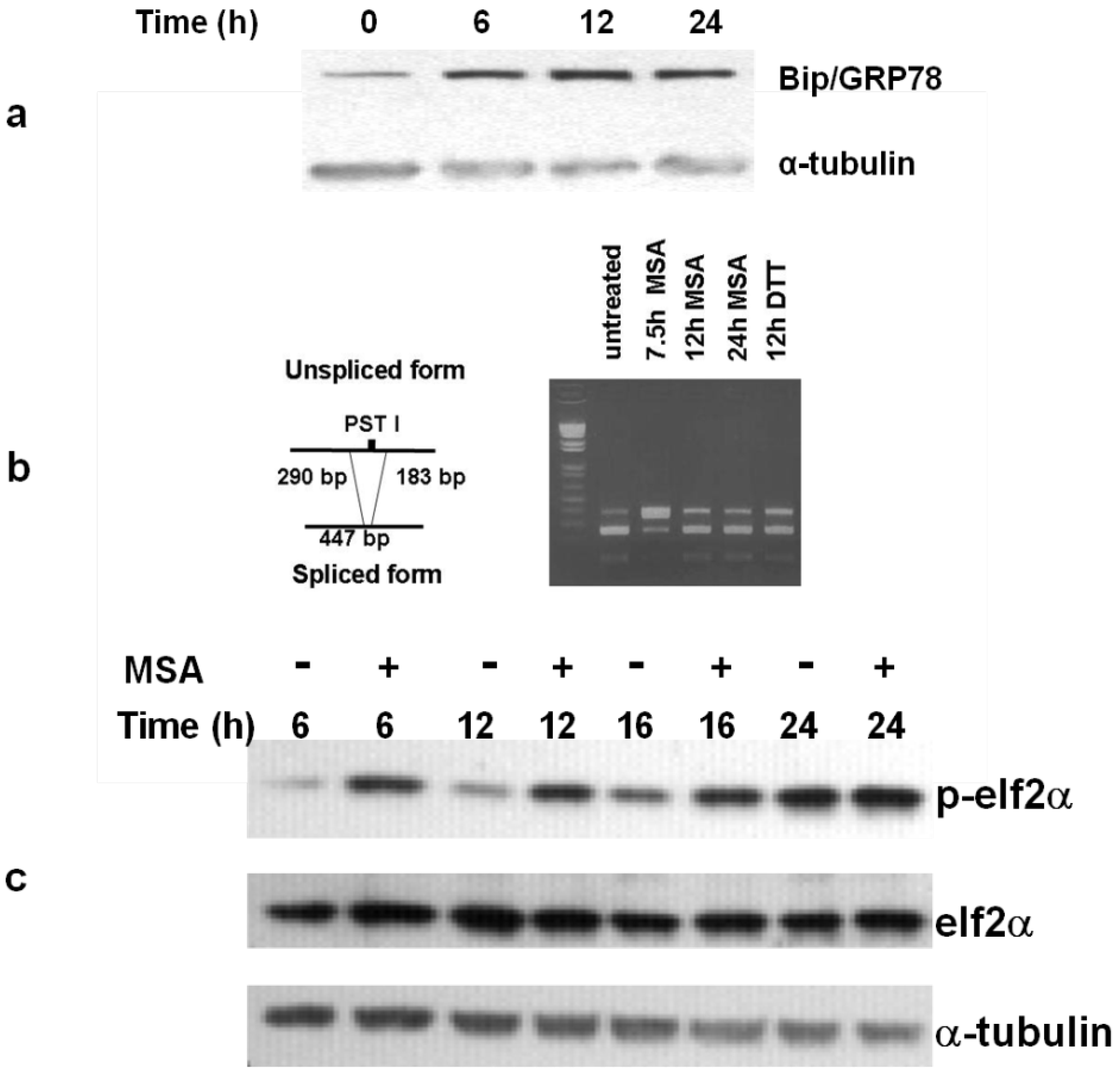
3.4. The Unfolded Protein Response (UPR) is Induced by MSA

3.5. MSA Induces Activation of Caspases

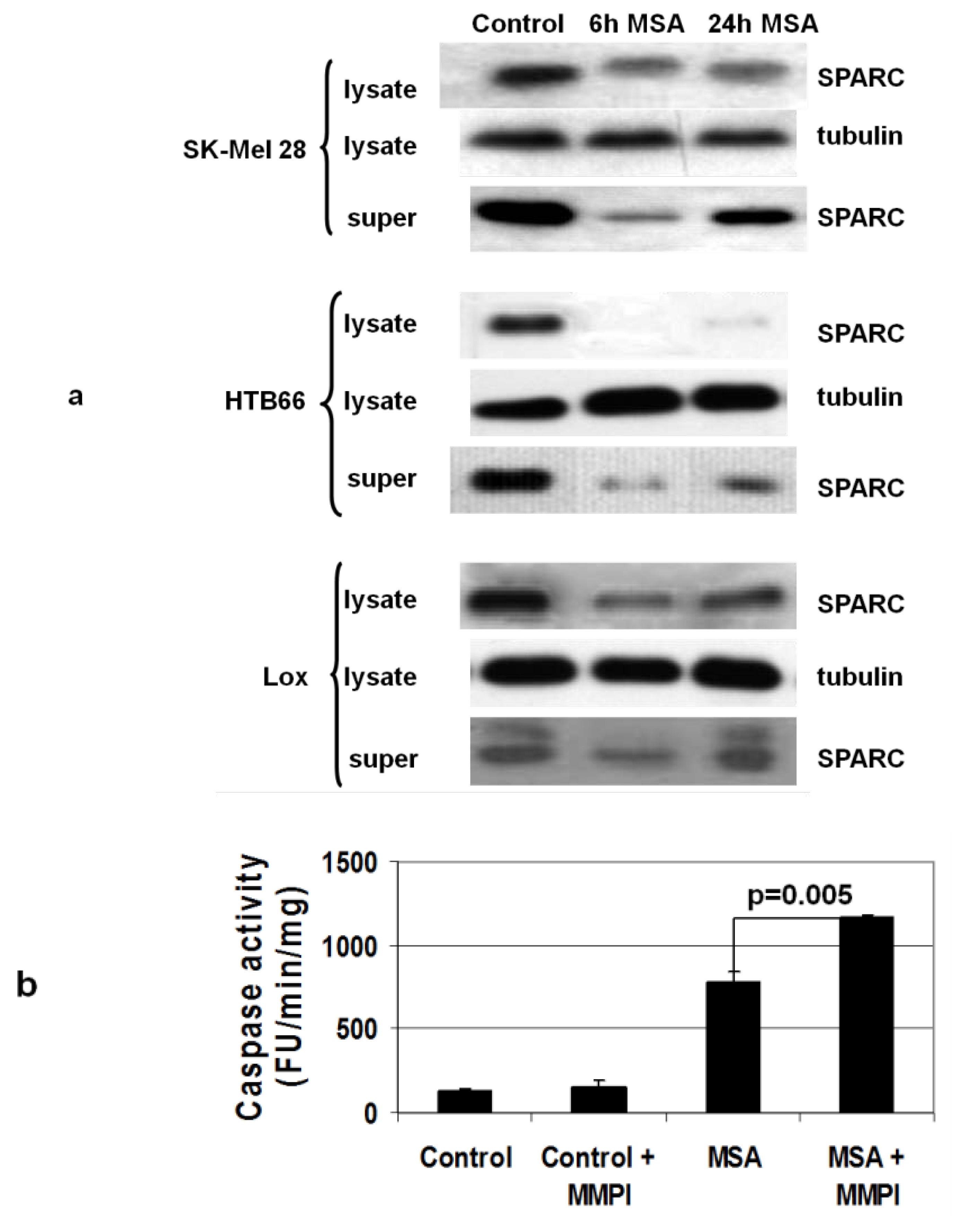
3.6. MSA Decreases Levels of Secreted Proteins

3.7. MSA Decreases Growth of Human Melanoma Xenograft Tumors

4. Discussion
5. Conclusions
Acknowledgments
Conflict of Interest
References
- Schrauzer, G.N.; Surai, P.F. Selenium in human and animal nutrition: Resolved and unresolved issues. Crit. Rev. Biotechnol. 2009, 29, 2–9. [Google Scholar] [CrossRef]
- Zhang, Y.; Zhou, Y.; Schweizer, U.; Savaskan, N.E.; Hua, D.; Kipnis, J.; Hatfield, D.L.; Gladyshev, V.N. Comparative analysis of selenocysteine machinery and selenoproteome gene expression in mouse brain identifies neurons as key functional sites of selenium in mammals. J. Biol. Chem. 2008, 283, 2427–2438. [Google Scholar]
- Walshe, J.; Serewko-Auret, M.M.; Teakle, N.; Cameron, S.; Minto, K.; Smith, L.; Burcham, P.C.; Russell, T.; Strutton, G.; Griffin, A.; et al. Inactivation of glutathione peroxidase activity contributes to UV-induced squamous cell carcinoma formation. Cancer Res. 2007, 67, 4751–4758. [Google Scholar] [CrossRef]
- Wenk, J.; Schuller, J.; Hinrichs, C.; Syrovets, T.; Azoitei, N.; Podda, M.; Wlaschek, M.; Brenneisen, P.; Schneider, L.A.; Sabiwalsky, A.; et al. Overexpression of phospholipid-hydroperoxide glutathione peroxidase in human dermal fibroblasts abrogates uva irradiation-induced expression ofinterstitial collagenase/matrix metalloproteinase-1 by suppression of phosphatidylcholine hydroperoxide-mediated nfkappab activation and interleukin-6 release. J. Biol. Chem. 2004, 279, 45634–45642. [Google Scholar] [CrossRef]
- Lee, J.B.; Yun, S.J.; Chae, H.Z.; Won, Y.H.; Kim, Y.P.; Lee, S.C. Expression of peroxiredoxin and thioredoxin in dermatological disorders. Br. J. Dermatol. 2002, 146, 710–712. [Google Scholar] [CrossRef]
- Velicer, C.M.; Ulrich, C.M. Vitamin and mineral supplementation among US adults after cancer diagnosis: A systematic review. J. Clin. Oncol. 2008, 28, 665–673. [Google Scholar] [CrossRef]
- Blot, W.J.; Li, J.Y.; Taylor, P.R.; Guo, W.; Dawsey, S.; Wang, G.Q.; Yang, C.S.; Zheng, S.F.; Gail, M.; Li, G.Y.; et al. Nutrition intervention trials in Linxian, China: Supplementation with specific vitamin/mineral combinations, cancer incidence, and disease-specific mortality in the general population. J. Natl. Cancer Inst. 1993, 85, 1483–1492. [Google Scholar] [CrossRef]
- Clark, L.C.; Combs, G.F., Jr.; Turnbull, B.W.; Slate, E.H.; Chalker, D.K.; Chow, J.; Davis, L.S.; Glover, R.A.; Graham, G.F.; Gross, E.G.; et al. Effects of selenium supplementation for cancer prevention in patients with carcinoma of the skin: A randomized controlled trial. JAMA 1996, 276, 1957–1963. [Google Scholar] [CrossRef]
- The Alpha-Tocopherol, Beta Carotene Cancer Prevention Study Group. The effect of vitamin E and beta carotene on the incidence of lung cancer and other cancers in male smokers. N. Engl. J. Med. 1994, 330, 1029–1035. [Google Scholar] [CrossRef]
- Lippman, S.M.; Goodman, P.J.; Klein, E.A.; Parnes, H.L.; Thompson, I.M., Jr.; Kristal, A.R.; Santella, R.M.; Probstfield, J.L.; Moinpour, C.M.; Albanes, D.; et al. Designing the selenium and vitamin E cancer prevention trial (SELECT). J. Natl. Cancer Inst. 2005, 97, 94–102. [Google Scholar] [CrossRef]
- Klein, E.A.; Thompson, I.M., Jr.; Tangen, C.M.; Crowley, J.J.; Lucia, M.S.; Goodman, P.J.; Minasian, L.M.; Ford, L.G.; Parnes, H.L.; Gaziano, J.M.; et al. Vitamin E and the risk of prostate cancer: The selenium and vitamin E cancer prevention trial (SELECT). JAMA 2011, 306, 1549–1556. [Google Scholar] [CrossRef]
- Gaziano, J.M.; Glynn, R.J.; Christen, W.G.; Kurth, T.; Belanger, C.; MacFadyen, J.; Bubes, V.; Manson, J.E.; Sesso, H.D.; Buring, J.E. Vitamins E and C in the prevention of prostate and total cancer in men: The physicians’ health study II randomized controlled trial. JAMA 2009, 301, 52–62. [Google Scholar]
- Gann, P.H. Randomized trials of antioxidant supplementation for cancer prevention: First bias, now chance—next, caus. JAMA 2009, 301, 102–103. [Google Scholar] [CrossRef]
- Lippman, S.M.; Klein, E.A.; Goodman, P.J.; Lucia, M.S.; Thompson, I.M.; Ford, L.G.; Parnes, H.L.; Minasian, L.M.; Gaziano, J.M.; Hartline, J.A.; et al. Effect of selenium and vitamin E on risk of prostate cancer and other cancers: The selenium and vitamin E cancer prevention trial (SELECT). JAMA 2009, 301, 39–51. [Google Scholar]
- Marshall, J.R.; Ip, C.; Romano, K.; Fetterly, G.; Fakih, M.; Jovanovic, B.; Perloff, M.; Crowell, J.; Davis, W.; French-Christy, R.; et al. Methyl selenocysteine: Single-dose pharmacokinetics in men. Cancer Prev. Res. 2011, 4, 1938–1944. [Google Scholar] [CrossRef]
- Grossman, D.; Leffell, D.J. The molecular basis of nonmelanoma skin cancer: New understanding. Arch. Dermatol. 1997, 133, 1263–1270. [Google Scholar] [CrossRef]
- Gilchrest, B.A.; Eller, M.S.; Geller, A.C.; Yaar, M. The pathogenesis of melanoma induced by ultraviolet radiation. N. Engl. J. Med. 1999, 340, 1341–1348. [Google Scholar] [CrossRef]
- Hodis, E.; Watson, I.R.; Kryukov, G.V.; Arold, S.T.; Imielinski, M.; Theurillat, J.P.; Nickerson, E.; Auclair, D.; Li, L.; Place, C.; et al. A landscape of driver mutations in melanoma. Cell 2012, 150, 251–263. [Google Scholar] [CrossRef]
- Krauthammer, M.; Kong, Y.; Ha, B.H.; Evans, P.; Bacchiocchi, A.; McCusker, J.P.; Cheng, E.; Davis, M.J.; Goh, G.; Choi, M.; et al. Exome sequencing identifies recurrent somatic RAC1 mutations in melanoma. Nat. Genet. 2012, 44, 1006–1014. [Google Scholar] [CrossRef]
- Meyskens, F.L., Jr.; Farmer, P.; Fruehauf, J.P. Redox regulation in human melanocytes and melanoma. Pigment Cell Res. 2001, 14, 148–154. [Google Scholar]
- Mitra, D.; Luo, X.; Morgan, A.; Wang, J.; Hoang, M.P.; Lo, J.; Guerrero, C.R.; Lennerz, J.K.; Mihm, M.C.; Wargo, J.A.; et al. An ultraviolet-radiation-independent pathway to melanoma carcinogenesis in the red hair/fair skin background. Nature 2012, 491, 449–453. [Google Scholar] [CrossRef]
- Bruner, S.D.; Norman, D.P.; Verdine, G.L. Structural basis for recognition and repair of the endogenous mutagen 8-oxoguanine in DNA. Nature 2000, 403, 859–866. [Google Scholar] [CrossRef]
- Katiyar, S.K.; Afaq, F.; Perez, A.; Mukhtar, H. Green tea polyphenol (−)-epigallocatechin-3-gallate treatment of human skin inhibits ultraviolet radiation-induced oxidative stress. Carcinogenesis 2001, 22, 287–294. [Google Scholar] [CrossRef]
- Cotter, M.A.; Thomas, J.; Cassidy, P.; Robinette, K.; Jenkins, N.; Florell, S.R.; Leachman, S.; Samlowski, W.E.; Grossman, D. N-acetylcysteine protects melanocytes against oxidative stress/damage and delays onset of ultraviolet-induced melanoma in mice. Clin. Cancer Res. 2007, 13, 5952–5958. [Google Scholar] [CrossRef]
- Francis, S.O.; Mahlberg, M.J.; Johnson, K.R.; Ming, M.E.; Dellavalle, R.P. Melanoma chemoprevention. J. Am. Acad. Dermatol. 2006, 55, 849–861. [Google Scholar] [CrossRef]
- Ip, C.; Thompson, H.J.; Zhu, Z.; Ganther, H.E. In vitro and in vivo studies of methylseleninic acid: Evidence that a monomethylated selenium metabolite is critical for cancer chemoprevention. Cancer Res. 2000, 60, 2882–2886. [Google Scholar]
- Menter, D.G.; Sabichi, A.L.; Lippman, S.M. Selenium effects on prostate cell growth. Cancer Epidemiol. Biomark. Prev. 2000, 9, 1171–1182. [Google Scholar]
- Halaban, R.; Cheng, E.; Zhang, Y.; Moellmann, G.; Hanlon, D.; Michalak, M.; Setaluri, V.; Hebert, D.N. Aberrant retention of tyrosinase in the endoplasmic reticulum mediates accelerated degradation of the enzyme and contributes to the dedifferentiated phenotype of amelanotic melanoma cells. Proc. Natl. Acad. Sci. USA 1997, 94, 6210–6215. [Google Scholar] [CrossRef]
- Bennett, D.C.; Cooper, P.J.; Hart, I.R. A line of non-tumorigenic mouse melanocytes, syngeneic with the b16 melanoma and requiring a tumour promoter for growth. Int. J. Cancer 1987, 39, 414–418. [Google Scholar] [CrossRef]
- Zu, K.; Bihani, T.; Lin, A.; Park, Y.M.; Mori, K.; Ip, C. Enhanced selenium effect on growth arrest by BIP/GRP78 knockdown in p53-null human prostate cancer cells. Oncogene 2006, 25, 546–554. [Google Scholar]
- Jenkins, N.C.; Liu, T.; Cassidy, P.; Leachman, S.A.; Boucher, K.M.; Goodson, A.G.; Samadashwily, G.; Grossman, D. The p16(INK4A) tumor suppressor regulates cellular oxidative stress. Oncogene 2011, 30, 265–274. [Google Scholar] [CrossRef]
- Brigelius-Flohe, R.; Wingler, K.; Muller, C. Estimation of individual types of glutathione peroxidases. Methods Enzymol. 2002, 347, 101–112. [Google Scholar] [CrossRef]
- Goodson, A.G.; Cotter, M.A.; Cassidy, P.; Wade, M.; Florell, S.R.; Liu, T.; Boucher, K.M.; Grossman, D. Use of oral N-acetylcysteine for protection of melanocytic nevi against UV-induced oxidative stress: Towards a novel paradigm for melanoma chemoprevention. Clin. Cancer Res. 2009, 15, 7434–7440. [Google Scholar] [CrossRef]
- Moos, P.J.; Fitzpatrick, F.A. Taxanes propagate apoptosis via two cell populations with distinctive cytological and molecular traits. Cell Growth Differ. 1998, 9, 687–697. [Google Scholar]
- Noonan, F.P.; Dudek, J.; Merlino, G.; De Fabo, E.C. Animal models of melanoma: An HGF/SF transgenic mouse model may facilitate experimental access to uv initiating events. Pigment Cell Res. 2003, 16, 16–25. [Google Scholar] [CrossRef]
- Grossman, D.; Kim, P.J.; Schechner, J.S.; Altieri, D.C. Inhibition of melanoma tumor growth in vivo by survivin targeting. Proc. Natl. Acad. Sci. USA 2001, 98, 635–640. [Google Scholar] [CrossRef]
- R: A Language and Environment for Statistical Computing; R Foundation for Statistical Computing: Vienna, Austria, 2011.
- Ip, C.; Dong, Y.; Ganther, H.E. New concepts in selenium chemoprevention. Cancer Metastasis Rev. 2002, 21, 281–289. [Google Scholar] [CrossRef]
- Short, M.D.; Xie, Y.; Li, L.; Cassidy, P.B.; Roberts, J.C. Characteristics of selenazolidine prodrugs of selenocysteine: Toxicity and glutathione peroxidase induction in V79 cells. J. Med. Chem. 2003, 46, 3308–3313. [Google Scholar] [CrossRef]
- Moos, P.J.; Edes, K.; Cassidy, P.; Massuda, E.; Fitzpatrick, F.A. Electrophilic prostaglandins and lipid aldehydes repress redox-sensitive transcription factors p53 and hypoxia-inducible factor by impairing the selenoprotein thioredoxin reductase. J. Biol. Chem. 2003, 278, 745–750. [Google Scholar]
- Cassidy, P.B.; Edes, K.; Nelson, C.C.; Parsawar, K.; Fitzpatrick, F.A.; Moos, P.J. Thioredoxin reductase is required for the inactivation of tumor suppressor p53 and for apoptosis induced by endogenous electrophiles. Carcinogenesis 2006, 27, 2538–2549. [Google Scholar] [CrossRef]
- Burke, K.E.; Combs, G.F., Jr.; Gross, E.G.; Bhuyan, K.C.; Abu-Libdeh, H. The effects of topical and oral l-selenomethionine on pigmentation and skin cancer induced by ultraviolet irradiation. Nutr. Cancer 1992, 17, 123–137. [Google Scholar] [CrossRef]
- Novoselov, S.V.; Calvisi, D.F.; Labunskyy, V.M.; Factor, V.M.; Carlson, B.A.; Fomenko, D.E.; Moustafa, M.E.; Hatfield, D.L.; Gladyshev, V.N. Selenoprotein deficiency and high levels of selenium compounds can effectively inhibit hepatocarcinogenesis in transgenic mice. Oncogene 2005, 24, 8003–8011. [Google Scholar] [CrossRef]
- Jiang, C.C.; Chen, L.H.; Gillespie, S.; Kiejda, K.A.; Mhaidat, N.; Wang, Y.F.; Thorne, R.; Zhang, X.D.; Hersey, P. Tunicamycin sensitizes human melanoma cells to tumor necrosis factor-related apoptosis-inducing ligand-induced apoptosis by up-regulation of TRAIL-R2 via the unfolded protein response. Cancer Res. 2007, 67, 5880–5888. [Google Scholar] [CrossRef]
- Li, H.; Zhu, H.; Xu, C.J.; Yuan, J. Cleavage of bid by caspase 8 mediates the mitochondrial damage in the fas pathway of apoptosis. Cell 1998, 94, 491–501. [Google Scholar] [CrossRef]
- Ikuta, Y.; Nakatsura, T.; Kageshita, T.; Fukushima, S.; Ito, S.; Wakamatsu, K.; Baba, H.; Nishimura, Y. Highly sensitive detection of melanoma at an early stage based on the increased serum secreted protein acidic and rich in cysteine and glypican-3 levels. Clin. Cancer Res. 2005, 11, 8079–8088. [Google Scholar] [CrossRef]
- Ledda, M.F.; Adris, S.; Bravo, A.I.; Kairiyama, C.; Bover, L.; Chernajovsky, Y.; Mordoh, J.; Podhajcer, O.L. Suppression of SPARC expression by antisense RNA abrogates the tumorigenicity of human melanoma cells. Nat. Med. 1997, 3, 171–176. [Google Scholar] [CrossRef]
- Juliger, S.; Goenaga-Infante, H.; Lister, T.A.; Fitzgibbon, J.; Joel, S.P. Chemosensitization of B-cell lymphomas by methylseleninic acid involves nuclear factor-kappab inhibition and the rapid generation of other selenium species. Cancer Res. 2007, 67, 10984–10992. [Google Scholar] [CrossRef]
- McClung, H.M.; Thomas, S.L.; Osenkowski, P.; Toth, M.; Menon, P.; Raz, A.; Fridman, R.; Rempel, S.A. SPARC upregulates MT1-MMP expression, MMP-2 activation, and the secretion and cleavage of galectin-3 in U87MG glioma cell. Neurosci. Lett. 2007, 419, 172–177. [Google Scholar] [CrossRef]
- Rizki, A.; Weaver, V.M.; Lee, S.Y.; Rozenberg, G.I.; Chin, K.; Myers, C.A.; Bascom, J.L.; Mott, J.D.; Semeiks, J.R.; Grate, L.R.; et al. A human breast cell model of preinvasive to invasive transition. Cancer Res. 2008, 68, 1378–1387. [Google Scholar] [CrossRef]
- Scid mice overview. Available online: http://jaxmice.jax.org/support/straindata/scid.html (accessed on 26 February 2013).
- Burk, R.F.; Norsworthy, B.K.; Hill, K.E.; Motley, A.K.; Byrne, D.W. Effects of chemical form of selenium on plasma biomarkers in a high-dose human supplementation trial. Cancer Epidemiol. Biomark. Prev. 2006, 15, 804–810. [Google Scholar] [CrossRef]
- Wang, L.; Bonorden, M.J.; Li, G.X.; Lee, H.J.; Hu, H.; Zhang, Y.; Liao, J.D.; Cleary, M.P.; Lu, J. Methyl-selenium compounds inhibit prostate carcinogenesis in the transgenic adenocarcinoma of mouse prostate model with survival benefit. Cancer Prev. Res. 2009, 2, 484–495. [Google Scholar] [CrossRef]
- Li, G.X.; Lee, H.J.; Wang, Z.; Hu, H.; Liao, J.D.; Watts, J.C.; Combs, G.F., Jr.; Lu, J. Superior in vivo inhibitory efficacy of methylseleninic acid against human prostate cancer over selenomethionine or selenite. Carcinogenesis 2008, 29, 1005–1012. [Google Scholar] [CrossRef]
- Syed, D.N.; Mukhtar, H. Botanicals for the prevention and treatment of cutaneous melanoma. Pigment Cell Melanoma Res. 2011, 24, 688–702. [Google Scholar] [CrossRef]
- Jackson, M.I.; Combs, G.F., Jr. Selenium and anticarcinogenesis: Underlying mechanisms. Curr. Opin. Clin. Nutr. Metab. Care 2008, 11, 718–726. [Google Scholar] [CrossRef]
- Ip, C. Lessons from basic research in selenium and cancer prevention. J. Nutr. 1998, 128, 1845–1854. [Google Scholar]
- Pinto, J.T.; Lee, J.I.; Sinha, R.; Macewan, M.E.; Cooper, A.J. Chemopreventive mechanisms of α-keto acid metabolites of naturally occurring organoselenium compounds. Amino Acids 2011, 41, 29–41. [Google Scholar]
- Ahonen, M.; Poukkula, M.; Baker, A.H.; Kashiwagi, M.; Nagase, H.; Eriksson, J.E.; Kahari, V.M. Tissue inhibitor of metalloproteinases-3 induces apoptosis in melanoma cells by stabilization of death receptors. Oncogene 2003, 22, 2121–2134. [Google Scholar] [CrossRef]
- Chetty, C.; Bhoopathi, P.; Lakka, S.S.; Rao, J.S. MMP-2 siRNA induced Fas/CD95-mediated extrinsic II apoptotic pathway in the A549 lung adenocarcinoma cell line. Oncogene 2007, 26, 7675–7683. [Google Scholar] [CrossRef]
- Reid, M.E.; Stratton, M.S.; Lillico, A.J.; Fakih, M.; Natarajan, R.; Clark, L.C.; Marshall, J.R. A report of high-dose selenium supplementation: Response and toxicities. J. Trace Elem. Med. Biol. 2004, 18, 69–74. [Google Scholar] [CrossRef]
- Reagan-Shaw, S.; Nihal, M.; Ahmad, N. Dose translation from animal to human studies revisited. FASEB J. 2008, 22, 659–661. [Google Scholar] [CrossRef]
- Hurst, R.; Armah, C.N.; Dainty, J.R.; Hart, D.J.; Teucher, B.; Goldson, A.J.; Broadley, M.R.; Motley, A.K.; Fairweather-Tait, S.J. Establishing optimal selenium status: Results of a randomized, double-blind, placebo-controlled trial. Am. J. Clin. Nutr. 2010, 91, 923–931. [Google Scholar] [CrossRef]
- Johnson, W.D.; Morrissey, R.L.; Kapetanovic, I.; Crowell, J.A.; McCormick, D.L. Subchronic oral toxicity studies of se-methylselenocysteine, an organoselenium compound for breast cancer prevention. Food Chem. Toxicol. 2008, 46, 1068–1078. [Google Scholar] [CrossRef]
- Zhang, J.; Wang, L.; Li, G.; Anderson, L.B.; Xu, Y.; Witthuhn, B.; Lu, J. Mouse prostate proteomes are differentially altered by supranutritional intake of four selenium compounds. Nutr. Cancer 2011, 63, 778–789. [Google Scholar] [CrossRef]
© 2013 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Cassidy, P.B.; Fain, H.D.; Cassidy, J.P., Jr.; Tran, S.M.; Moos, P.J.; Boucher, K.M.; Gerads, R.; Florell, S.R.; Grossman, D.; Leachman, S.A. Selenium for the Prevention of Cutaneous Melanoma. Nutrients 2013, 5, 725-749. https://doi.org/10.3390/nu5030725
Cassidy PB, Fain HD, Cassidy JP Jr., Tran SM, Moos PJ, Boucher KM, Gerads R, Florell SR, Grossman D, Leachman SA. Selenium for the Prevention of Cutaneous Melanoma. Nutrients. 2013; 5(3):725-749. https://doi.org/10.3390/nu5030725
Chicago/Turabian StyleCassidy, Pamela B., Heidi D. Fain, James P. Cassidy, Jr., Sally M. Tran, Philip J. Moos, Kenneth M. Boucher, Russell Gerads, Scott R. Florell, Douglas Grossman, and Sancy A. Leachman. 2013. "Selenium for the Prevention of Cutaneous Melanoma" Nutrients 5, no. 3: 725-749. https://doi.org/10.3390/nu5030725
APA StyleCassidy, P. B., Fain, H. D., Cassidy, J. P., Jr., Tran, S. M., Moos, P. J., Boucher, K. M., Gerads, R., Florell, S. R., Grossman, D., & Leachman, S. A. (2013). Selenium for the Prevention of Cutaneous Melanoma. Nutrients, 5(3), 725-749. https://doi.org/10.3390/nu5030725