Influence of microRNA on the Maintenance of Human Iron Metabolism

Abstract
:1. Introduction
2. microRNA Nomenclature
3. Iron and Heme in microRNA Processing

4. The Pathologic and Physiologic Roles of miRNA in Humans
- (1)
- investigation of the contributions of miRNA regulation/dysregulation in disease pathology;
- (2)
- the use of chemical modifiers to antagonize or restore key miRNA related to disease pathology;
- (3)
- the identification of miRNA as serum biomarkers for disease diagnosis and prognosis.
5. Regulation of Iron Metabolism
5.1. Control of Systemic Iron Homeostasis


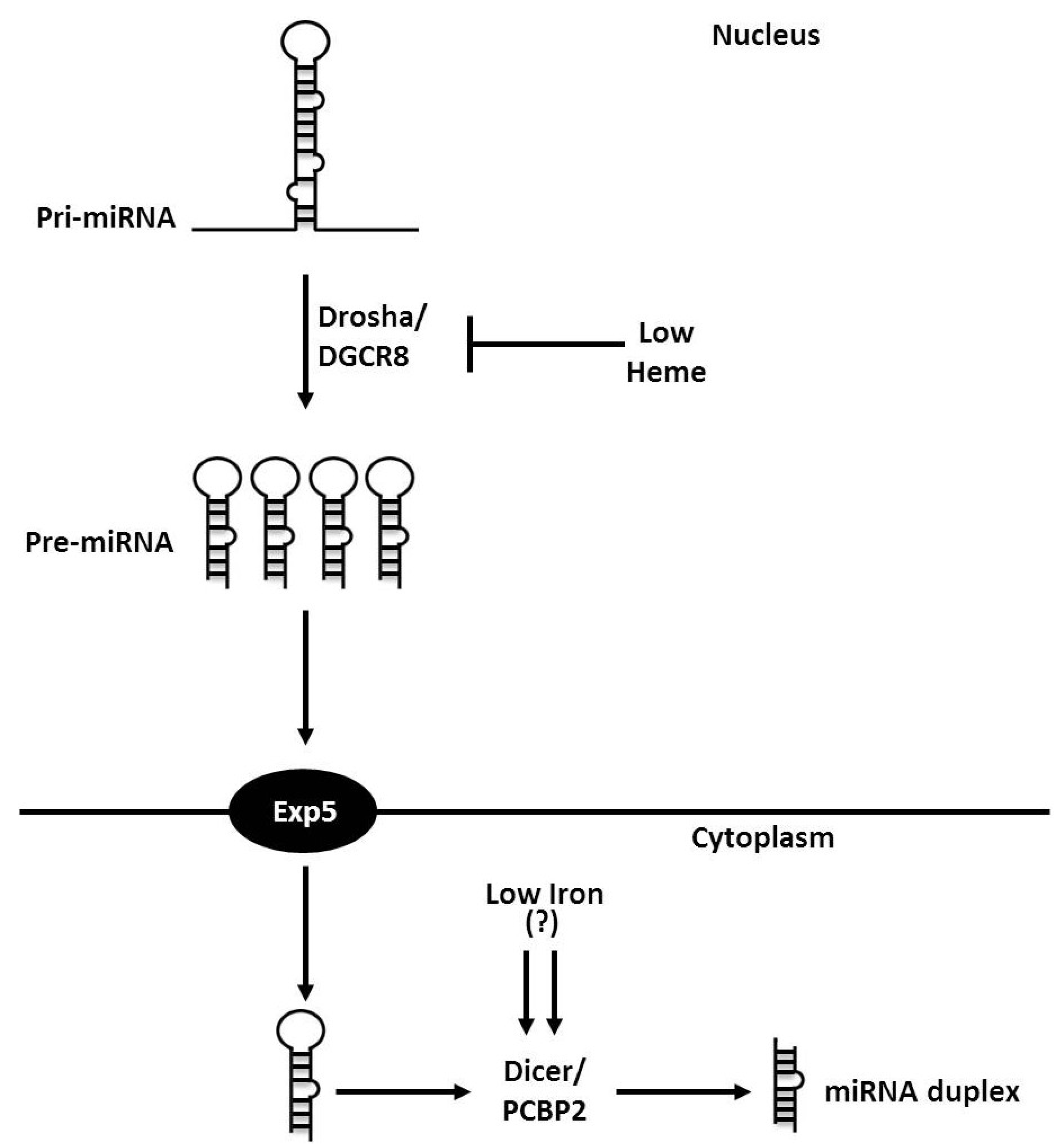{kind=link}
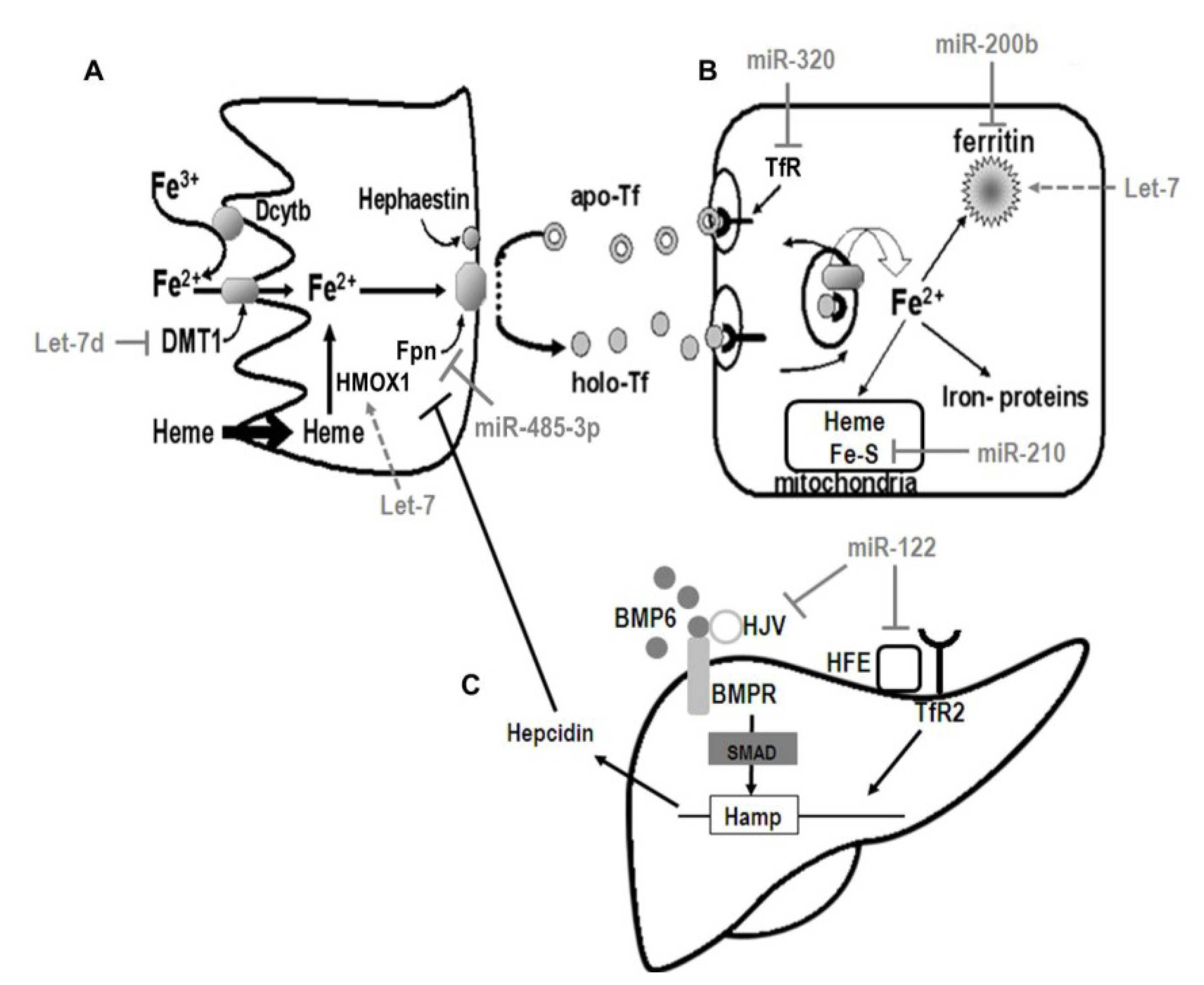{kind=link}
| miRNA | Target mRNA | Reference(s) |
|---|---|---|
| miR-Let-7d | DMT (∆IRE), BACH1 | Andolfo et al. (2010) [42], Hou et al. (2012) [43] |
| miR-122 | HFE, HJV | Castoldi et al. (2009) [44] |
| miR-196 | BACH1 | Hou et al. (2010) [45] |
| miR-200b | FTH | Shpyleva et al. (2009) [46] |
| miR-210 | ISCU, TFR | Chan et al. (2009) [47], Yoshioka et al. (2012) [48] |
| miR-214 | Lactoferrin | Liao et al. (2010) [49] |
| miR-320 | TFR | Schaar et al. (2009) [50] |
| miR-485-3p | FPN | Sangokoya et al. (2013) [51] |
| miR-584 | Lactoferrin Receptor | Liao et al. (2010) [52] |
5.2. Regulation of Cellular Iron Metabolism
6. The Molecular Coordination of Iron Homeostasis by miRNA
7. microRNA Mediate the Reciprocal Relationship between Iron and Oxygen Homeostasis
8. Future Developments in the Role of miRNA in the Regulation of Iron Homeostasis
9. Conclusions
Acknowledgments
Abbreviations
| Ago | Argonaute |
| BMP | bone morphogenetic protein |
| c-acon | cytosolic aconitase |
| Dcytb | duodenal cytochrome b |
| DGCR8 | DiGeorge syndrome critical region 8 |
| DMT1 | divalent metal transporter 1 |
| FBXL5 | F-box and leucine-rich repeat protein5 |
| Fe–S | iron-sulfur |
| Fpn | ferroportin |
| Hamp | hepcidin |
| HFE | hemochromatosis gene |
| HIF | hypoxia inducible factor |
| Hjv | hemojuvelin |
| Hmox1 | heme oxygenase 1 |
| IRE | iron responsive element |
| IRP | iron regulatory protein |
| Iscu | iron-sulfur cluster scaffold homolog |
| LNA | locked nucleic acid |
| m-acon | mitochondrial aconitase |
| miRNA | microRNA |
| Pcbp2 | poly(rC) binding protein 2 |
| pre-miRNA | precursor miRNA |
| pri-miRNA | primary miRNA |
| RES | reticuloendothial system |
| RISC | RNA-induced silencing complex |
| ROS | reactive oxygen species |
| Srebp2 | sterol regulatory element binding protein 2 |
| Tf | transferrin |
| TfR | transferrin receptor |
| UTR | untranslated region |
Conflict of Interest
References
- McLean, E.; Cogswell, M.; Egli, I.; Wojdyla, D.; de Benoist, B. Worldwide prevalence of anaemia, WHO vitamin and mineral nutrition information system, 1993–2005. Public Health Nutr. 2009, 12, 444–454. [Google Scholar]
- Weaver, C.M.; Rajaram, S. Exercise and iron status. J. Nutr. 1992, 122 (3 Suppl.), 782–787. [Google Scholar]
- Barr, I.; Smith, A.T.; Chen, Y.; Senturia, R.; Burstyn, J.N.; Guo, F. Ferric, not ferrous, heme activates RNA-binding protein DGCR8 for primary microRNA processing. Proc. Natl. Acad. Sci. USA 2012, 109, 1919–1924. [Google Scholar]
- Faller, M.; Matsunaga, M.; Yin, S.; Loo, J.A.; Guo, F. Heme is involved in microRNA processing. Nat. Struct. Mol. Biol. 2007, 14, 23–29. [Google Scholar]
- Lee, R.C.; Feinbaum, R.L.; Ambros, V. The C. elegans heterochronic gene lin-4 encodes small RNAs with antisense complementarity to lin-14. Cell 1993, 75, 843–854. [Google Scholar]
- Reinhart, B.J.; Slack, F.J.; Basson, M.; Pasquinelli, A.E.; Bettinger, J.C.; Rougvie, A.E.; Horvitz, H.R.; Ruvkun, G. The 21-nucleotide let-7 RNA regulates developmental timing in Caenorhabditis elegans. Nature 2000, 403, 901–906. [Google Scholar]
- Pasquinelli, A.E.; Reinhart, B.J.; Slack, F.; Martindale, M.Q.; Kuroda, M.I.; Maller, B.; Hayward, D.C.; Ball, E.E.; Degnan, B.; Muller, P.; et al. Conservation of the sequence and temporal expression of let-7 heterochronic regulatory RNA. Nature 2000, 408, 86–89. [Google Scholar]
- Lagos-Quintana, M.; Rauhut, R.; Lendeckel, W.; Tuschl, T. Identification of novel genes coding for small expressed RNAs. Science 2001, 294, 853–858. [Google Scholar]
- Lau, N.C.; Lim, L.P.; Weinstein, E.G.; Bartel, D.P. An abundant class of tiny RNAs with probable regulatory roles in Caenorhabditis elegans. Science 2001, 294, 858–862. [Google Scholar] [CrossRef]
- Kozomara, A.; Griffiths-Jones, S. miRBase: Integrating microRNA annotation and deep-sequencing data. Nucl. Acids. Res. 2011, 39, D152–D157. [Google Scholar] [CrossRef]
- Ambros, V.; Bartel, B.; Bartel, D.P.; Burge, C.B.; Carrington, J.C.; Chen, X.; Dreyfuss, G.; Eddy, S.R.; Griffiths-Jones, S.; Marshall, M.; et al. A uniform system for microRNA annotation. RNA 2003, 9, 277–279. [Google Scholar] [CrossRef]
- Griffiths-Jones, S.; Grocock, R.J.; van Dongen, S.; Bateman, A.; Enright, A.J. miRBase: MicroRNA sequences, targets and gene nomenclature. Nucl. Acids. Res. 2006, 34, D140–D144. [Google Scholar] [CrossRef]
- miRBase: What do the names/identifiers mean? Available online: http://www.mirbase.org/help/nomenclature.shtml (accessed on 5 April 2013).
- Slezak-Prochazka, I.; Durmus, S.; Kroesen, B.J.; van den Berg, A. MicroRNAs, macrocontrol: Regulation of miRNA processing. RNA 2010, 16, 1087–1095. [Google Scholar] [CrossRef]
- Kim, V.N.; Han, J.; Siomi, M.C. Biogenesis of small RNAs in animals. Nat. Rev. 2009, 10, 126–139. [Google Scholar]
- Huntzinger, E.; Izaurralde, E. Gene silencing by microRNAs: Contributions of translational repression and mRNA decay. Nat. Rev. Genet. 2011, 12, 99–110. [Google Scholar] [CrossRef]
- Ender, C.; Meister, G. Argonaute proteins at a glance. J. Cell Sci. 2010, 123, 1819–1823. [Google Scholar]
- Kawamata, T.; Tomari, Y. Making RISC. Trends Biochem. Sci. 2010, 35, 368–376. [Google Scholar] [CrossRef]
- Grimson, A.; Farh, K.K.; Johnston, W.K.; Garrett-Engele, P.; Lim, L.P.; Bartel, D.P. MicroRNA targeting specificity in mammals: Determinants beyond seed pairing. Mol. Cell 2007, 27, 91–105. [Google Scholar] [CrossRef]
- Lytle, J.R.; Yario, T.A.; Steitz, J.A. Target mRNAs are repressed as efficiently by microRNA-binding sites in the 5′ UTR as in the 3′ UTR. Proc. Natl. Acad. Sci. USA 2007, 104, 9667–9672. [Google Scholar] [CrossRef]
- Okamura, K.; Phillips, M.D.; Tyler, D.M.; Duan, H.; Chou, Y.T.; Lai, E.C. The regulatory activity of microRNA* species has substantial influence on microRNA and 3′ UTR evolution. Nat. Struct. Mol. Biol. 2008, 15, 354–363. [Google Scholar]
- Yang, J.S.; Phillips, M.D.; Betel, D.; Mu, P.; Ventura, A.; Siepel, A.C.; Chen, K.C.; Lai, E.C. Widespread regulatory activity of vertebrate microRNA* species. RNA 2011, 17, 312–326. [Google Scholar] [CrossRef]
- Li, Y.; Lin, L.; Li, Z.; Ye, X.; Xiong, K.; Aryal, B.; Xu, Z.; Paroo, Z.; Liu, Q.; He, C.; et al. Iron homeostasis regulates the activity of the microRNA pathway through poly(C)-binding protein 2. Cell Metab. 2012, 15, 895–904. [Google Scholar] [CrossRef]
- Calin, G.A.; Dumitru, C.D.; Shimizu, M.; Bichi, R.; Zupo, S.; Noch, E.; Aldler, H.; Rattan, S.; Keating, M.; Rai, K.; et al. Frequent deletions and down-regulation of micro-RNA genes miR15 and miR16 at 13q14 in chronic lymphocytic leukemia. Proc. Natl. Acad. Sci. USA 2002, 99, 15524–15529. [Google Scholar] [CrossRef]
- He, L.; Thomson, J.M.; Hemann, M.T.; Hernando-Monge, E.; Mu, D.; Goodson, S.; Powers, S.; Cordon-Cardo, C.; Lowe, S.W.; Hannon, G.J.; et al. A microRNA polycistron as a potential human oncogene. Nature 2005, 435, 828–833. [Google Scholar] [CrossRef]
- Calin, G.A.; Croce, C.M. MicroRNA-cancer connection: The beginning of a new tale. Cancer Res. 2006, 66, 7390–7394. [Google Scholar] [CrossRef]
- Jopling, C.L.; Yi, M.; Lancaster, A.M.; Lemon, S.M.; Sarnow, P. Modulation of hepatitis C virus RNA abundance by a liver-specific MicroRNA. Science 2005, 309, 1577–1581. [Google Scholar] [CrossRef]
- Small, E.M.; Frost, R.J.; Olson, E.N. MicroRNAs add a new dimension to cardiovascular disease. Circulation 2010, 121, 1022–1032. [Google Scholar] [CrossRef]
- Alvarez-Garcia, I.; Miska, E.A. MicroRNA functions in animal development and human disease. Development 2005, 132, 4653–4662. [Google Scholar] [CrossRef]
- Friedman, R.C.; Farh, K.K.; Burge, C.B.; Bartel, D.P. Most mammalian mRNAs are conserved targets of microRNAs. Genome Res. 2009, 19, 92–105. [Google Scholar]
- Esau, C.; Davis, S.; Murray, S.F.; Yu, X.X.; Pandey, S.K.; Pear, M.; Watts, L.; Booten, S.L.; Graham, M.; McKay, R.; et al. MiR-122 regulation of lipid metabolism revealed by in vivo antisense targeting. Cell Metab. 2006, 3, 87–98. [Google Scholar] [CrossRef]
- Lindow, M.; Kauppinen, S. Discovering the first microRNA-targeted drug. J. Cell Biol. 2012, 199, 407–412. [Google Scholar] [CrossRef]
- Rayner, K.J.; Suarez, Y.; Davalos, A.; Parathath, S.; Fitzgerald, M.L.; Tamehiro, N.; Fisher, E.A.; Moore, K.J.; Fernandez-Hernando, C. MiR-33 contributes to the regulation of cholesterol homeostasis. Science 2010, 328, 1570–1573. [Google Scholar] [CrossRef]
- Carrer, M.; Liu, N.; Grueter, C.E.; Williams, A.H.; Frisard, M.I.; Hulver, M.W.; Bassel-Duby, R.; Olson, E.N. Control of mitochondrial metabolism and systemic energy homeostasis by microRNAs 378 and 378*. Proc. Natl. Acad. Sci. USA 2012, 109, 15330–15335. [Google Scholar] [CrossRef]
- Frost, R.J.; Olson, E.N. Control of glucose homeostasis and insulin sensitivity by the Let-7 family of microRNAs. Proc. Natl. Acad. Sci. USA 2011, 108, 21075–21080. [Google Scholar] [CrossRef]
- Parasramka, M.A.; Ho, E.; Williams, D.E.; Dashwood, R.H. MicroRNAs, diet, and cancer: New mechanistic insights on the epigenetic actions of phytochemicals. Mol. Carcinog. 2012, 51, 213–230. [Google Scholar] [CrossRef]
- Anderson, C.P.; Shen, M.; Eisenstein, R.S.; Leibold, E.A. Mammalian iron metabolism and its control by iron regulatory proteins. Biochim. Biophys. Acta 2012, 1823, 1468–1483. [Google Scholar] [CrossRef]
- Andrews, N.C. Forging a field: The golden age of iron biology. Blood 2008, 112, 219–230. [Google Scholar] [CrossRef]
- Hentze, M.W.; Muckenthaler, M.U.; Galy, B.; Camaschella, C. Two to tango: Regulation of Mammalian iron metabolism. Cell 2010, 142, 24–38. [Google Scholar] [CrossRef]
- Ganz, T. Hepcidin and its role in regulating systemic iron metabolism. Hematology Am. Soc. Hematol. Educ. Program 2006, 29–35. [Google Scholar] [CrossRef]
- Collins, J.F.; Wessling-Resnick, M.; Knutson, M.D. Hepcidin regulation of iron transport. J. Nutr. 2008, 138, 2284–2288. [Google Scholar] [CrossRef]
- Andolfo, I.; de Falco, L.; Asci, R.; Russo, R.; Colucci, S.; Gorrese, M.; Zollo, M.; Iolascon, A. Regulation of divalent metal transporter 1 (DMT1) non-IRE isoform by the microRNA Let-7d in erythroid cells. Haematologica 2010, 95, 1244–1252. [Google Scholar] [CrossRef]
- Hou, W.; Tian, Q.; Steuerwald, N.M.; Schrum, L.W.; Bonkovsky, H.L. The let-7 microRNA enhances heme oxygenase-1 by suppressing Bach1 and attenuates oxidant injury in human hepatocytes. Biochim. Biophys. Acta 2012, 1819, 1113–1122. [Google Scholar] [CrossRef]
- Castoldi, M.; Vujic Spasic, M.; Altamura, S.; Elmen, J.; Lindow, M.; Kiss, J.; Stolte, J.; Sparla, R.; D’Alessandro, L.A.; Klingmuller, U.; et al. The liver-specific microRNA miR-122 controls systemic iron homeostasis in mice. J. Clin. Investig. 2011, 121, 1386–1396. [Google Scholar] [CrossRef]
- Hou, W.; Tian, Q.; Zheng, J.; Bonkovsky, H.L. MicroRNA-196 represses Bach1 protein and hepatitis C virus gene expression in human hepatoma cells expressing hepatitis C viral proteins. Hepatology 2010, 51, 1494–1504. [Google Scholar] [CrossRef]
- Shpyleva, S.I.; Tryndyak, V.P.; Kovalchuk, O.; Starlard-Davenport, A.; Chekhun, V.F.; Beland, F.A.; Pogribny, I.P. Role of ferritin alterations in human breast cancer cells. Breast Cancer Res. Treat. 2011, 126, 63–71. [Google Scholar] [CrossRef]
- Chan, S.Y.; Zhang, Y.Y.; Hemann, C.; Mahoney, C.E.; Zweier, J.L.; Loscalzo, J. MicroRNA-210 controls mitochondrial metabolism during hypoxia by repressing the iron-sulfur cluster assembly proteins ISCU1/2. Cell Metab. 2009, 10, 273–284. [Google Scholar] [CrossRef]
- Yoshioka, Y.; Kosaka, N.; Ochiya, T.; Kato, T. Micromanaging Iron Homeostasis: Hypoxia-inducible micro-RNA-210 suppresses iron homeostasis-related proteins. J. Biol. Chem. 2012, 287, 34110–34119. [Google Scholar] [CrossRef]
- Liao, Y.; Du, X.; Lonnerdal, B. miR-214 regulates lactoferrin expression and pro-apoptotic function in mammary epithelial cells. J. Nutr. 2010, 140, 1552–1556. [Google Scholar] [CrossRef]
- Schaar, D.G.; Medina, D.J.; Moore, D.F.; Strair, R.K.; Ting, Y. miR-320 targets transferrin receptor 1 (CD71) and inhibits cell proliferation. Exp. Hematol. 2009, 37, 245–255. [Google Scholar] [CrossRef]
- Sangokoya, C.; Doss, J.F.; Chi, J.T. Iron-responsive miR-485-3p regulates cellular iron homeostasis by targeting ferroportin. PLoS Genet. 2013, 9, e1003408. [Google Scholar] [CrossRef]
- Liao, Y.; Lonnerdal, B. miR-584 mediates post-transcriptional expression of lactoferrin receptor in Caco-2 cells and in mouse small intestine during the perinatal period. Int. J. Biochem. Cell Biol. 2010, 42, 1363–1369. [Google Scholar] [CrossRef]
- Eisenstein, R.S. Iron regulatory proteins and the molecular control of mammalian iron metabolism. Ann. Rev. Nutr. 2000, 20, 627–662. [Google Scholar] [CrossRef]
- Wallander, M.L.; Leibold, E.A.; Eisenstein, R.S. Molecular control of vertebrate iron homeostasis by iron regulatory proteins. Biochim. Biophys. Acta 2006, 1763, 668–689. [Google Scholar] [CrossRef]
- Guo, B.; Yu, Y.; Leibold, E.A. Iron regulates cytoplasmic levels of a novel iron-responsive element-binding protein without aconitase activity. J. Biol. Chem. 1994, 269, 24252–24260. [Google Scholar]
- Iwai, K.; Drake, S.K.; Wehr, N.B.; Weissman, A.M.; LaVaute, T.; Minato, N.; Klausner, R.D.; Levine, R.L.; Rouault, T.A. Iron-dependent oxidation, ubiquitination, and degradation of iron regulatory protein 2: Implications for degradation of oxidized proteins. Proc. Natl. Acad. Sci. USA 1998, 95, 4924–4928. [Google Scholar] [CrossRef]
- Salahudeen, A.A.; Thompson, J.W.; Ruiz, J.C.; Ma, H.W.; Kinch, L.N.; Li, Q.; Grishin, N.V.; Bruick, R.K. An E3 ligase possessing an iron-responsive hemerythrin domain is a regulator of iron homeostasis. Science 2009, 326, 722–726. [Google Scholar] [CrossRef]
- Vashisht, A.A.; Zumbrennen, K.B.; Huang, X.; Powers, D.N.; Durazo, A.; Sun, D.; Bhaskaran, N.; Persson, A.; Uhlen, M.; Sangfelt, O.; et al. Control of iron homeostasis by an iron-regulated ubiquitin ligase. Science 2009, 326, 718–721. [Google Scholar] [CrossRef]
- Lill, R. Function and biogenesis of iron-sulphur proteins. Nature 2009, 460, 831–838. [Google Scholar] [CrossRef]
- Rouault, T.A.; Tong, W.H. Iron-sulfur cluster biogenesis and human disease. Trends Genet. 2008, 24, 398–407. [Google Scholar] [CrossRef]
- Sheftel, A.; Stehling, O.; Lill, R. Iron-sulfur proteins in health and disease. Trends Endocrinol. Metab. 2010, 21, 302–314. [Google Scholar] [CrossRef]
- Ryu, M.S.; Langkamp-Henken, B.; Chang, S.M.; Shankar, M.N.; Cousins, R.J. Genomic analysis, cytokine expression, and microRNA profiling reveal biomarkers of human dietary zinc depletion and homeostasis. Proc. Natl. Acad. Sci. USA 2011, 108, 20970–20975. [Google Scholar]
- Hintze, K.J.; Katoh, Y.; Igarashi, K.; Theil, E.C. Bach1 repression of ferritin and thioredoxin reductase1 is heme-sensitive in cells and in vitro and coordinates expression with heme oxygenase1, β-globin, and NADP(H) quinone (oxido) reductase1. J. Biol. Chem. 2007, 282, 34365–343271. [Google Scholar]
- Siaj, R.; Sauer, V.; Stoppeler, S.; Gerss, J.; Spiegel, H.U.; Kohler, G.; Zibert, A.; Schmidt, H.H. Longitudinal analysis of serum miR-122 in a rat model of Wilson’s disease. Hepatol. Int. 2012, 6, 770–777. [Google Scholar] [CrossRef]
- Lagos-Quintana, M.; Rauhut, R.; Yalcin, A.; Meyer, J.; Lendeckel, W.; Tuschl, T. Identification of tissue-specific microRNAs from mouse. Curr. Biol. 2002, 12, 735–739. [Google Scholar]
- Listowski, M.A.; Heger, E.; Boguslawska, D.M.; Machnicka, B.; Kuliczkowski, K.; Leluk, J.; Sikorski, A.F. microRNAs: Fine tuning of erythropoiesis. Cell. Mol. Biol. Lett. 2013, 18, 34–46. [Google Scholar] [CrossRef]
- Azzouzi, I.; Moest, H.; Winkler, J.; Fauchere, J.C.; Gerber, A.P.; Wollscheid, B.; Stoffel, M.; Schmugge, M.; Speer, O. MicroRNA-96 directly inhibits gamma-globin expression in human erythropoiesis. PLoS One 2011, 6, e22838. [Google Scholar] [CrossRef] [Green Version]
- Winterbourn, C.C. Toxicity of iron and hydrogen peroxide: The Fenton reaction. Toxicol. Lett. 1995, 82–83, 969–974. [Google Scholar] [CrossRef]
- Borel, M.J.; Smith, S.H.; Brigham, D.E.; Beard, J.L. The impact of varying degrees of iron nutriture on several functional consequences of iron deficiency in rats. J. Nutr. 1991, 121, 729–736. [Google Scholar]
- Wang, G.L.; Semenza, G.L. Desferrioxamine induces erythropoietin gene expression and hypoxia-inducible factor 1 DNA-binding activity: Implications for models of hypoxia signal transduction. Blood 1993, 82, 3610–3615. [Google Scholar]
- Sanchez, M.; Galy, B.; Muckenthaler, M.U.; Hentze, M.W. Iron-regulatory proteins limit hypoxia-inducible factor-2α expression in iron deficiency. Nat. Struct. Mol. Biol. 2007, 14, 420–426. [Google Scholar] [CrossRef]
- Davis, M.R.; Shawron, K.M.; Rendina, E.; Peterson, S.K.; Lucas, E.A.; Smith, B.J.; Clarke, S.L. Hypoxia inducible factor-2 α is translationally repressed in response to dietary iron deficiency in Sprague-Dawley rats. J. Nutr. 2011, 141, 1590–1596. [Google Scholar] [CrossRef]
- Anderson, S.A.; Nizzi, C.P.; Chang, Y.I.; Deck, K.M.; Schmidt, P.J.; Galy, B.; Damnernsawad, A.; Broman, A.T.; Kendziorski, C.; Hentze, M.W.; et al. The IRP1-HIF-2α axis coordinates iron and oxygen sensing with erythropoiesis and iron absorption. Cell Metab. 2013, 17, 282–290. [Google Scholar] [CrossRef]
- Semenza, G.L. Hypoxia-inducible factor 1 (HIF-1) pathway. Sci. STKE 2007, 2007, cm8. [Google Scholar] [CrossRef]
- Huang, X.; Ding, L.; Bennewith, K.L.; Tong, R.T.; Welford, S.M.; Ang, K.K.; Story, M.; Le, Q.T.; Giaccia, A.J. Hypoxia-inducible mir-210 regulates normoxic gene expression involved in tumor initiation. Mol. Cell 2009, 35, 856–867. [Google Scholar] [CrossRef]
© 2013 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Davis, M.; Clarke, S. Influence of microRNA on the Maintenance of Human Iron Metabolism. Nutrients 2013, 5, 2611-2628. https://doi.org/10.3390/nu5072611
Davis M, Clarke S. Influence of microRNA on the Maintenance of Human Iron Metabolism. Nutrients. 2013; 5(7):2611-2628. https://doi.org/10.3390/nu5072611
Chicago/Turabian StyleDavis, McKale, and Stephen Clarke. 2013. "Influence of microRNA on the Maintenance of Human Iron Metabolism" Nutrients 5, no. 7: 2611-2628. https://doi.org/10.3390/nu5072611
APA StyleDavis, M., & Clarke, S. (2013). Influence of microRNA on the Maintenance of Human Iron Metabolism. Nutrients, 5(7), 2611-2628. https://doi.org/10.3390/nu5072611