Does Vitamin C Deficiency Affect Cognitive Development and Function?



Abstract
:1. Introduction
2. Vitamin C

3. The Functions of Vitamin C in the Brain
3.1. The Antioxidant Role of Vitamin C in the Brain
3.2. Vitamin C as a Neuromodulator
3.3. Vitamin C in Angiogenesis
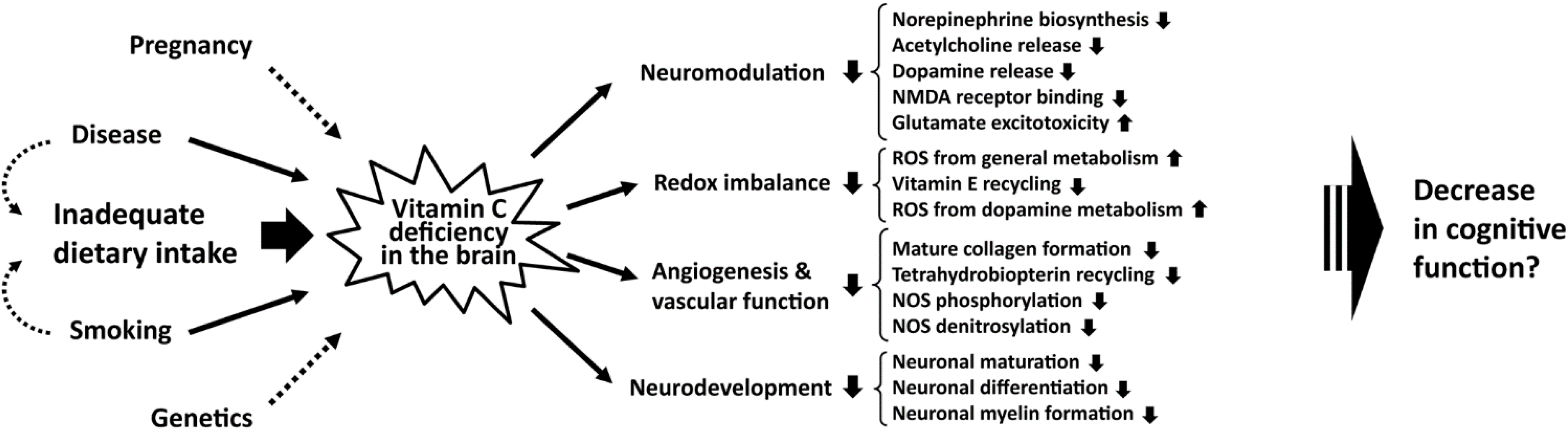
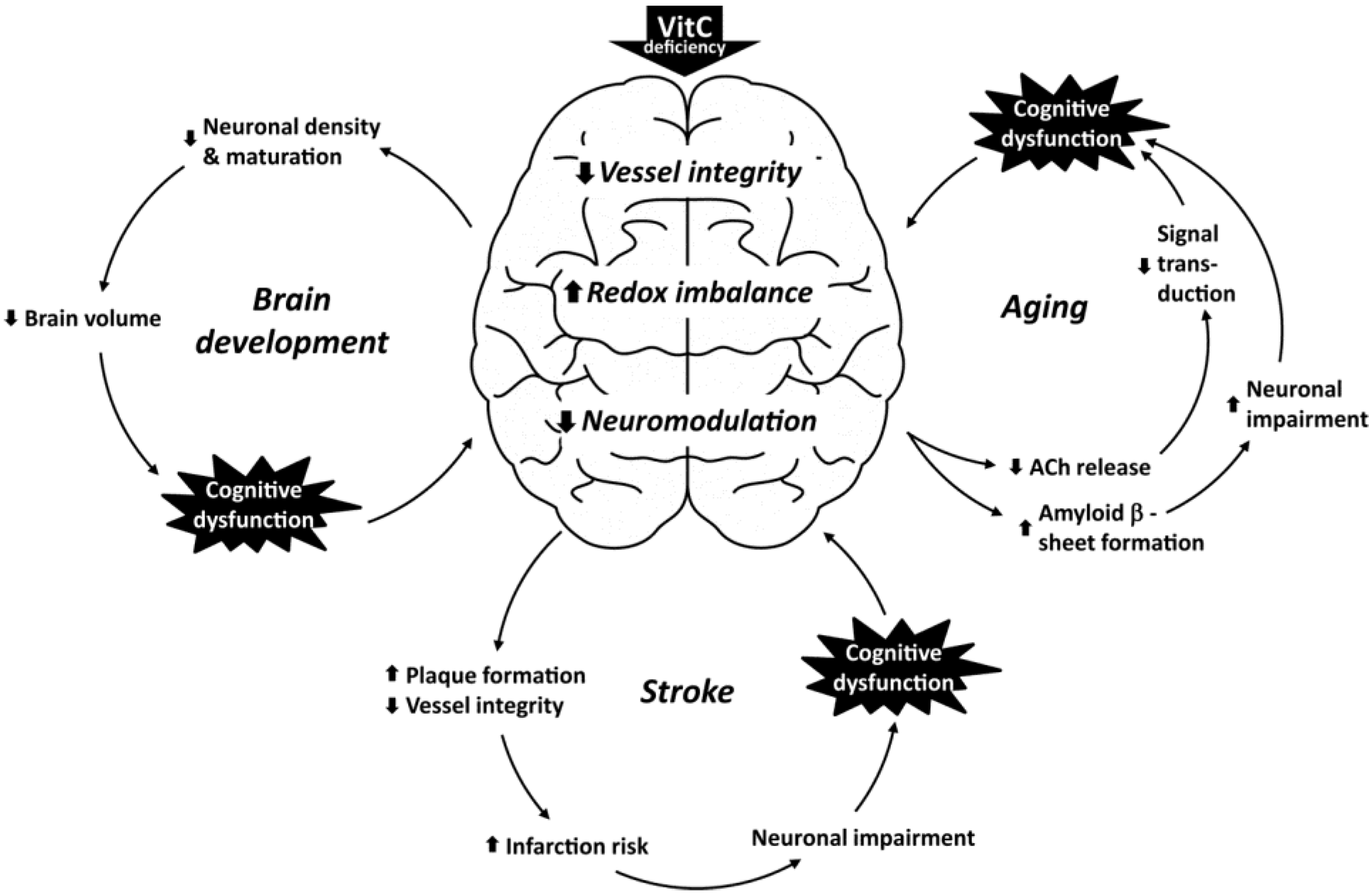
4. Vitamin C Deficiency and Cognitive Dysfunction

4.1. Putative Consequences of Vitamin C Deficiency in the Developing Brain

{kind=link}
{kind=link}
| Species | Intervention | Measurements | Outcome | Reference | |
|---|---|---|---|---|---|
| In Vivo Studies | |||||
| SVCT2(−/−) mice (ED: 18.5–19.5). | Dams: 0.33 g/L VitC in drinking water. | VitC content, MDA, F2-isoprostanes and F4-neuroprostanes in brain (cortex). Additional IHC. | The SVCT2(−/−) fetuses: Increased F2-isoprostanes (p < 0.001), F4-neuroprostanesincreased (p < 0.05), isoketal staining, apoptotic cells and hemorrhages. Decreased collagen-IV staining and VitC content (p < 0.001). | [7] | |
| Dunkin Hartley guinea pigs (6/7 to 60/61days). Postnatal deficiency. | VitC in diet: 923 mg/kg or 100 mg/kg feed. | Asc, DHA, glutathione, MDA and SOD in brain. Quantitation of hippocampal neurons. Functional assessment in MWM. | Decreased performance in MWM (p < 0.05) and reduced number of neurons in hippocampus (p < 0.05) in VitC deficient animals. | [1] | |
| Dunkin Hartley guinea pigs (GD: 18). Prenatal deficiency. | VitC prenatal: 900 mg/kg diet or 100 mg/kg diet. Postnatal: 750 mg/kg or 100 mg/kg. | Asc, DHA and MDA in brain. Hippocampal neurogenesis and volume. Functional assessment in MWM. | Significant and persistent lower hippocampal volume (p < 0.001). | [9] | |
| Dunkin Hartley guinea pigs (2 days to 3 weeks). | VitC in diet: 1036 mg/kg or 36 mg/kg. | Asc, DHA, glutathione, SOD, MDA, α- and γ-tocopherol, protein carbonyls, 8-oxo-deoxyguanosine and base excision repair in brain. | VitC deficiency caused significant reductions in Asc (p < 0.001), DHA (p = 0.034), MDA (p < 0.001) and protein carbonyls (p = 0.003) and an increase in base excision repair (p = 0.014). | [5] | |
| Design and Subjects | Intervention | Measurements | Outcome | Reference | |
| Clinical Studies | |||||
| Cohort Studies | 92 preterm children (7.86 ± 0.7 years, birth weight: 1475.13 ± 556.44 g) 40 age-matched controls. | Cognitive testing: Spatial pattern/Recognition, Intradimensional/Extra Dimensional Set-Shifting task, Tower of London task, Spatial Working Memory task, Spatial Memory Span task, and a Psychomotor screening. | Preterm children had decreased performance in Psychomotor test (p < 0.01), Recognition test (p < 0.01), Spatial Memory Span (p < 0.01), and Spatial Working Memory (p < 0.001) | [90] | |
| 13 IUGR preterm infants (gestational age 33–34 weeks) and 12 controls. | Maternal blood, umbilical cord blood and placental samples: SOD, GSH-Px, MDA, AOP, ADA, CAT and XO. | All markers, except GSH-Px and AOP were elevated in umbilical cord blood. IUGR mothers differed significantly in all markers other than CAT. Placental samples were significantly changed in all markers, except SOD and ADA (p < 0.01 or less). | [96] | ||
| Controlled trials | Randomized clinical trial: 160 women in high risk for pre-eclampsia (16–22 weeks pregnant) and 32 controls. | VitC (1000 mg/day) and VitE (400 IU/day) or placebo. | Plasma VitC, PAI-2, placenta growth factor, 8-epi-prostaglandin F2α, leptin, PAI-1/2 ratio. | Vitamin supplemented: VitC, 8-epi-prostaglandin F2α, leptin, and PAI-1/-2 equal to controls; whereas placebo-treated displayed decreased VitC, PAI-2, and placenta growth factor and increased 8-epi-prostaglandin F2α, leptin, and PAI 1/-2 ratio. | [104] |
| Randomized clinical trial: 283 women with high risk of pre-eclampsia (16–22 weeks pregnant). | VitC (1000 mg/day) and VitE (400 IU/day) or placebo. | PAI-1 and -2 measured every month until delivery. Pre-eclampsia assessed by the development of proteinuric hypertension. | VitC + E supplementation was associated with a decrease in the PAI-1/PAI-2 ratio (p = 0.015) and a significantly decreased risk of pre-eclampsia (p = 0.002). | [107] | |
| Double blind randomized clinical trial: 100 women in high risk of pre-eclampsia (14–20 weeks pregnant). | VitC (1000 mg/day) and VitE (400 IU/day) or placebo. | Incidence of pre-eclampsia. | No significant effect of VitC + E treatment. | [105] | |
| Double blind randomized clinical trial: 1365 women in high risk for pre-eclampsia (14–22 weeks pregnant). | VitC (1000 mg/day) and VitE (400 IU/day) or placebo. | Occurrence of pre-eclampsia defined as hypertension and onset of proteinuria. | Supplementation with VitC + E did not reduce risk of pre-eclampsia. | [106] | |
4.2. Vitamin C and Aging
| Species | Intervention | Measurement | Outcome | Reference | |
|---|---|---|---|---|---|
| In vivo studies | |||||
| APP/PSEN1 and B6C3F1/J mice (6–10 months). | VitC in diet (1 g/kg) and high or low dose VitE (750/400 IU/kg). | Functional assessment, amyloid, F4-neuroprostanes and MDA. | Supplementation with VitC and low VitE decreased markers of oxidative stress in transgenic mice (p < 0.05). Improvement of MWM performance was seen in low VitE group (0.05 > p < 0.001). | [127] | |
| Swiss mice (3 and 7 months). | IP injection of 60 and 120 mg/kg VitC for three or eight consecutive days. | Elevated plus maze, passive avoidance test. | Treatment improved performance in young animals (p < 0.05) and reversed performance deficits in old animals (p < 0.05). | [111] | |
| Dunkin Hartley guinea pig (3–9 months and 36–42 months). | Diet containing 325 mg VitC/kg or 100 mg VitC/kg. | VitC, MDA, glutathione, 8-oxodG and SOD in brain. SVCT2 mRNA expression in brain. | Deficiency did not cause significant changes in oxidative stress markers but aging per se showed a significant effect (p < 0.05). No detectable effect on SVCT mRNA expression in deficient. | [116] | |
| AβPP mice (6–12 months). | 1333 mg/kg/day VitC in drinking water. | IHC for anti-Aβ, Western blot, Aβ-ELISA, OxyBlot, glutathione, functional assessment by MVM and elevated plus maze. | VitC prevented some behavioral abnormalities in AβPP mice (0.05 > p < 0.02), down-regulated amyloid (p < 0.05), significant difference of Aβ42/Aβ40 ratio (p < 0.02) and increased in synaptophysin (p < 0.05). Phosphorylated tau was decreased (p < 0.05). | [129] | |
| Female ovariectomized Wistar rats (80 days). | VitE (40 mg/kg) and VitC (100 mg/kg) IP once daily for 30 days. | MWM, open field test. | Vitamin C + E treatment prevented deficits in reference memory in MWM (0.01 > p < 0.05). | [114] | |
| Swiss mice (3 months). | VitC (60 mg/kg) IP injection of for three consecutive days. | Elevated plus maze and passive avoidance. | VitC injection reversed amnesia induced by scopolamine (0.4 mg/kg) and diazepam (1 mg/kg) (p < 0.05). | [111] | |
| B6C3F1/J mice (12 weeks). | VitC (125 mg/kg) IP. | Behavioral testing, MDA and Asc content in cortex, AChE activity, brain glutathione. | VitC treatment reversed some of the memory deficits induced by scopolamine (1 mg/kg IP) (0.05 > p < 0.001) and increased medial forebrain AChE acticity (p < 0.001). | [130] | |
| CD1 mice (16 months). | Oxiracetam (62.5/125/250 mg/kg), VitC (50/100/200 mg/kg), VitC (125 mg/kg) + oxiracetam (100 mg/kg) IP for three consecutive days. | Light-dark aversion test. | VitC alone or in combination with oxiracetam significantly reduced scopolamine-induced (0.25 mg/kg IP, day 4) amnesia (p < 0.01). | [136] | |
| Dunkin Hartley guinea pigs (3–9 and 8–14 months). | Diet with 325 mg/kg VitC. | VitC in brain and CSF. | Concentrations of VitC significantly increased in CSF with age (p < 0.05). Elevated Asc oxidation ratio in young compared to old animals (p < 0.05). | [115] | |
| Dunkin Hartley guinea pigs (3–9 and 36–42 months). | Diet with 325 mg VitC/kg or 100 mg VitC/kg. | VitC in CSF and 8-oxodG, MDA, glutathione and SOD in brain. | No effect was observed besides on VitC concentration in brain and CSF in deficient animals. | [116] | |
| Design and Subjects | Intervention | Measurements | Outcome | Reference | |
| Clinical Studies | |||||
| Cohort studies | 12 AD or dementia patients (71 ± 11 years) and healthy controls (35 ± 5 years). | Blood samples of VitC and DHA. | Dementia and AD patients had significantly lower Asc and DHA levels (p < 0.001). | [137] | |
| Patients (n = 134) (AD, vascular dementia or Parkinson’s) and 58 matching controls. | Plasma content of: α-carotene, β-carotene, lycopene, VitA, VitC, VitE and TAC. | VitC was significantly lower in AD (p < 0.001), vascular dementia (p < 0.001) and Parkinson’s disease with dementia (p < 0.01). | [117] | ||
| Prospective cohort study: 633 participants age ≥65 years. | Direct inspection of ingested supplements (two weeks of base-line). Participants were followed for a mean of 4.3 years. | None of the VitE or VitC users developed AD despite a predicted incidence of 3.9 and 3.2, respectively (p = 0.04). | [132] | ||
| Nurses’ Health study: 14,968 women age 70–79 in 1995–2000. | Semi-quantitative questionnaire on lifestyle, supplemental use and medical history biennially from 1980. | TICS, 10-word list, immediate and delayed recall, verbal fluency, digit span backwards test. | Long-term VitC + E supplementation was associated with better cognitive function (p = 0.03) and a trend toward better performance (p = 0.04) | [118] | |
| The Honolulu-Asia Aging Study: 3385 men age 71–93 years. | Questionnaires on vitamin supplementation in 1982/1988. | Assessment of cognitive performance by CASI in 1991–1993. | VitC and/or VitE supplementation decreased the incidence of vascular (OR: 0.12) and mixed/other type dementia (OR: 0.31) and was associated with a higher cognitive performance (OR: 1.25). | [119] | |
| The Rotterdam Prospective Study: 5395 participants age ≥55 years in 1990–1993 | Interview of dietary intake of VitC, VitE, β-carotene, supplements, educational level, etc. | Clinical examination and MMSE, GMS, CAMDEX in 1993–1994 and 1997–1999. | High dietary intake of VitC and VitE may lower the risk of Alzheimer’s disease. RR = +0.82/standard deviation increase in VitC intake. | [16] | |
| Prospective cohort study: 32 patients with mild to moderate AD age 71 ± 7 years. | Physical examination. | ADAS-cog, MMSE, CDR and geriatric depression. CSF and blood samples at baseline. | CSF/Plasma VitC content predicted cognitive decline partially due to a compromised blood brain barrier integrity. | [15] | |
| Nurses’ Health Study: 16,010 women age ≥70 years in 1995–2000. | Food-questionnaire in 1980 and expanded version in 1984, 1986, and every four years thereafter. FRAP assessment. | TICS scores and ten word list, global composite scores, East Boston Memory test on three occations. | No significant association between FRAP scores and cognitive function, when adjusted for confounders. | [138] | |
| Adult Changes in Thought Prospective Study: 2969 participants age ≥65 years. | Self-reported VitC, VitE or multivitamin supplement. Participants were followed for a mean of 5.5 years. | Health and lifestyle parameters (e.g., BMI, smoking and alcohol consumption) CASI score every second year. | Neither VitC, VitE nor multivitamin use was associated with a decreased incidence of AD or dementia. | [133] | |
| Prospective cohort study: 137 elderly age 66–90 years. | Nutritional data collected in 1980 and 1986. | Cognitive evaluation in Logical Memory, Abstraction and Visual Reproduction trials in 1986. | Plasma concentrations of VitC were positively correlated with Rey-Osterrieth Copy test performance and Visual Reproduction (p < 0.05). | [139] | |
| Prospective cohort study: 921 elderly age ≥65 years. | A one week food diary or interviews to quantify consumer habits. Participants were followed for 20 years. | Medical examination including Hodkinson Abbreviated Mental test. | Participants with the lowest dietary/plasma VitC status had the poorest cognitive function (OR: 1.6). | [3] | |
| Clinical trials | Randomized open-label clinical trial: 23 AD patients receiving cholinergic treatment. | 400 IU VitE and 1000 mg VitC per day or no vitamin treatment. CSF samples at baseline, one month and twelve months. | Clinical and neuropsychological assessment. | Significant increases in VitC content in CSF and decreases in autoxidation (p < 0.05). No neuropsychological differences. | [140] |
4.3. Vitamin C and Stroke
| Species | Intervention | Measurement | Outcome | Reference | |
|---|---|---|---|---|---|
| In vivo studies | |||||
| C57BL/6J mice. | DHA (40/250/500 mg/kg) or Asc (250/500 mg/kg) IV on three time points following MCAO. | Cortical cerebral blood flow, infarct volume, neurological assessment, mortality. | DHA improved cerebral blood flow dose-dependent. Decreased infarct size and mortality (p < 0.05). Asc did not show these effects. | [51] | |
| SHR and SHR-SP rats (4–5 months old). | VitC (200 mg/kg) and VitE (100 mg/kg) PO once daily for 4 weeks MCAO of 24 h duration. | 2D Western blot of antioxidative protein expression, TAC, GSH-Px and MDA in brain. Cerebral infarct area. | VitC + E treatment significantly reduced oxidative stress and infarct area in SHR-SP (p < 0.01). | [52] | |
| Male Sprague-Dawley rats (4 weeks old) with or without STZ-induced diabetes for six weeks | VitC (100 mg/kg) PO once daily for 2 weeks following MCAO/Re | Infarct volume and edema, neurological score. | VitC treatment significantly reduced infarct area, edema and neurological score in both non-diabetic and diabetic animals compared to untreated controls (p < 0.01). | [143] | |
| Maccaca radiata monkey. | Ascorbate (500 mg/kg up to 2 g IV) immediately before MCAO of 4 h duration. | Cerebral infarct area. | VitC treatment significantly reduced infarct area (p = 0.0003). | [142] | |
| Design and Subjects | Intervention | Measurements | Outcome | Reference | |
| Clinical Studies | |||||
| Cohort studies | Department of Health and Social Security nutritional survey: 730 participants age ≥65 years. | Food diary and interviews. Participants were followed for 20 years. | Plasma VitC, physical examination. | Participants in the highest third of VitC intake had a RR = 0.5, when compared with the lowest third. | [146] |
| The Nurses’ Health Study: 85,118 participants age 30–55 years. | Semi-quantitative questionnaire on lifestyle, supplemental use and medical history. The participants were followed for 16 years. | VitC supplemental use is significantly associated with lower risk of coronary heart disease (RR = 0.72). | [147] | ||
| Cancer-Norfolk prospective study: 20,649 participants age 40–79 years. | Health and lifestyle questionnaire, socioeconomic data | Physical examination, plasma VitC content. | Plasma VitC was inversely related to risk of stroke. Participants in top quantile had a RR = 0.58. | [145] | |
| Basel prospective study 2974 men | The participants were followed for 12 years. | Baseline values of VitC and β-carotene in plasma. | Low levels of VitC and β-carotene were related to an increased risk of dying from ischemic heart disease or stroke. | [50] | |
| Clinical trials | Double-blind randomized clinical trial: 40 patients (0–2 years after cardiac transplant). | 500 mg VitC and 400 IU VitE twice daily for one year or placebo. | Plasma VitC and VitE content. Average intimal index, coronary endothelium-dependent vasoreactivity. | Supplementation with VitC + E caused retardation of early signs of atherosclerosis associated with heart transplantation (p = 0.008). | [148] |
| Sixty ischemic stroke patients (72.8 ± 10.4 years), VitC vs. non-VitC group and 20 controls (69.8 ± 10.5 years). | 500 mg/day VitC IV in addition to standard stroke treatment for ten days starting the day after stroke. | NIHSS neurological status and bilirubin, creatinine, uric acid, and TAC day one, three, five and ten of treatment. NIHSS three months after stroke. | No difference in clinical status of patients during the ten day treatment or after the three months follow-up. | [144] | |
5. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Tveden-Nyborg, P.; Johansen, L.K.; Raida, Z.; Villumsen, C.K.; Larsen, J.O.; Lykkesfeldt, J. Vitamin C deficiency in early postnatal life impairs spatial memory and reduces the number of hippocampal neurons in guinea pigs. Am. J. Clin. Nutr. 2009, 90, 540–546. [Google Scholar] [PubMed]
- Tveden-Nyborg, P.; Lykkesfeldt, J. Does vitamin C deficiency result in impaired brain development in infants? Redox Rep. 2009, 14, 1–6. [Google Scholar]
- Gale, C.R.; Martyn, C.N.; Cooper, C. Cognitive impairment and mortality in a cohort of elderly people. Br. Med. J. 1996, 312, 608–611. [Google Scholar] [CrossRef]
- Rice, M.E. Ascorbate regulation and its neuroprotective role in the brain. Trends Neurosci. 2000, 23, 209–216. [Google Scholar] [CrossRef] [PubMed]
- Lykkesfeldt, J.; Trueba, G.P.; Poulsen, H.E.; Christen, S. Vitamin C deficiency in weanling guinea pigs: Differential expression of oxidative stress and DNA repair in liver and brain. Br. J. Nutr. 2007, 98, 1116–1119. [Google Scholar] [CrossRef] [PubMed]
- Hughes, R.E.; Hurley, R.J.; Jones, P.R. Retention of ascorbic acid by guinea pig tissues. Br. J. Nutr. 1971, 26, 433–438. [Google Scholar] [CrossRef] [PubMed]
- Harrison, F.E.; Dawes, S.M.; Meredith, M.E.; Babaev, V.R.; Li, L.; May, J.M. Low vitamin C and increased oxidative stress and cell death in mice that lack the sodium-dependent vitamin C transporter SVCT2. Free Radic. Biol. Med. 2010, 49, 821–829. [Google Scholar] [CrossRef] [PubMed]
- Sotiriou, S.; Gispert, S.; Cheng, J.; Wang, Y.H.; Chen, A.; Hoogstraten-Miller, S.; Miller, G.F.; Kwon, O.; Levine, M.; Guttentag, S.H.; et al. Ascorbic acid transporter Slc23a1 is essential for vitamin C transport into the brain and for perinatal survival. Nat. Med. 2002, 8, 514–517. [Google Scholar] [CrossRef] [PubMed]
- Tveden-Nyborg, P.; Vogt, L.; Schjoldager, J.G.; Jeannet, N.; Hasselholt, S.; Paidi, M.D.; Christen, S.; Lykkesfeldt, J. Maternal vitamin C deficiency during pregnancy persistently impairs hippocampal neurogenesis in offspring of guinea pigs. PLoS One 2012, 7, e48488. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hampl, J.S.; Taylor, C.A.; Johnston, C.S. Vitamin C deficiency and depletion in the United States: The Third National Health and Nutrition Examination Survey, 1988 to 1994. Am. J. Public Health 2004, 94, 870–875. [Google Scholar] [CrossRef] [PubMed]
- Lykkesfeldt, J.; Michels, A.J.; Frei, B. Vitamin C. Adv. Nutr. 2014, 5, 16–18. [Google Scholar] [CrossRef] [PubMed]
- Smith, J.L.; Hodges, R.E. Serum levels of vitamin C in relation to dietary and supplemental intake of vitamin C in smokers and nonsmokers. Ann. N. Y. Acad. Sci. 1987, 498, 144–152. [Google Scholar] [CrossRef] [PubMed]
- Tveden-Nyborg, P.; Lykkesfeldt, J. Does vitamin C deficiency increase lifestyle associated vascular disease progression? Evidence based on experimental and clinical studies. Antioxid. Redox Signal. 2013, 19, 2084–2104. [Google Scholar] [CrossRef] [PubMed]
- Rebec, G.V.; Barton, S.J.; Marseilles, A.M.; Collins, K. Ascorbate treatment attenuates the Huntington behavioral phenotype in mice. Neuroreport 2003, 14, 1263–1265. [Google Scholar] [CrossRef] [PubMed]
- Bowman, G.L.; Dodge, H.; Frei, B.; Calabrese, C.; Oken, B.S.; Kaye, J.A.; Quinn, J.F. Ascorbic acid and rates of cognitive decline in Alzheimer’s disease. J. Alzheimers Dis. 2009, 16, 93–98. [Google Scholar] [PubMed]
- Engelhart, M.J.; Geerlings, M.I.; Ruitenberg, A.; van Swieten, J.C.; Holman, A.; Witteman, J.C.M.; Breteler, M.M.B. Dietary intake of antioxidants and risk of Alzheimer disease. J. Am. Med. Assoc. 2002, 287, 3223–3229. [Google Scholar] [CrossRef]
- Valencia, A.; Sapp, E.; Kimm, J.S.; McClory, H.; Reeves, P.B.; Alexander, J.; Ansong, K.A.; Masso, N.; Frosch, M.P.; Kegel, K.B.; et al. Elevated NADPH oxidase activity contributes to oxidative stress and cell death in Huntington’s disease. Hum. Mol. Genet. 2013, 22, 1112–1131. [Google Scholar]
- Hercberg, S.; Galan, P.; Preziosi, P.; Bertrais, S.; Mennen, L.; Malvy, D.; Roussel, A.M.; Favier, P.; Briancon, S. The Su.Vi.Max study—A randomized, placebo-controlled trial of the health effects of antioxidant vitamins and minerals. Arch. Int. Med. 2004, 164, 2335–2342. [Google Scholar] [CrossRef]
- Mosdol, A.; Erens, B.; Brunner, E.J. Estimated prevalence and predictors of vitamin C deficiency within UK’s low-income population. J. Public Health 2008, 30, 456–460. [Google Scholar] [CrossRef]
- Wrieden, W.L.; Hannah, M.K.; Bolton-Smith, C.; Tavendale, R.; Morrison, C.; Tunstall-Pedoe, H. Plasma vitamin C and food choice in the third Glasgow Monica population survey. J. Epidemiol. Community Health 2000, 54, 355–360. [Google Scholar] [CrossRef] [PubMed]
- De Oliveira, A.M.; Rondo, P.H.D.; Barros, S.B.D. Concentrations of ascorbic acid in the plasma of pregnant smokers and nonsmokers and their newborns. Int. J. Vitam. Nutr. Res. 2004, 74, 193–198. [Google Scholar] [CrossRef] [PubMed]
- Ortega, R.M.; Lopez-Sobaler, A.M.; Quintas, M.E.; Martinez, R.M.; Andres, P. The influence of smoking on vitamin C status during the third trimester of pregnancy and on vitamin C levels in maternal milk. J. Am. Coll. Nutr. 1998, 17, 379–384. [Google Scholar] [CrossRef] [PubMed]
- Johnston, C.S.; Thompson, L.L. Vitamin C status of an outpatient population. J. Am. Coll. Nutr. 1998, 17, 366–370. [Google Scholar] [CrossRef] [PubMed]
- Dhariwal, K.R.; Hartzell, W.O.; Levine, M. Ascorbic acid and dehydroascorbic acid measurements in human plasma and serum. Am. J. Clin. Nutr. 1991, 54, 712–716. [Google Scholar] [PubMed]
- Nishikimi, M.; Kawai, T.; Yagi, K. Guinea pigs possess a highly mutated gene for l-gulono-gamma-lactone oxidase, the key ezyme for l-ascorbic acid biosynthesis missing in this species. J. Biol. Chem. 1992, 267, 21967–21972. [Google Scholar] [PubMed]
- Nishikimi, M.; Fukuyama, R.; Minoshima, S.; Shimizu, N.; Yagi, K. Cloning and chromosomal mapping of the human nonfunctional gene for l-gulono-gamma-lactone oxidase, the enzyme for l-ascorbic acid biosynthesis missing in man. J. Biol. Chem. 1994, 269, 13685–13688. [Google Scholar] [PubMed]
- Chatterjee, I. Evolution and biosynthesis of ascorbic acid. Science 1973, 182, 1271–1272. [Google Scholar] [PubMed]
- Nandi, A.; Mukhopadhyay, C.K.; Ghosh, M.K.; Chattopadhyay, D.J.; Chatterjee, I.B. Evolutionary significance of vitamin C biosynthesis in terrestrial vertebrates. Free Radic. Biol. Med. 1997, 22, 1047–1054. [Google Scholar] [CrossRef] [PubMed]
- Fischer, H.; Schwarzer, C.; Illek, B. Vitamin C controls the cystic fibrosis transmembrane conductance regulator chloride channel. Proc. Natl. Acad. Sci. USA 2004, 101, 3691–3696. [Google Scholar] [CrossRef] [PubMed]
- Corti, A.; Casini, A.F.; Pompella, A. Cellular pathways for transport and efflux of ascorbate and dehydroascorbate. Arch. Biochem. Biophys. 2010, 500, 107–115. [Google Scholar] [CrossRef] [PubMed]
- Lindblad, M.; Tveden-Nyborg, P.; Lykkesfeldt, J. Regulation of vitamin C homeostasis during deficiency. Nutrients 2013, 5, 2860–2879. [Google Scholar] [CrossRef] [PubMed]
- Tsukaguchi, H.; Tokui, T.; Mackenzie, B.; Berger, U.V.; Chen, X.Z.; Wang, Y.X.; Brubaker, R.F.; Hediger, M.A. A family of mammalian Na+-dependent l-ascorbic acid transporters. Nature 1999, 399, 70–75. [Google Scholar] [CrossRef] [PubMed]
- Lykkesfeldt, J.; Loft, S.; Nielsen, J.B.; Poulsen, H.E. Ascorbic acid and dehydroascorbic acid as biomarkers of oxidative stress caused by smoking. Am. J. Clin. Nutr. 1997, 65, 959–963. [Google Scholar] [PubMed]
- Lykkesfeldt, J.; Viscovich, M.; Poulsen, H.E. Ascorbic acid recycling in human erythrocytes is induced by smoking in vivo. Free Radic. Biol. Med. 2003, 35, 1439–1447. [Google Scholar] [PubMed]
- Berger, U.V.; Hediger, M.A. The vitamin C transporter SVCT2 is expressed by astrocytes in culture but not in situ. Neuroreport 2000, 11, 1395–1399. [Google Scholar] [PubMed]
- Harrison, F.E.; Green, R.J.; Dawes, S.M.; May, J.M. Vitamin C distribution and retention in the mouse brain. Brain Res. 2010, 1348, 181–186. [Google Scholar] [CrossRef] [PubMed]
- Milby, K.; Oke, A.; Adams, R.N. Detailed mapping of ascorbate distribution in rat brain. Neurosci. Lett. 1982, 28, 15–20. [Google Scholar] [CrossRef] [PubMed]
- Dobbing, J. The later growth of the brain and its vulnerability. Pediatrics 1974, 53, 2–6. [Google Scholar] [PubMed]
- Rice, M.E.; Russo-Menna, I. Differential compartmentalization of brain ascorbate and glutathione between neurons and glia. Neuroscience 1998, 82, 1213–1223. [Google Scholar] [CrossRef] [PubMed]
- Erecinska, M.; Cherian, S.; Silver, I.A. Energy metabolism in mammalian brian during development. Prog. Neurobiol. 2004, 73, 397–445. [Google Scholar] [CrossRef] [PubMed]
- Mun, G.H.; Kim, M.J.; Lee, J.H.; Kim, H.J.; Chung, Y.H.; Chung, Y.B.; Kang, J.S.; Hwang, Y.I.; Oh, S.H.; Kim, J.G.; et al. Immunohistochemical study of the distribution of sodium-dependent vitamin C transporters in adult rat brain. J. Neurosci. Res. 2006, 83, 919–928. [Google Scholar] [CrossRef] [PubMed]
- Mefford, I.N.; Oke, A.F.; Adams, R.N. Regional distribution of ascorbate in human brain. Brain Res. 1981, 212, 223–226. [Google Scholar] [CrossRef] [PubMed]
- Grant, M.M.; Barber, V.S.; Griffiths, H.R. The presence of ascorbate induces expression of brain derived neurotrophic factor in SH-SY5Y neuroblastorna cells after peroxide insult, which is associated with increased survival. Proteomics 2005, 5, 534–540. [Google Scholar] [CrossRef] [PubMed]
- Halliwell, B. Reactive oxygen species and the central nervous system. J. Neurochem. 1992, 59, 1609–1623. [Google Scholar] [CrossRef] [PubMed]
- Bendich, A.; Machlin, L.J.; Scandurra, O.; Burton, G.W.; Wayner, D.D.M. The antioxidant role of vitamin C. Adv. Free Radic. Biol. Med. 1986, 2, 419–444. [Google Scholar] [CrossRef]
- Seregi, A.; Schaefer, A.; Komlos, M. Protective role of brain ascorbic acid content against lipid peroxidation. Experientia 1978, 34, 1056–1057. [Google Scholar] [CrossRef] [PubMed]
- Qiu, S.; Li, L.; Weeber, E.J.; May, J.M. Ascorbate transport by primary cultured neurons and its role in neuronal function and protection against excitotoxicity. J. Neurosci. Res. 2007, 85, 1046–1056. [Google Scholar] [CrossRef] [PubMed]
- Buettner, G.R.S.; Schafer, F.Q. Ascorbate as an antioxidant. In Vitamin C—Function and Biochemistry in Animals and Plants; Asard, H., May, J., Smirnoff, N., Eds.; Garland Science/BIOS Scientific Publishers: Bodmin, UK, 2004; pp. 173–188. [Google Scholar]
- Abramov, A.Y.; Scorziello, A.; Duchen, M.R. Three distinct mechanisms generate oxygen free radicals in neurons and contribute to cell death during anoxia and reoxygenation. J. Neurosci. 2007, 27, 1129–1138. [Google Scholar] [CrossRef] [PubMed]
- Gey, K.F.; Stahelin, H.B.; Eichholzer, M. Poor plasma status of carotene and vitamin C is associated with higher mortality from ischaemic heart disease and stroke: Basel Prospective Study. Clin. Investig. 1993, 71, 3–6. [Google Scholar] [PubMed]
- Huang, J.; Agus, D.B.; Winfree, C.J.; Kiss, S.; Mack, W.J.; McTaggart, R.A.; Choudhri, T.F.; Kim, L.J.; Mocco, J.; Pinsky, D.J.; et al. Dehydroascorbic acid, a blood-brain barrier transportable form of vitamin C, mediates potent cerebroprotection in experimental stroke. Proc. Natl. Acad. Sci. USA 2001, 98, 11720–11724. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.H.; Lei, H.; Liu, A.J.; Zou, Y.X.; Shen, F.M.; Su, D.F. Increased oxidative stress is responsible for severer cerebral infarction in stroke-prone spontaneously hypertensive rats. CNS Neurosci. Ther. 2011, 17, 590–598. [Google Scholar] [CrossRef] [PubMed]
- Bogdanov, M.B.; Andreassen, O.A.; Dedeoglu, A.; Ferrante, R.J.; Beal, M.F. Increased oxidative damage to DNA in a transgenic mouse model of Huntington’s disease. J. Neurochem. 2001, 79, 1246–1249. [Google Scholar] [CrossRef] [PubMed]
- Heo, J.-H.; Hyon, L.; Lee, K.-M. The possible role of antioxidant vitamin C in Alzheimer’s disease treatment and prevention. Am. J. Alzheimers Dis. Dement. 2013, 28, 120–125. [Google Scholar] [CrossRef]
- Browne, S.E.; Bowling, A.C.; MacGarvey, U.; Baik, M.J.; Berger, S.C.; Muqit, M.M.K.; Bird, E.D.; Beal, M.F. Oxidative damage and metabolic dysfunction in Huntington’s disease: Selective vulnerability of the basal ganglia. Ann. Neurol. 1997, 41, 646–653. [Google Scholar] [CrossRef] [PubMed]
- Do, K.Q.; Trabesinger, A.H.; Kirsten-Kruger, M.; Lauer, C.J.; Dydak, U.; Hell, D.; Holsboer, F.; Boesiger, P.; Cuenod, M. Schizophrenia: Glutathione deficit in cerebrospinal fluid and prefrontal cortex in vivo. Eur. J. Neurosci. 2000, 12, 3721–3728. [Google Scholar] [CrossRef] [PubMed]
- Akyol, O.; Herken, H.; Uz, E.; Fadillioglu, E.; Unal, S.; Sogut, S.; Ozyurt, H.; Savas, H.A. The indices of endogenous oxidative and antioxidative processes in plasma from schizophrenic patients the possible role of oxidant/antioxidant imbalance. Prog. Neuro-Psychopharmacol. Biol. Psychiatry 2002, 26, 995–1005. [Google Scholar]
- Kulak, A.; Cuenod, M.; Do, K.Q. Behavioral phenotyping of glutathione-deficient mice: Relevance to schizophrenia and bipolar disorder. Behav. Brain Res. 2012, 226, 563–570. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.Y.; Chang, M.Y.; Park, C.H.; Kim, H.Y.; Kim, J.H.; Son, H.; Lee, Y.S.; Lee, S.H. Ascorbate-induced differentiation of embryonic cortical precursors into neurons and astrocytes. J. Neurosci. Res. 2003, 73, 156–165. [Google Scholar] [CrossRef] [PubMed]
- Eldridge, C.F.; Bunge, M.B.; Bunge, R.P.; Wood, P.M. Differentiation of axon-related schwann-cells in vitro. Ascorbic acid regulates basal lamina assembly and myelin formation. J. Cell Biol. 1987, 105, 1023–1034. [Google Scholar]
- Jaber, M.; Robinson, S.W.; Missale, C.; Caron, M.G. Dopamine receptors and brain function. Neuropharmacology 1996, 35, 1503–1519. [Google Scholar] [PubMed]
- Beaulieu, J.M.; Gainetdinov, R.R. The physiology, signaling, and pharmacology of dopamine receptors. Mol. Cell. Biol. 2014, 34, 182–217. [Google Scholar]
- Diliberto, E.J.; Allen, P.L. Semidehydroascorbate as a product of the enzymic conversion of dopamine to norepinephrine—Coupling of semidehydroascorbate reductase to dopamine-β-hydroxylase. Mol. Pharmacol. 1980, 17, 421–426. [Google Scholar] [PubMed]
- Levine, M.; Asher, A.; Pollard, H.; Zinder, O. Ascorbic acid and catecholamine secretion from cultured chromaffin cells. J. Biol. Chem. 1983, 258, 3111–3115. [Google Scholar]
- Desole, M.S.; Miele, M.; Enrico, P.; Esposito, G.; Fresu, L.; Denatale, G.; Miele, E. Investigations into the relationship between the dopaminergic system and ascorbic acid in rat striatum. Neurosci. Lett. 1991, 127, 34–38. [Google Scholar] [CrossRef] [PubMed]
- Berman, S.B.; Zigmond, M.J.; Hastings, T.G. Modification of dopamine transporter function: Effect of reactive oxygen species and dopamine. J. Neurochem. 1996, 67, 593–600. [Google Scholar] [CrossRef] [PubMed]
- Hastings, T.G.; Lewis, D.A.; Zigmond, M.J. Role of oxidation in the neurotoxic effects of intrastriatal dopamine injections. Proc. Natl. Acad. Sci. USA 1996, 93, 1956–1961. [Google Scholar] [PubMed]
- Pierce, R.C.; Rebec, G.V. Stimulation of both D1-dopamine and D2-dopamine receptors increases behavioral activation and ascorbate release in the neostriatum of freely moving rats. Eur. J. Pharmacol. 1990, 191, 295–302. [Google Scholar] [CrossRef] [PubMed]
- Bornstein, S.R.; Yoshida-Hiroi, M.; Sotiriou, S.; Levine, M.; Hartwig, H.G.; Nussbaum, R.L.; Eisenhofer, G. Impaired adrenal catecholamine system function in mice with deficiency of the ascorbic acid transporter (SVCT2). FASEB J. 2003, 17, 1928–1930. [Google Scholar] [PubMed]
- Kuo, C.H.; Hata, F.; Yoshida, H.; Yamatodani, A.; Wada, H. Effect of ascorbic acid on release of acetylcholine from synaptic vesicles prepared from different species of animals and release of noradrenaline from synaptic vesicles of rat brain. Life Sci. 1979, 24, 911–916. [Google Scholar] [CrossRef] [PubMed]
- Sandstrom, M.I.; Rebec, G.V. Extracellular ascorbate modulates glutamate dynamics: Role of behavioral activation. BMC Neurosci. 2007, 8, 32. [Google Scholar] [CrossRef]
- Majewska, M.D.; Bell, J.A.; London, E.D. Regulation of the NMDA receptor by redox phenomena—Inhibitory role of ascorbate. Brain Res. 1990, 537, 328–332. [Google Scholar] [CrossRef] [PubMed]
- Yusa, T. Increased extracellular ascorbate release reflects glutamate re-uptake during the early stage of reperfusion after forebrain ischemia in rats. Brain Res. 2001, 897, 104–113. [Google Scholar] [CrossRef] [PubMed]
- Hara, K.; Akiyama, Y. Collagen-related abnormalities, reduction in bone quality, and effects of menatetrenone in rats with a congenital ascorbic acid deficiency. J. Bone Miner. Metab. 2009, 27, 324–332. [Google Scholar] [CrossRef] [PubMed]
- Telang, S.; Clem, A.L.; Eaton, J.W.; Chesney, J. Depletion of ascorbic acid restricts angiogenesis and retards tumor growth in a mouse model. Neoplasia 2007, 9, 47–56. [Google Scholar] [CrossRef]
- Peterkofsky, B. Ascorbate requirement for hydroxylation and secretion of procollagen—Relationship to inhibition of collagen synthesis in scurvy. Am. J. Clin. Nutr. 1991, 54, S1135–S1140. [Google Scholar]
- May, J.M.; Harrison, F.E. Role of vitamin C in the function of the vascular endothelium. Antioxid. Redox Signal. 2013, 19, 2068–2083. [Google Scholar] [CrossRef] [PubMed]
- Barnes, M.J.; Constabl, B.J.; Kodicek, E. Excretion of hydroxyproline and other amino acids in scorbutic guinea pigs. Biochim. Biophys. Acta 1969, 184, 358–365. [Google Scholar] [CrossRef] [PubMed]
- Schofield, C.J.; Ratcliffe, P.J. Oxygen sensing by HIF hydroxylases. Nat. Rev. Mol. Cell Biol. 2004, 5, 343–354. [Google Scholar] [CrossRef] [PubMed]
- Flashman, E.; Davies, S.L.; Yeoh, K.K.; Schofield, C.J. Investigating the dependence of the hypoxia-inducible factor hydroxylases (factor inhibiting HIF and prolyl hydroxylase domain 2) on ascorbate and other reducing agents. Biochem. J. 2010, 427, 135–142. [Google Scholar] [CrossRef] [PubMed]
- Tomita, S.; Ueno, M.; Sakamoto, M.; Kitahama, Y.; Ueki, M.; Maekawa, N.; Sakamoto, H.; Gassmann, M.; Kageyama, R.; Ueda, N.; et al. Defective brain development in mice lacking the HIF-1 alpha gene in neural cells. Mol. Cell. Biol. 2003, 23, 6739–6749. [Google Scholar] [CrossRef] [PubMed]
- Iyer, N.V.; Kotch, L.E.; Agani, F.; Leung, S.W.; Laughner, E.; Wenger, R.H.; Gassmann, M.; Gearhart, J.D.; Lawler, A.M.; Yu, A.Y.; et al. Cellular and developmental control of O2 homeostasis by hypoxia-inducible factor 1 alpha. Genes Dev. 1998, 12, 149–162. [Google Scholar] [CrossRef] [PubMed]
- Pate, S.K.; Lukert, B.P.; Kipp, D.E. Tissue vitamin C levels of guinea pig offspring are influenced by maternal vitamin C intake during pregnancy. J. Nutr. Biochem. 1996, 7, 524–528. [Google Scholar] [CrossRef]
- Schjoldager, J.G.; Tveden-Nyborg, P.; Lykkesfeldt, J. Prolonged maternal vitamin C deficiency overrides preferential fetal ascorbate transport but does not influence perinatal survival in guinea pigs. Br. J. Nutr. 2013, 110, 1573–1579. [Google Scholar] [CrossRef] [PubMed]
- Scaife, A.R.; McNeill, G.; Campbell, D.M.; Martindale, S.; Devereux, G.; Seaton, A. Maternal intake of antioxidant vitamins in pregnancy in relation to maternal and fetal plasma levels at delivery. Br. J. Nutr. 2006, 95, 771–778. [Google Scholar] [CrossRef] [PubMed]
- Ikonomidou, C.; Kaindl, A.M. Neuronal death and oxidative stress in the developing brain. Antioxid. Redox Signal. 2011, 14, 1535–1550. [Google Scholar] [CrossRef] [PubMed]
- Harrison, F.E.; Yu, S.S.; van den Bossche, K.L.; Li, L.; May, J.M.; McDonald, M.P. Elevated oxidative stress and sensorimotor deficits but normal cognition in mice that cannot synthesize ascorbic acid. J. Neurochem. 2008, 106, 1198–1208. [Google Scholar] [CrossRef] [PubMed]
- Burgess, N.; Maguire, E.A.; O’Keefe, J. The human hippocampus and spatial and episodic memory. Neuron 2002, 35, 625–641. [Google Scholar] [CrossRef] [PubMed]
- Giap, B.T.; Jong, C.N.; Ricker, J.H.; Cullen, N.K.; Zafonte, R.D. The hippocampus: Anatomy, pathophysiology, and regenerative capacity. J. Head Trauma Rehabil. 2000, 15, 875–894. [Google Scholar] [CrossRef] [PubMed]
- Luciana, M.; Lindeke, L.; Georgieff, M.; Mills, M.; Nelson, C.A. Neurobehavioral evidence for working memory deficits in school-aged children with histories of prematurity. Dev. Med. Child. Neurol. 1999, 41, 521–533. [Google Scholar] [CrossRef] [PubMed]
- Isaacs, E.B.; Lucas, A.; Chong, W.K.; Wood, S.J.; Johnson, C.L.; Marshall, C.; Vargha-Khadem, F.; Gadian, D.G. Hippocampal volume and everyday memory in children of very low birth weight. Pediatr. Res. 2000, 47, 713–720. [Google Scholar] [CrossRef] [PubMed]
- Tolsa, C.B.; Zimine, S.; Warfield, S.K.; Freschi, M.; Rossignol, A.S.; Lazeyras, F.; Hanquinet, S.; Pfizenmaier, M.; Hüppi, P.S. Early alteration of structural and functional brain development in premature infants born with intrauterine growth restriction. Pediatr. Res. 2004, 56, 132–138. [Google Scholar] [CrossRef] [PubMed]
- Kratzing, C.C.; Kelly, J.D.; Kratzing, J.E. Ascorbic acid in fetal rat brain. J. Neurochem. 1985, 44, 1623–1624. [Google Scholar] [CrossRef] [PubMed]
- Adlard, B.P.F.; Desouza, S.W.; Moon, S. Ascorbic acid in fetal human brain. Arch. Dis. Child. 1974, 49, 278–282. [Google Scholar] [CrossRef] [PubMed]
- Zalani, S.; Rajalakshmi, R.; Parekh, L.J. Ascorbic acid concentration of human fetal tissues in relation to fetal size and gestational age. Br. J. Nutr. 1989, 61, 601–606. [Google Scholar] [CrossRef] [PubMed]
- Biri, A.; Bozkurt, N.; Turp, A.; Kavutcu, M.; Himmetoglu, O.; Durak, I. Role of oxidative stress in intrauterine growth restriction. Gynecol. Obstet. Investig. 2007, 64, 187–192. [Google Scholar] [CrossRef]
- Berger, T.M.; Polidori, M.C.; Dabbagh, A.; Evans, P.J.; Halliwell, B.; Morrow, J.D.; Roberts, L.J.; Frei, B. Antioxidant activity of vitamin C in iron-overloaded human plasma. J. Biol. Chem. 1997, 272, 15656–15660. [Google Scholar] [CrossRef]
- Redman, C.W.; Sargent, I.L. Latest advances in understanding preeclampsia. Science 2005, 308, 1592–1594. [Google Scholar] [CrossRef] [PubMed]
- Ehrenstein, V.; Rothman, K.J.; Pedersen, L.; Hatch, E.E.; Sorensen, H.T. Pregnancy-associated hypertensive disorders and adult cognitive function among Danish conscripts. Am. J. Epidemiol. 2009, 170, 1025–1031. [Google Scholar] [CrossRef] [PubMed]
- Tuovinen, S.; Raikkonen, K.; Kajantie, E.; Leskinen, J.T.; Henriksson, M.; Pesonen, A.K.; Heinonen, K.; Osmond, C.; Barker, D.; Eriksson, J.G. Hypertensive disorders in pregnancy and intellectual abilities in the offspring in young adulthood: The Helsinki Birth Cohort Study. Ann. Med. 2012, 44, 394–403. [Google Scholar] [CrossRef] [PubMed]
- Cheng, S.W.; Chou, H.C.; Tsou, K.I.; Fang, L.J.; Tsao, P.N. Delivery before 32 weeks of gestation for maternal preeclampsia: Neonatal outcome and 2-year developmental outcome. Early Hum. Dev. 2004, 76, 39–46. [Google Scholar] [CrossRef] [PubMed]
- Many, A.; Fattal, A.; Leitner, Y.; Kupferminc, M.J.; Harel, S.; Jaffa, A. Neurodevelopmental and cognitive assessment of children born growth restricted to mothers with and without preeclampsia. Hypertens. Pregnancy 2003, 22, 25–29. [Google Scholar] [CrossRef] [PubMed]
- Mikhail, M.S.; Anyaegbunam, A.; Garfinkel, D.; Palan, P.R.; Basu, J.; Romney, S.L. Preeclampsia and antioxidant nutrients—Decreased plasma levels of reduced ascorbic acid, alpha-tocopherol and beta-carotene in women with preeclampsia. Am. J. Obstet. Gynecol. 1994, 171, 150–157. [Google Scholar] [CrossRef] [PubMed]
- Chappell, L.C.; Seed, P.T.; Kelly, F.J.; Briley, A.; Hunt, B.J.; Charnock-Jones, D.S.; Mallet, A.; Poston, L. Vitamin C and E supplementation in women at risk of preeclampsia is associated with changes in indices of oxidative stress and placental function. Am. J. Obstet. Gynecol. 2002, 187, 777–784. [Google Scholar] [CrossRef] [PubMed]
- Beazley, D.; Ahokas, R.; Livingston, J.; Griggs, M.; Sibai, B.M. Vitamin C and E supplementation in women at high risk for preeclampsia: A double-blind, placebo-controlled trial. Am. J. Obstet. Gynecol. 2005, 192, 520–521. [Google Scholar] [CrossRef] [PubMed]
- Villar, J.; Purwar, M.; Merialdi, M.; Zavaleta, N.; Ngoc, N.T.N.; Anthony, J.; de Greeff, A.; Poston, L.; Shennan, A.; WHO vitamin C and vitamin E trial group. World Health Organisation multicentre randomised trial of supplementation with vitamins C and E among pregnant women at high risk for preeclampsia in populations of low nutritional status from developing countries. BJOG 2009, 116, 780–788. [Google Scholar]
- Chappell, L.C.; Seed, P.T.; Briley, A.L.; Kelly, F.J.; Lee, R.; Hunt, B.J.; Parmar, K.; Bewley, S.J.; Shennan, A.H.; Steer, P.J.; et al. Effect of antioxidants on the occurrence of preeclampsia in women at increased risk: A randomised trial. Lancet 1999, 354, 810–816. [Google Scholar] [CrossRef] [PubMed]
- Harman, D. The aging process. Proc. Natl. Acad. Sci. USA 1981, 78, 7124–7128. [Google Scholar] [CrossRef] [PubMed]
- Mecocci, P.; Macgarvey, U.; Kaufman, A.E.; Koontz, D.; Shoffner, J.M.; Wallace, D.C.; Beal, M.F. Oxidative damage to mitochondrial-DNA shows marked age-dependent increases in human brain. Ann. Neurol. 1993, 34, 609–616. [Google Scholar] [CrossRef] [PubMed]
- Bowling, A.C.; Mutisya, E.M.; Walker, L.C.; Price, D.L.; Cork, L.C.; Beal, M.F. Age-dependend impairment of mitochondrial-function in primate brain. J. Neurochem. 1993, 60, 1964–1967. [Google Scholar] [CrossRef] [PubMed]
- Parle, M.; Dhingra, D. Ascorbic acid: A promising memory enhancer in mice. J. Pharmacol. Sci. 2003, 93, 129–135. [Google Scholar] [CrossRef] [PubMed]
- Matthews, K.; Cauley, J.; Yaffe, K.; Zmuda, J.M. Estrogen replacement therapy and cognitive decline in older community women. J. Am. Geriatr. Soc. 1999, 47, 518–523. [Google Scholar] [PubMed]
- Jacobs, D.M.; Tang, M.X.; Stern, Y.; Sano, M.; Marder, K.; Bell, K.L.; Schofield, P.; Dooneief, G.; Gurland, B.; Mayeux, R. Cognitive function in nondemented older women who took estrogen after menopause. Neurology 1998, 50, 368–373. [Google Scholar] [CrossRef] [PubMed]
- Monteiro, S.C.; Matté, C.; Bavaresco, C.S.; Netto, C.A.; Wyse, A.T.S. Vitamins E and C pretreatment prevents ovariectomy-induced memory deficits in water maze. Neurobiol. Learn. Mem. 2005, 84, 192–199. [Google Scholar] [CrossRef] [PubMed]
- Lykkesfeldt, J.; Moos, T. Age-dependent change in vitamin C status: A phenomenon of maturation rather than of ageing. Mech. Ageing Dev. 2005, 126, 892–898. [Google Scholar] [CrossRef] [PubMed]
- Tveden-Nyborg, P.; Hasselholt, S.; Miyashita, N.; Moos, T.; Poulsen, H.E.; Lykkesfeldt, J. Chronic vitamin C deficiency does not accelerate oxidative stress in ageing brains of guinea pigs. Basic Clin. Pharmacol. Toxicol. 2012, 110, 524–529. [Google Scholar] [CrossRef] [PubMed]
- Foy, C.J.; Passmore, A.P.; Vahidassr, M.D.; Young, I.S.; Lawson, J.T. Plasma chain-breaking antioxidants in Alzheimer’s disease, vascular dementia and Parkinson’s disease. QJM Mon. J. Assoc. Physicians 1999, 92, 39–45. [Google Scholar] [CrossRef]
- Grodstein, F.; Chen, J.; Willett, W.C. High-dose antioxidant supplements and cognitive function in community-dwelling elderly women. Am. J. Clin. Nutr. 2003, 77, 975–984. [Google Scholar]
- Masaki, K.H.; Losonczy, K.G.; Izmirlian, G.; Foley, D.J.; Ross, G.W.; Petrovitch, H.; Havlik, R.; White, L.R. Association of vitamin E and C supplement use with cognitive function and dementia in elderly men. Neurology 2000, 54, 1265–1272. [Google Scholar] [CrossRef] [PubMed]
- Fillenbaum, G.G.; Kuchibhatla, M.N.; Hanlon, J.T.; Artz, M.B.; Pieper, C.F.; Schmader, K.E.; Dysken, M.W.; Gray, S.L. Dementia and Alzheimer’s disease in community-dwelling elders taking vitamin C and/or vitamin E. Ann. Pharmacother. 2005, 39, 2009–2014. [Google Scholar] [CrossRef] [PubMed]
- Harrison, F.E.; Bowman, G.L.; Polidori, M.C. Ascorbic acid and the brain: Rationale for the use against cognitive decline. Nutrients 2014, 6, 1752–1781. [Google Scholar] [PubMed]
- Brookmeyer, R.; Johnson, E.; Ziegler-Graham, K.; Arrighi, H.M. Forecasting the global burden of Alzheimer’s disease. Alzheimers Dement. 2007, 3, 186–191. [Google Scholar] [CrossRef] [PubMed]
- Bowman, G.L. Ascorbic acid, cognitive function, and Alzheimer’s disease: A current review and future direction. Biofactors 2012, 38, 114–122. [Google Scholar] [CrossRef] [PubMed]
- Polidori, M.C.; Mecocci, P. Plasma susceptibility to free radical-induced antioxidant consumption and lipid peroxidation is increased in very old subjects with Alzheimer’s disease. J. Alzheimers Dis. 2002, 4, 517–522. [Google Scholar] [PubMed]
- Riviere, S.; Birlouez-Aragon, I.; Nourhashemi, F.; Vellas, B. Low plasma vitamin C in Alzheimer patients despite an adequate diet. Int. J. Geriatr. Psychiatr. 1998, 13, 749–754. [Google Scholar] [CrossRef]
- Harrison, F.E.; Allard, J.; Bixler, R.; Usoh, C.; Li, L.; May, J.M.; McDonald, M.P. Antioxidants and cognitive training interact to affect oxidative stress and memory in APP/PSEN1 mice. Nutr. Neurosci. 2009, 12, 203–218. [Google Scholar] [CrossRef] [PubMed]
- Harrison, F.E.; Hosseini, A.H.; McDonald, M.P.; May, J.M. Vitamin C reduces spatial learning deficits in middle-aged and very old APP/PSEN1 transgenic and wild-type mice. Pharmacol. Biochem. Behav. 2009, 93, 443–450. [Google Scholar] [CrossRef] [PubMed]
- Shahidi, S.; Komaki, A.; Mahmoodi, M.; Atrvash, N.; Ghodrati, M. Ascorbic acid supplementation could affect passive avoidance learning and memory in rat. Brain Res. Bull. 2008, 76, 109–113. [Google Scholar] [CrossRef] [PubMed]
- Murakami, K.; Murata, N.; Ozawa, Y.; Kinoshita, N.; Irie, K.; Shirasawa, T.; Shimizu, T. Vitamin C restores behavioral deficits and amyloid-beta oligomerization without affecting plaque formation in a mouse model of Alzheimer’s disease. J. Alzheimers Dis. 2011, 26, 7–18. [Google Scholar] [CrossRef] [PubMed]
- Harrison, F.E.; Hosseini, A.H.; Dawes, S.M.; Weaver, S.; May, J.M. Ascorbic acid attenuates scopolamine-induced spatial learning deficits in the water maze. Behav. Brain Res. 2009, 205, 550–558. [Google Scholar] [CrossRef] [PubMed]
- Ward, M.S.; Lamb, J.; May, J.M.; Harrison, F.E. Behavioral and monoamine changes following severe vitamin C deficiency. J. Neurochem. 2013, 124, 363–375. [Google Scholar] [CrossRef] [PubMed]
- Morris, M.C.; Beckett, L.A.; Scherr, P.A.; Hebert, L.E.; Bennett, D.A.; Field, T.S.; Evans, D.A. Vitamin E and vitamin C supplement use and risk of incident Alzheimer’s disease. Alzheimer Dis. Assoc. Dis. 1998, 12, 121–126. [Google Scholar] [CrossRef]
- Gray, S.L.; Anderson, M.L.; Crane, P.K.; Breitner, J.C.S.; McCormick, W.; Bowen, J.D.; Teri, L.; Larson, E. Antioxidant vitamin supplement use and risk of dementia or Alzheimer’s disease in older adults. J. Am. Geriatr. Soc. 2008, 56, 291–295. [Google Scholar] [CrossRef] [PubMed]
- Lykkesfeldt, J.; Poulsen, H.E. Is vitamin C supplementation beneficial? Lessons learned from randomised controlled trials. Br. J. Nutr. 2010, 103, 1251–1259. [Google Scholar] [CrossRef] [PubMed]
- Frei, B.; Birlouez-Aragon, I.; Lykkesfeldt, J. Authors’ perspective: What is the optimum intake of vitamin C in humans? Crit. Rev. Food Sci. Nutr. 2012, 52, 815–829. [Google Scholar] [CrossRef]
- DeAngelis, L.; Furlan, C. The effects of ascorbic acid and oxiracetam on scopolamine-induced amnesia in a habituation test in aged mice. Neurobiol. Learn. Mem. 1995, 64, 119–124. [Google Scholar] [PubMed]
- Barabás, J.; Nagy, E.; Degrell, I. Ascorbic acid in cerebrospinal fluid—A possible protection against free radicals in the brain. Arch. Gerontol. Geriatr. 1995, 21, 43–48. [Google Scholar] [CrossRef] [PubMed]
- Devore, E.E.; Kang, J.H.; Stampfer, M.J.; Grodstein, F. Total antioxidant capacity of diet in relation to cognitive function and decline. Am. J. Clin. Nutr. 2010, 92, 1157–1164. [Google Scholar] [CrossRef] [PubMed]
- LaRue, A.; Koehler, K.M.; Wayne, S.J.; Chiulli, S.J.; Haaland, K.Y.; Garry, P.J. Nutritional status and cognitive functioning in a normally aging sample: A 6-year reassessment. Am. J. Clin. Nutr. 1997, 65, 20–29. [Google Scholar] [PubMed]
- Arlt, S.; Mueller-Thomsen, T.; Beisiegel, U.; Kontush, A. Effect of one-year vitamin C and E supplementation on cerebrospinal fluid oxidation parameters and clinical course in Alzheimer’s disease. Neurochem. Res. 2012, 37, 2706–2714. [Google Scholar] [PubMed]
- Landolt, H.; Lutz, T.W.; Langemann, H.; Stauble, D.; Mendelowitsch, A.; Gratzl, O.; Honegger, C.G. Extracellular antioxidants and amino acids in the cortex of the rat: Monitoring by microdialysis of early ischemic changes. J. Cereb. Blood Flow Metab. 1992, 12, 96–102. [Google Scholar] [CrossRef] [PubMed]
- Henry, P.T.; Chandy, M.J. Effect of ascorbic acid on infarct size in experimental focal cerebral ischaemia and reperfusion in a primate model. Acta Neurochir. 1998, 140, 977–980. [Google Scholar] [CrossRef] [PubMed]
- Iwata, N.; Okazaki, M.; Xuan, M.Y.; Kamiuchi, S.; Matsuzaki, H.; Hibino, Y. Orally administrated ascorbic acid suppresses neuronal damage and modifies expression of SVCT2 and GLUT1 in the brain of diabetic rats with cerebral ischemia-reperfusion. Nutrients 2014, 6, 1554–1577. [Google Scholar] [CrossRef] [PubMed]
- Lagowska-Lenard, M.; Stelmasiak, Z.; Bartosik-Psujek, H. Influence of vitamin C on markers of oxidative stress in the earliest period of ischemic stroke. Pharmacol. Rep. 2010, 62, 751–756. [Google Scholar] [CrossRef] [PubMed]
- Myint, P.K.; Luben, R.N.; Welch, A.A.; Bingham, S.A.; Wareham, N.J.; Khaw, K.T. Plasma vitamin C concentrations predict risk of incident stroke over 10-year in 20,649 participants of the European prospective investigation into Cancer-Norfolk Prospective Population Study. Am. J. Clin. Nutr. 2008, 87, 64–69. [Google Scholar] [PubMed]
- Gale, C.R.; Martyn, C.N.; Winter, P.D.; Cooper, C. Vitamin C and risk of death from stroke and coronary heart disease in cohort of elderly people. Br. Med. J. 1995, 310, 1563–1566. [Google Scholar] [CrossRef]
- Osganian, S.K.; Stampfer, M.J.; Rimm, E.; Spiegelman, D.; Hu, F.B.; Manson, J.E.; Willett, W.C. Vitamin C and risk of coronary heart disease in women. J. Am. Coll. Cardiol. 2003, 42, 246–252. [Google Scholar] [CrossRef] [PubMed]
- Fang, J.C.; Kinlay, S.; Beltrame, J.; Hikiti, H.; Wainstein, M.; Behrendt, D.; Suh, J.; Frei, B.; Mudge, G.H.; Selwyn, A.P.; et al. Effect of vitamins C and E on progression of transplant-associated arteriosclerosis: A randomised trial. Lancet 2002, 359, 1108–1113. [Google Scholar] [CrossRef] [PubMed]
© 2014 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Hansen, S.N.; Tveden-Nyborg, P.; Lykkesfeldt, J. Does Vitamin C Deficiency Affect Cognitive Development and Function? Nutrients 2014, 6, 3818-3846. https://doi.org/10.3390/nu6093818
Hansen SN, Tveden-Nyborg P, Lykkesfeldt J. Does Vitamin C Deficiency Affect Cognitive Development and Function? Nutrients. 2014; 6(9):3818-3846. https://doi.org/10.3390/nu6093818
Chicago/Turabian StyleHansen, Stine Normann, Pernille Tveden-Nyborg, and Jens Lykkesfeldt. 2014. "Does Vitamin C Deficiency Affect Cognitive Development and Function?" Nutrients 6, no. 9: 3818-3846. https://doi.org/10.3390/nu6093818
APA StyleHansen, S. N., Tveden-Nyborg, P., & Lykkesfeldt, J. (2014). Does Vitamin C Deficiency Affect Cognitive Development and Function? Nutrients, 6(9), 3818-3846. https://doi.org/10.3390/nu6093818