Sulphate in Pregnancy


{kind=link}
{kind=link}
Abstract
:1. Introduction
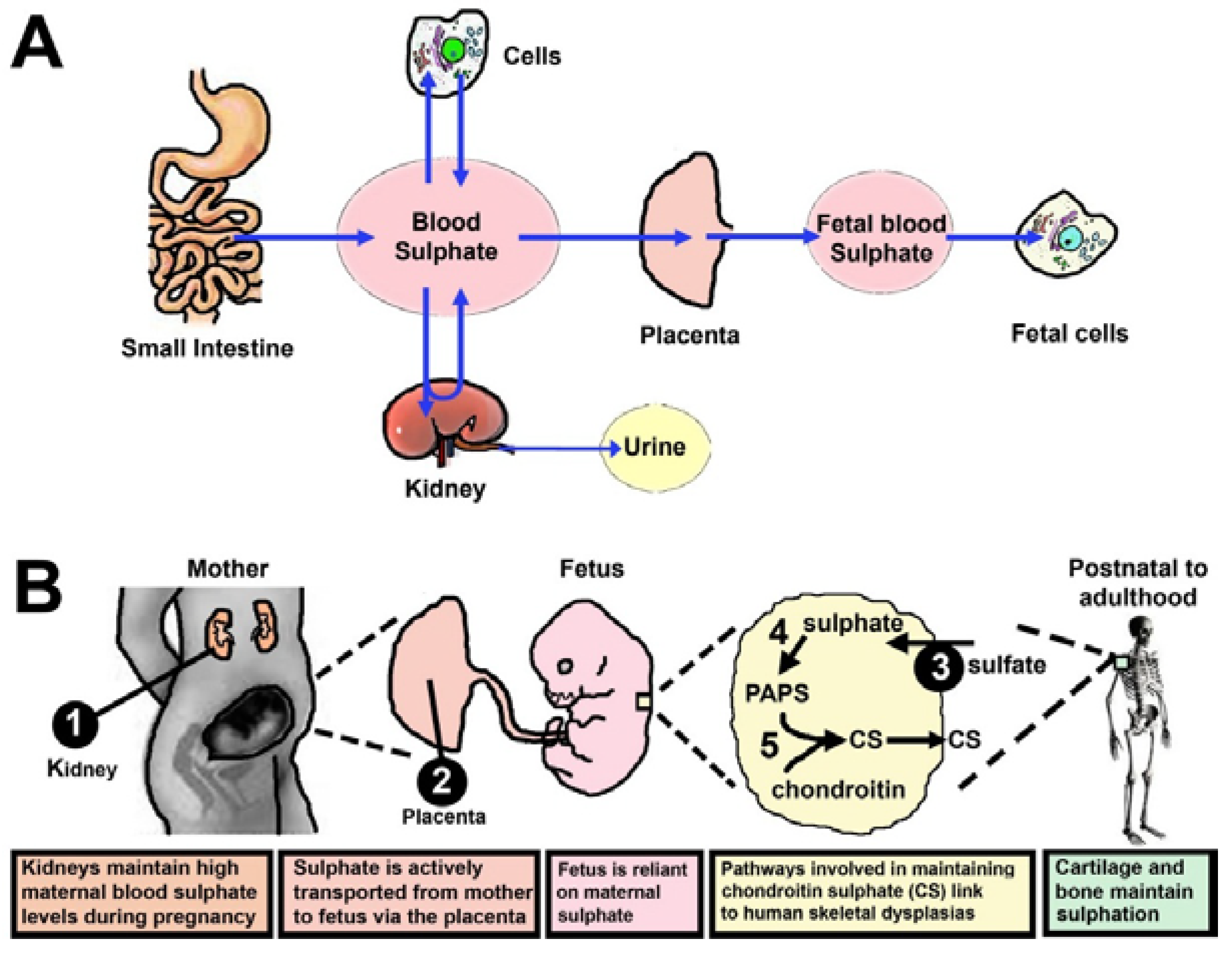
2. Sulphate is Obtained from the Diet
3. Generation of Sulphate from Intracellular Metabolism


4. Sulphate Is Supplied from Mother to Foetus
5. Reduced Sulphonation Capacity Perturbs Skeletal Growth and Development
6. Conclusion
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Dawson, P.A. Sulfate in fetal development. Semin. Cell Dev. Biol. 2011, 22, 653–659. [Google Scholar] [CrossRef] [PubMed]
- Darras, V.M.; Hume, R.; Visser, T.J. Regulation of thyroid hormone metabolism during fetal development. Mol. Cell. Endocrinol. 1999, 151, 37–47. [Google Scholar] [CrossRef] [PubMed]
- Dawson, P.A. The biological roles of steroid sulfonation. In Steroids—From Physiology to Clinical Medicine; Ostojic, S.M., Ed.; Intech: Rijeka, Croatia, 2012; pp. 45–64. [Google Scholar]
- Richard, K.; Hume, R.; Kaptein, E.; Stanley, E.L.; Visser, T.J.; Coughtrie, M.W. Sulfation of thyroid hormone and dopamine during human development: Ontogeny of phenol sulfotransferases and arylsulfatase in liver, lung, and brain. J. Clin. Endocrinol. Metab. 2001, 86, 2734–2742. [Google Scholar] [PubMed]
- Mulder, G.J.; Jakoby, W.B. Sulfation. In Conjugation Reactions in Drug Metabolism: An Integrated Approach: Substrates, Co-substrates, Enzymes and Their Interactions in Vivo and in Vitro; Mulder, G.J., Ed.; Taylor and Francis: London, UK, 1990; pp. 107–161. [Google Scholar]
- Yamaguchi, Y. Heparan sulfate proteoglycans in the nervous system: Their diverse roles in neurogenesis, axon guidance, and synaptogenesis. Semin. Cell Dev. Biol. 2001, 12, 99–106. [Google Scholar] [CrossRef] [PubMed]
- Alnouti, Y.; Klaassen, C.D. Tissue distribution and ontogeny of sulfotransferase enzymes in mice. Toxicol. Sci. 2006, 93, 242–255. [Google Scholar] [CrossRef] [PubMed]
- Stanley, E.L.; Hume, R.; Coughtrie, M.W. Expression profiling of human fetal cytosolic sulfotransferases involved in steroid and thyroid hormone metabolism and in detoxification. Mol. Cell. Endocrinol. 2005, 240, 32–42. [Google Scholar] [CrossRef] [PubMed]
- Hines, R.N.; McCarver, D.G. The ontogeny of human drug-metabolizing enzymes: Phase I oxidative enzymes. J. Pharmacol. Exp. Ther. 2002, 300, 355–360. [Google Scholar] [CrossRef] [PubMed]
- McCarver, D.G.; Hines, R.N. The ontogeny of human drug-metabolizing enzymes: Phase II conjugation enzymes and regulatory mechanisms. J. Pharmacol. Exp. Ther. 2002, 300, 361–366. [Google Scholar] [CrossRef] [PubMed]
- Dawson, P.A. Role of sulphate in development. Reproduction 2013, 146, R81–R89. [Google Scholar] [CrossRef] [PubMed]
- Smith, J.T.; Acuff, R.V. An effect of dietary sulfur on liver inorganic sulfate in the rat. Ann. Nutr. Metab. 1983, 27, 345–348. [Google Scholar] [CrossRef] [PubMed]
- Allen, H.E.; Halley-Henderson, M.A.; Hass, C.N. Chemical composition of bottled mineral water. Arch. Environ. Health 1989, 44, 102–116. [Google Scholar] [CrossRef] [PubMed]
- Florin, T.; Neale, G.; Gibson, G.R.; Christl, S.U.; Cummings, J.H. Metabolism of dietary sulphate: Absorption and excretion in humans. Gut 1991, 32, 766–773. [Google Scholar] [CrossRef] [PubMed]
- Florin, T.H.J.; Neale, G.; Goretski, S.; Cummings, J.H. The sulfate content of foods and beverages. J. Food Compos. Anal. 1993, 6, 140–151. [Google Scholar] [CrossRef]
- National Research Council. Sulfate. In Dietary Reference Intakes for Water, Potassium, Sodium, Chloride, and Sulfate; The National Academies Press: Washington, DC, USA, 2005; pp. 424–448. [Google Scholar]
- Health Canada. Sulphate. Available online: http://www.hc-sc.gc.ca/ewh-semt/pubs/water-eau/sulphate-sulfates/index-eng.php (accessed on 17 February 2015).
- U.S. Environmental Protection Agency. Drinking Water Advisory: Consumer Acceptability Advice and Health Effects Analysis on Sulfate. 2003. Available online: http://water.epa.gov/action/advisories/drinking/upload/2008_01_10_support_cc1_sulfate_healtheffects.pdf (accessed on 17 February 2015). [Google Scholar]
- Chien, L.; Robertson, H.; Gerrard, J.W. Infantile gastroenteritis due to water with high sulfate content. Can. Med. Assoc. J. 1968, 99, 102–104. [Google Scholar] [PubMed]
- Cocchetto, D.M.; Levy, G. Absorption of orally administered sodium sulfate in humans. J. Pharm. Sci. 1981, 70, 331–333. [Google Scholar] [CrossRef] [PubMed]
- Paterson, D.W.; Wahlstrom, R.C.; Libal, G.W.; Olson, O.E. Effects of sulfate in water on swine reproduction and young pig performance. J. Anim. Sci. 1979, 49, 664–667. [Google Scholar] [PubMed]
- Izzo, A.A.; Gaginella, T.S.; Capasso, F. The osmotic and intrinsic mechanisms of the pharmacological laxative action of oral high doses of magnesium sulphate. Importance of the release of digestive polypeptides and nitric oxide. Magnes. Res. 1996, 9, 133–138. [Google Scholar] [PubMed]
- Morris, M.E.; LeRoy, S.; Sutton, S.C. Absorption of magnesium from orally administered magnesium sulfate in man. J. Toxicol. Clin. Toxicol. 1987, 25, 371–382. [Google Scholar] [CrossRef] [PubMed]
- Doyle, L.W.; Crowther, C.A.; Middleton, P.; Marret, S.; Rouse, D. Magnesium sulphate for women at risk of preterm birth for neuroprotection of the fetus. Cochrane Database Syst. Rev. 2007, 18, CD004661. [Google Scholar]
- Panarelli, N.C. Drug-induced injury in the gastrointestinal tract. Semin. Diagn. Pathol. 2014, 31, 165–175. [Google Scholar] [CrossRef] [PubMed]
- Proudfoot, A.T.; Simpson, D.; Dyson, E.H. Management of acute iron poisoning. Med. Toxicol. 1986, 1, 83–100. [Google Scholar] [PubMed]
- Toblli, J.E.; Cao, G.; Oliveri, L.; Angerosa, M. Effects of iron polymaltose complex, ferrous fumarate and ferrous sulfate treatments in anemic pregnant rats, their fetuses and placentas. Inflamm. Allergy Drug Targets 2013, 12, 190–198. [Google Scholar] [CrossRef] [PubMed]
- Daniels, A.L.; Rich, J.K. The role of inorganic sulfates in nutrition. J. Biol. Chem. 1918, 36, 27–32. [Google Scholar]
- Hou, C.; Wykes, L.J.; Hoffer, L.J. Urinary sulfur excretion and the nitrogen/sulfur balance ratio reveal nonprotein sulfur amino acid retention in piglets. J. Nutr. 2003, 133, 766–772. [Google Scholar] [PubMed]
- McGarry, P.C.; Roe, D.A. Development of sulfur depletion in pregnant and fetal rats: Interaction of protein restriction and indole or salicylamide administration. J. Nutr. 1973, 103, 1279–1290. [Google Scholar] [PubMed]
- Pecora, F.; Gualeni, B.; Forlino, A.; Superti-Furga, A.; Tenni, R.; Cetta, G.; Rossi, A. In vivo contribution of amino acid sulfur to cartilage proteoglycan sulfation. Biochem. J. 2006, 398, 509–514. [Google Scholar] [CrossRef] [PubMed]
- Price, V.F.; Jollow, D.J. Effects of sulfur-amino acid-deficient diets on acetaminophen metabolism and hepatotoxicity in rats. Toxicol. Appl. Pharmacol. 1989, 101, 356–369. [Google Scholar] [CrossRef] [PubMed]
- Dawson, P.A.; Gardiner, B.; Lee, S.; Grimmond, S.; Markovich, D. Kidney transcriptome reveals altered steroid homeostasis in NaS1 sulfate transporter null mice. J. Steroid Biochem. Mol. Biol. 2008, 112, 55–62. [Google Scholar] [CrossRef] [PubMed]
- Hindmarsh, K.W.; Mayers, D.J.; Wallace, S.M.; Danilkewich, A.; Ernst, A. Increased serum sulfate concentrations in man due to environmental factors: Effects on acetaminophen metabolism. Vet. Hum. Toxicol. 1991, 33, 441–445. [Google Scholar] [PubMed]
- Morris, M.E.; Levy, G. Serum concentration and renal excretion by normal adults of inorganic sulfate after acetaminophen, ascorbic acid, or sodium sulfate. Clin. Pharmacol. Ther. 1983, 33, 529–536. [Google Scholar] [CrossRef] [PubMed]
- Ricci, J.; Oster, J.R.; Gutierrez, R.; Schlessinger, F.B.; Rietberg, B.; O’Sullivan, M.J.; Clerch, A.R.; Vaamonde, C.A. Influence of magnesium sulfate-induced hypermagnesemia on the anion gap: Role of hypersulfatemia. Am. J. Nephrol. 1990, 10, 409–411. [Google Scholar] [CrossRef] [PubMed]
- Slattery, J.T.; Levy, G. Reduction of acetaminophen toxicity by sodium sulfate in mice. Res. Commun. Chem. Pathol. Pharmacol. 1977, 18, 167–170. [Google Scholar] [PubMed]
- Waring, R.; Klovrza, L.V. Sulphur metabolism in autism. J. Nutr. Environ. Med. 2000, 10, 25–32. [Google Scholar] [CrossRef]
- Wu, Y.; Zhang, X.; Bardag-Gorce, F.; Robel, R.C.; Aguilo, J.; Chen, L.; Zeng, Y.; Hwang, K.; French, S.W.; Lu, S.C.; et al. Retinoid X receptor alpha regulates glutathione homeostasis and xenobiotic detoxification processes in mouse liver. Mol. Pharmacol. 2004, 65, 550–557. [Google Scholar] [CrossRef] [PubMed]
- Brand, E. Amino acid composition of simple proteins. Ann. NY Acad. Sci. 1946, 47, 187–228. [Google Scholar] [CrossRef] [PubMed]
- Australian Government. Nutrient Reference Values for Australia and New Zealand; NHMRC: Canberra, Australia, 2014. [Google Scholar]
- Ueki, I.; Roman, H.B.; Valli, A.; Fieselmann, K.; Lam, J.; Peters, R.; Hirschberger, L.L.; Stipanuk, M.H. Knockout of the murine cysteine dioxygenase gene results in severe impairment in ability to synthesize taurine and an increased catabolism of cysteine to hydrogen sulfide. Am. J. Physiol. Endocrinol. Metab. 2011, 301, E668–E684. [Google Scholar] [CrossRef] [PubMed]
- Gaull, G.; Sturman, J.A.; Raiha, N.C. Development of mammalian sulfur metabolism: Absence of cystathionase in human fetal tissues. Pediatr. Res. 1972, 6, 538–547. [Google Scholar] [CrossRef] [PubMed]
- Loriette, C.; Chatagner, F. Cysteine oxidase and cysteine sulfinic acid decarboxylase in developing rat liver. Experientia 1978, 34, 981–982. [Google Scholar] [CrossRef] [PubMed]
- Strott, C.A. Steroid sulfotransferases. Endocr. Rev. 1996, 17, 670–697. [Google Scholar] [CrossRef] [PubMed]
- Strott, C.A. Sulfonation and molecular action. Endocr. Rev. 2002, 23, 703–732. [Google Scholar] [CrossRef] [PubMed]
- Dawson, P.A.; McIntyre, H.D.; Petersen, S.; Gibbons, K.; Bowling, F.G.; Hurrion, E. Sulfate in human pregnancy and preterm babies: What we ought to know. J. Paed. Child Health 2014, 50, 46. [Google Scholar]
- Dawson, P.A.; Sim, P.; Simmons, D.G.; Markovich, D. Fetal loss and hyposulfataemia in pregnant NaS1 transporter null mice. J. Reprod. Dev. 2011, 57, 444–449. [Google Scholar] [CrossRef] [PubMed]
- Lind, T. Clinical chemistry of pregnancy. Adv. Clin. Chem. 1980, 21, 1–24. [Google Scholar] [PubMed]
- Dawson, P.A.; Rakoczy, J.; Simmons, D.G. Placental, renal, and ileal sulfate transporter gene expression in mouse gestation. Biol. Reprod. 2012, 87, 1–9. [Google Scholar] [CrossRef]
- Bowling, F.G.; Heussler, H.S.; McWhinney, A.; Dawson, P.A. Plasma and urinary sulfate determination in a cohort with autism. Biochem. Genet. 2012, 51, 147–153. [Google Scholar] [CrossRef] [PubMed]
- Dawson, P.A.; Beck, L.; Markovich, D. Hyposulfatemia, growth retardation, reduced fertility and seizures in mice lacking a functional NaSi-1 gene. Proc. Natl. Acad. Sci. USA 2003, 100, 13704–13709. [Google Scholar] [CrossRef] [PubMed]
- Dawson, P.A.; Gardiner, B.; Grimmond, S.; Markovich, D. Transcriptional profile reveals altered hepatic lipid and cholesterol metabolism in hyposulfatemic NaS1 null mice. Physiol. Genomics 2006, 26, 116–124. [Google Scholar] [CrossRef] [PubMed]
- Dawson, P.A.; Huxley, S.; Gardiner, B.; Tran, T.; McAuley, J.L.; Grimmond, S.; McGuckin, M.A.; Markovich, D. Reduced mucin sulfonation and impaired intestinal barrier function in the hyposulfataemic NaS1 null mouse. Gut 2009, 58, 910–919. [Google Scholar] [CrossRef] [PubMed]
- Dawson, P.A.; Steane, S.E.; Markovich, D. Behavioural abnormalities of the hyposulfataemic Nas1 knock-out mouse. Behav. Brain Res. 2004, 154, 457–463. [Google Scholar] [CrossRef] [PubMed]
- Dawson, P.A.; Steane, S.E.; Markovich, D. Impaired memory and olfactory performance in NaSi-1 sulphate transporter deficient mice. Behav. Brain Res. 2005, 159, 15–20. [Google Scholar] [CrossRef] [PubMed]
- Lee, S.; Dawson, P.A.; Hewavitharana, A.K.; Shaw, P.N.; Markovich, D. Disruption of NaS1 sulfate transport function in mice leads to enhanced acetaminophen-induced hepatotoxicity. Hepatology 2006, 43, 1241–7124. [Google Scholar] [CrossRef] [PubMed]
- Lee, S.; Kesby, J.P.; Muslim, M.D.; Steane, S.E.; Eyles, D.W.; Dawson, P.A.; Markovich, D. Hyperserotonaemia and reduced brain serotonin levels in NaS1 sulphate transporter null mice. Neuroreport 2007, 18, 1981–1985. [Google Scholar] [CrossRef] [PubMed]
- Simmons, D.G.; Rakoczy, J.; Jefferis, J.; Lourie, R.; McIntyre, H.D.; Dawson, P.A. Human placental sulfate transporter mRNA profiling identifies abundant SLC13A4 in syncytiotrophoblasts and SLC26A2 in cytotrophoblasts. Placenta 2013, 34, 381–384. [Google Scholar] [CrossRef] [PubMed]
- Rakoczy, J.; Dawson, P.A.; Simmons, D.G. Loss of placental sulphate transporter SLC13A4 causes severe developmental defects and embryonic lethality. Placenta 2014, 35, A96–A97. [Google Scholar] [CrossRef]
- Tong, M.H.; Jiang, H.; Liu, P.; Lawson, J.A.; Brass, L.F.; Song, W.C. Spontaneous fetal loss caused by placental thrombosis in estrogen sulfotransferase-deficient mice. Nat. Med. 2005, 11, 153–159. [Google Scholar] [CrossRef] [PubMed]
- Cole, D.E.; Evrovski, J. The clinical chemistry of inorganic sulfate. Crit. Rev. Clin. Lab. Sci. 2000, 37, 299–344. [Google Scholar] [CrossRef] [PubMed]
- Cole, D.E.; Baldwin, L.S.; Stirk, L.J. Increased inorganic sulfate in mother and fetus at parturition: Evidence for a fetal-to-maternal gradient. Am. J. Obstet. Gynecol. 1984, 148, 596–599. [Google Scholar] [CrossRef] [PubMed]
- Cole, D.E.; Baldwin, L.S.; Stirk, L.J. Increased serum sulfate in pregnancy: Relationship to gestational age. Clin. Chem. 1985, 31, 866–867. [Google Scholar] [PubMed]
- Tallgren, L.G. Inorganic sulphate in relation to the serum thyroxine level and in renal failure. Acta Med. Scand. 1980, 640, 1–100. [Google Scholar]
- Habuchi, H.; Habuchi, O.; Kimata, K. Sulfation pattern in glycosaminoglycan: Does it have a code? Glycoconj. J. 2004, 21, 47–52. [Google Scholar] [CrossRef] [PubMed]
- Klüppel, M. The roles of chondroitin-4-sulfotransferase-1 in development and disease. Prog. Mol. Biol. Transl. Sci. 2010, 93, 113–132. [Google Scholar] [PubMed]
- Cortes, M.; Baria, A.T.; Schwartz, N.B. Sulfation of chondroitin sulfate proteoglycans is necessary for proper Indian hedgehog signaling in the developing growth plate. Development 2009, 136, 1697–1706. [Google Scholar] [CrossRef] [PubMed]
- Rossi, A.; Bonaventure, J.; Delezoide, A.L.; Superti-Furga, A.; Cetta, G. Undersulfation of cartilage proteoglycans ex vivo and increased contribution of amino acid sulfur to sulfation in vitro in McAlister dysplasia/atelosteogenesis type 2. Eur. J. Biochem. 1997, 248, 741–747. [Google Scholar] [CrossRef] [PubMed]
- Dawson, P.A.; Markovich, D. Pathogenetics of the human SLC26 transporters. Curr. Med. Chem. 2005, 12, 385–396. [Google Scholar] [CrossRef] [PubMed]
- Cornaglia, A.I.; Casasco, A.; Casasco, M.; Riva, F.; Necchi, V. Dysplastic histogenesis of cartilage growth plate by alteration of sulphation pathway: A transgenic model. Connect. Tissue Res. 2009, 50, 232–242. [Google Scholar] [CrossRef] [PubMed]
- Forlino, A.; Piazza, R.; Tiveron, C.; Della Torre, S.; Tatangelo, L.; Bonafe, L.; Gualeni, B.; Romano, A.; Pecora, F.; Superti-Furga, A.; et al. A diastrophic dysplasia sulfate transporter (SLC26A2) mutant mouse: Morphological and biochemical characterization of the resulting chondrodysplasia phenotype. Hum. Mol. Genet. 2005, 14, 859–871. [Google Scholar] [CrossRef] [PubMed]
- Hästbacka, J.; de la Chapelle, A.; Mahtani, M.; Clines, G.; Reeve-Daly, M.P.; Daly, M.; Hamilton, B.A.; Kusumi, K.; Trivedi, B.; Weaver, A.; et al. The diastrophic dysplasia gene encodes a novel sulfate transporter: Positional cloning by fine-structure linkage disequilibrium mapping. Cell 1994, 78, 1073–1087. [Google Scholar] [CrossRef] [PubMed]
- Sugahara, K.; Schwartz, N.B. Defect in 3′-phosphoadenosine 5′-phosphosulfate synthesis in brachymorphic mice. I. Characterization of the defect. Arch. Biochem. Biophys. 1982, 214, 589–601. [Google Scholar] [CrossRef] [PubMed]
- Klassen, C.D.; Boles, J. The importance of 3′-phosphoadenosine 5′-phosphosulfate (PAPS) in the regulation of sulfation. FASEB J. 1997, 11, 404–418. [Google Scholar] [PubMed]
- Faiyaz ul Haque, M.; King, L.M.; Krakow, D.; Cantor, R.M.; Rusiniak, M.E.; Swank, R.T.; Superti-Furga, A.; Haque, S.; Abbas, H.; Ahmad, W.; et al. Mutations in orthologous genes in human spondyloepimetaphyseal dysplasia and the brachymorphic mouse. Nat. Genet. 1998, 20, 157–162. [Google Scholar] [CrossRef] [PubMed]
- Kurima, K.; Warman, M.L.; Krishnan, S.; Domowicz, M.; Krueger, R.C., Jr.; Deyrup, A.; Schwartz, N.B. A member of a family of sulfate-activating enzymes causes murine brachymorphism. Proc. Natl. Acad. Sci. USA 1998, 95, 8681–8685. [Google Scholar] [CrossRef] [PubMed]
- Xu, Z.H.; Otterness, D.M.; Freimuth, R.R.; Carlini, E.J.; Wood, T.C.; Mitchell, S.; Moon, E.; Kim, U.J.; Xu, J.P.; Siciliano, M.J.; et al. Human 3′-phosphoadenosine 5′-phosphosulfate synthetase 1 (PAPSS1) and PAPSS2: Gene cloning, characterization and chromosomal localization. Biochem. Biophys. Res. Commun. 2000, 268, 437–444. [Google Scholar] [CrossRef] [PubMed]
- Clément, A.; Wiweger, M.; von der Hardt, S.; Rusch, M.A.; Selleck, S.B.; Chien, C.B.; Roehl, H.H. Regulation of zebrafish skeletogenesis by ext2/dackel and papst1/pinscher. PLoS Genet. 2008, 4, e1000136. [Google Scholar] [CrossRef] [PubMed]
- Thiele, H.; Sakano, M.; Kitagawa, H.; Sugahara, K.; Rajab, A.; Höhne, W.; Ritter, H.; Leschik, G.; Nürnberg, P.; Mundlos, S. Loss of chondroitin 6-O-sulfotransferase-1 function results in severe human chondrodysplasia with progressive spinal involvement. Proc. Natl. Acad. Sci. USA 2004, 101, 10155–10160. [Google Scholar] [CrossRef] [PubMed]
- Ikeda, T.; Mabuchi, A.; Fukuda, A.; Hiraoka, H.; Kawakami, A.; Yamamoto, S.; Machida, H.; Takatori, Y.; Kawaguchi, H.; Nakamura, K.; Ikegawa, S. Identification of sequence polymorphisms in two sulfation-related genes, PAPSS2 and SLC26A2, and an association analysis with knee osteoarthritis. J. Hum. Genet. 2001, 46, 538–543. [Google Scholar] [CrossRef] [PubMed]
- Stevenson, D.A.; Carey, J.C.; Byrne, J.L.; Srisukhumbowornchai, S.; Feldkamp, M.L. Analysis of skeletal dysplasias in the Utah population. Am. J. Med. Genet. A 2012, 158A, 1046–1054. [Google Scholar] [CrossRef]
- Neff, M.W.; Beck, J.S.; Koeman, J.M.; Boguslawski, E.; Kefene, L.; Borgman, A.; Ruhe, A.L. Partial deletion of the sulfate transporter SLC13A1 is associated with an osteochondrodysplasia in the Miniature Poodle breed. PLoS One 2012, 7, e51917. [Google Scholar] [CrossRef] [PubMed]
- Zhao, X.; Onteru, S.K.; Piripi, S.; Thompson, K.G.; Blair, H.T.; Garrick, D.J.; Rothschild, M.F. In a shake of a lamb’s tail: Using genomics to unravel a cause of chondrodysplasia in Texel sheep. Anim. Genet. 2012, 43, 9–18. [Google Scholar] [CrossRef] [PubMed]
- Orenius, T.; Krüger, L.; Kautiainen, H.; Hurri, H.; Pohjolainen, T. The sense of coherence and its relation to health factors among patients with diastrophic dysplasia. J. Pub. Health Epidem. 2012, 4, 305–310. [Google Scholar] [CrossRef]
- Rossi, A.; Kaitila, I.; Wilcox, W.R.; Rimoin, D.L.; Steinmann, B.; Cetta, G.; Superti-Furga, A. Proteoglycan sulfation in cartilage and cell cultures from patients with sulfate transporter chondrodysplasias: Relationship to clinical severity and indications on the role of intracellular sulfate production. Matrix Biol. 1998, 17, 361–369. [Google Scholar] [CrossRef] [PubMed]
© 2015 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Dawson, P.A.; Elliott, A.; Bowling, F.G. Sulphate in Pregnancy. Nutrients 2015, 7, 1594-1606. https://doi.org/10.3390/nu7031594
Dawson PA, Elliott A, Bowling FG. Sulphate in Pregnancy. Nutrients. 2015; 7(3):1594-1606. https://doi.org/10.3390/nu7031594
Chicago/Turabian StyleDawson, Paul A., Aoife Elliott, and Francis G. Bowling. 2015. "Sulphate in Pregnancy" Nutrients 7, no. 3: 1594-1606. https://doi.org/10.3390/nu7031594
APA StyleDawson, P. A., Elliott, A., & Bowling, F. G. (2015). Sulphate in Pregnancy. Nutrients, 7(3), 1594-1606. https://doi.org/10.3390/nu7031594